
Coordination polymer hydrogels through Ag(I)-mediated spontaneous self-assembly of unsubstituted nucleobases and their antimicrobial activity†
Bhagwati Sharma,
Arup Mahata,
Sonam Mandani,
Tridib K. Sarma* and
Biswarup Pathak*
Discipline of Chemistry, School of Basic Sciences, Indian Institute of Technology Indore, Simrol, Khandwa Road, Indore-452020, India. E-mail: tridib@iiti.ac.in; biswarup@iiti.ac.in
First published on 23rd June 2016
Abstract
Spontaneous formation of metallogels through the self-assembly of unsubstituted nucleobases with Ag(I) ions is reported. Whereas adenine, cytosine, thymine and uracil form metallogels under deprotonated conditions with nanofibrillar morphology, gelation of guanine with Ag(I) occurs only under acidic conditions through the self-organization of nanoscale metal–organic particles. In situ reduction of Ag salts occurs in Ag–pyrimidine gels to yield Ag nanoparticles decorated on the gel nanofibers. The Ag–nucleobase hydrogels showed excellent antimicrobial properties against both Gram positive as well as Gram negative bacteria.
Introduction
The self-assembly of small molecules into ordered structures such as rods, spheres, tapes or fibres constitutes the platform for functional nanostructured materials.1 Conventional coordination chemistry is a key enabler for the fabrication of such nano- and micro-superstructures.1,2 The formation of metallogels represents one of the most elegant bottom-up approaches towards these functional materials, where discrete metal coordination complexes of low-molecular-weight ligands or well defined coordination polymers enable entangled coordination networks that can immobilize large volumes of solvents and guest molecules by different mechanisms. These integrative self-assembled structures with metallic elements incorporated into the viscoelastic gel matrix harness catalytic, magnetic as well as stimuli-responsive properties that are crucial for various technological applications including catalysis, sensing and optics.3 Metal–nucleobase interactions are extensively studied for elucidating the role of metal ions in the structure and function of nucleic acids and genetic information transfer.4 Additionally, nucleobases offer multiple sites for metal binding and plethora of potential non-covalent interactions stimulating nucleobase–metal interactions as supramolecular motif for crystal engineering, bioMOFs as well as coordination polymer soft materials.5,6 Metal ion mediated self-assembly of guanosine and their derivatives into G-quadraplex and subsequent hydrogel formation have been exploited in supramolecular chemistry and nanotechnology.6 However, derivatization of nucleobases is generally required to induce gelation7 and metallogels involving pure nucleobases as ligands have not been realized.Here we report our serendipitous observation of Ag(I) ion induced spontaneous self-assembly of deprotonated or protonated nucleobases into coordination polymer hydrogels under ambient conditions. Simple addition of Ag+ ions to a deprotonated solution of adenine, cytosine, thymine or uracil resulted in 3D fibrous network through simultaneous coordination polymerization and hydrogen bonding interactions. Conversely, Ag(I) induced hydrogelation of guanine was observed only under acidic conditions.
Results and discussion
An opaque metallogel (Ag–A) was spontaneously formed when AgNO3 (100 mM) was added to a deprotonated adenine solution (80 mM) in aqueous medium at pH 10.5 and 25 °C, as ascertained physically by observing the absence of flow upon inversion of tube (Fig. S1†). The self-assembled formation of the metallogels ensued spontaneously within one minute and did not require the involvement of external stimuli such as heating/cooling or ultrasonication. The fibrous nanostructures were formed even at very low concentrations of the two components (Fig. S2†), however higher concentration of Ag+ salt and adenine was required for stable gel formation. In contrast, gelation did not take place in acidic medium owing to protonation of nitrogen atoms in adenine, clearly supporting the view that adenine forms the hydrogel mainly by coordination bonds. The Ag–A gels collapsed to form a clear solution under acidic conditions showing pH responsiveness. Upon adjusting the pH from acidic to basic the recovery of the hydrogel did not occur, instead a white precipitate of AgOH was obtained. Interestingly, Ag(I) induced hydrogelation was not observed in case of deprotonated guanine, as addition of AgNO3 to guanine (at pH 12.1) resulted only in a white precipitate (Ag–G) (Scheme 1).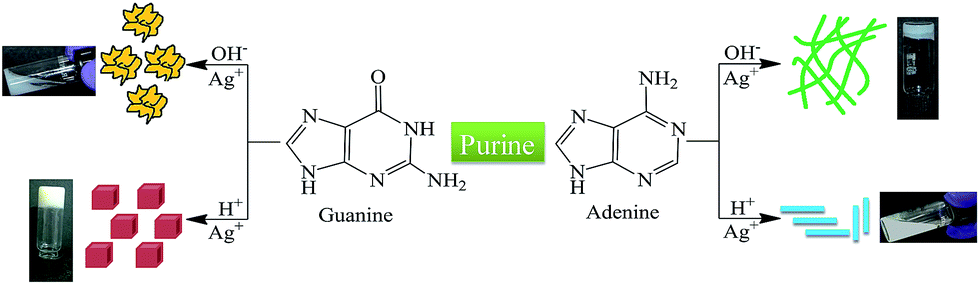 | ||
| Scheme 1 Schematic representation for the formation of coordination polymer superstructures with purine nucleobases. | ||
Transmission electron microscopy (TEM) image of the Ag–A xerogel revealed the formation of homogeneous short individual nanofibers (Fig. 1a) with thickness between 20 to 35 nm and several micrometer in length. Scanning electron microscopy (SEM) image of the Ag–A hydrogel showed highly entangled nanofibrillar morphology (Fig. 1b). Probably the fibers observed in the image consist of bundles of gelator aggregates that understandably immobilize water molecules in the network. On the other hand, the SEM image of Ag–G precipitate (Fig. 1c) revealed the formation of fibrous aggregates that assembled to give a large flower like morphology. However the formation of an entangled network primarily responsible for entrapping solvent molecules in hydrogels was not observed. The powder X-ray diffraction pattern (Fig. S3†) of the Ag–A hydrogel suggested high crystallinity with a layered assembly structure8 having an interlayer separation of 12.5 Å. This value is quite close to the value of the length of two adenine units connected through hydrogen bonding with water (12.7 Å). The infrared spectrum (FTIR) of the Ag–A complex suggested the involvement of N9 of adenine in binding with Ag+ ions (Fig. S4†). Similarly, the crystallinity and nature of bonding in Ag–G precipitate was evaluated using powder XRD and FTIR (Fig. S6†). From oscillatory strain rheometry studies of Ag–A metallogels, it was evident that initially G′ (storage moduli) was significantly higher than G′′ (loss moduli) upto a strain of 51%, beyond which a crossover to liquid like regime was evident (Fig. 1d). Dynamic frequency sweep rheological measurement showed that G′ is always greater than G′′ by several factors in the frequency range of 0.1 to 100 s−1, confirming the dominance of elastic behaviour of the gel over its viscous nature (Fig. 1e).
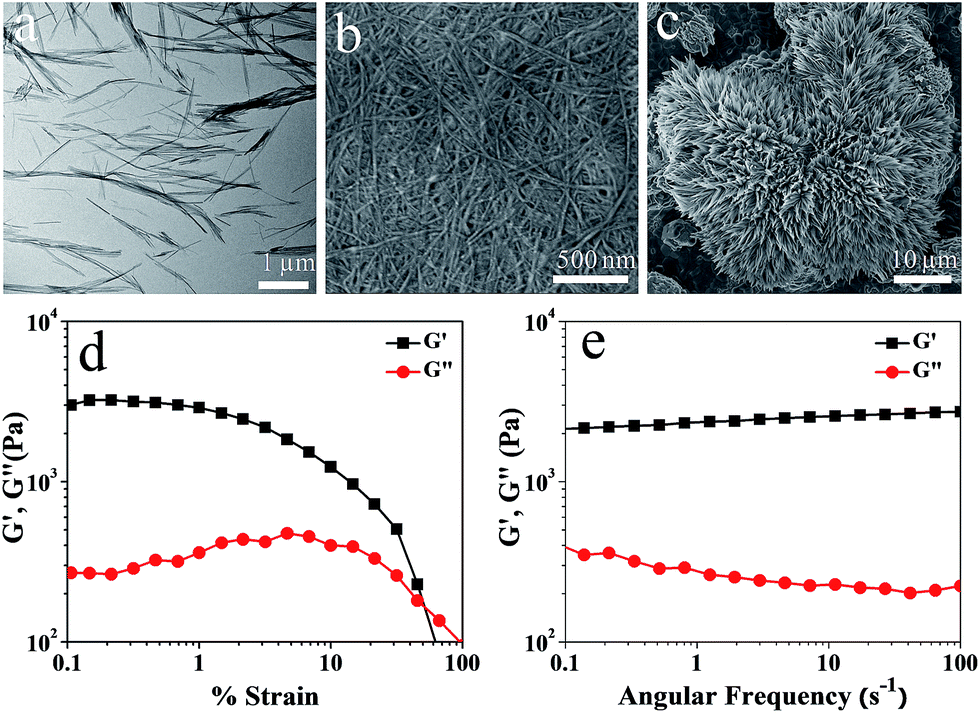 | ||
| Fig. 1 (a) TEM image and (b) FESEM image of the Ag–A hydrogel. (c) FESEM image of Ag–G precipitate. (d) Strain sweep and (e) frequency sweep rheological investigation (at a constant strain of 1%) of Ag–A hydrogel. | ||
Several control experiments were performed to realize the conditions for the Ag–A metallogel formation. It was observed that the molar concentration of Ag+ should be higher than adenine for stable gel formation (Table S1†). Further, counterions with hard base characteristics did not significantly influence the gelation (Fig. S7†). The role of water in the gel formation was crucial as only precipitation was observed in pure methanol, DMF or DMF–methanol mixture (Fig. S8 and S9†). On the other hand spontaneous gelation took place in mixed solvent systems having water as a component (Fig. S10, S11 and Table S2†) suggesting extensive hydrogen bonding network in the metallogels propagated by water molecules. Adenosine or N9-methyladenine could not form metallogels with AgNO3 under deprotonated conditions, suggesting that any substitution at the N9 position was detrimental towards hydrogel formation. The results suggest the probable involvement of N9 in adenine for coordination to metal ion that directs the coordination polymer formation.
Next, the Ag(I) induced formation of metallogels were evaluated for pyrimidine nucleobases. Under deprotonated conditions, all three bases cytosine (C), thymine (T) and uracil (U) formed stable self-standing hydrogels. TEM and SEM analyses of the Ag–C, Ag–T and Ag–U xerogels revealed the formation of dense entangled networks of nanofibers (Fig. S12†). The binding of Ag+ ions to nucleobases as well as crystallinity of the hydrogels were confirmed by FTIR (Fig. S13 and S14†) and X-ray diffraction studies (Fig. S15†) respectively. Rheological investigation of the three hydrogels suggested that each of the three materials had solid like viscoelastic properties (Fig. S16†). Further spontaneous gelation was observed in a tri- or tetra-component system involving combination of two or three nucleobase coordinating with Ag+ ions in aqueous alkaline medium, except for when guanine (G) was present. Electron microscopic studies (Fig. 2a, b, S19 and S20†) and rheological studies (Fig. S21–S24†) illustrated the formation of 3D entangled networks and solid like viscoelastic properties in these multicomponent metallogels. The presence of guanine in a mixture of nucleobases, when coordinated with Ag+, led to precipitation of the composite (Fig. S25 and S26†).
 | ||
| Fig. 2 FESEM image of (a) tri-component Ag–A–T hydrogel (b) tetra-component Ag–A–T–U hydrogel (c) hybrid nanoscale cubic particles in Ag–pG hydrogel and (d) Ag–pA precipitates with rod shaped particles. | ||
Conversely in acidic medium (pH < 1), protonated guanine formed a stable hydrogel spontaneously at room temperature in coordination with AgNO3 (Ag–pG). On the other hand, protonated adenine resulted in the formation of a white precipitate (Ag–pA), whereas clear solutions were obtained with the three protonated pyrimidine bases (cytosine, thymine and uracil). FESEM studies of the Ag–pG xerogel (Fig. 2c) suggested that cross-linking of irregularly interconnected cubic metal–organic particles (average dimension ∼ 200 nm) resulted in the gel formation instead of typical fibrous structures,9 spanning large void spaces and sustaining the gel matrix. On the other hand, the Ag–pA precipitate formed in acidic medium comprised of rod shaped particles with dimensions in the micrometer regime (Fig. 2d). Although solid-like (G′ > G′′) behavior in low strain amplitudes and little frequency dependence of the moduli in the linear viscoelastic regime was observed by rheology studies in case of Ag–pG hydrogels (Fig. S27†), the gels are noticeably softer compared to the Ag–A gels in alkaline medium (storage moduli of Ag–A and Ag–pG are 3.06 and 0.03 kPa in the linear regime respectively). This can be due to structural inhomogeneity in Ag–pG hydrogel rendered by cross-linked nanoscale particles rather than fibril networks observed in metallogels under alkaline medium.
Labile Ag(I)-based supramolecular complexes are known to demonstrate high anion responsiveness.10 Ag–nucleobase hydrogels also showed quick chemical response to several anions causing gel–sol transformations. For example, the addition of aqueous solutions of KBr, KI or Na2S to the Ag–thymine gel resulted in the formation of precipitates in the form of AgBr, AgI or Ag2S (Fig. S29†). This implies the breaking of Ag–T coordination bond in presence of halides or sulfides. However removal of precipitates by filtration followed by addition of AgNO3 to the supernatant solution resulted in the return of the gel state, showing the chemo-reversibility of the response.
The synthesis or incorporation of colloidal metal nanoparticles in a gel matrix has recently gained significance because generating such a system is an attempt to integrate nanoparticles into unconventional environments, with the aim of producing hybrid materials.11 The in situ formation of metallic nanoparticles into a hydrogel without an external reducing agent notably increases its potential applications.12 These metal nanoparticles–hydrogel nanocomposite materials can be used for various applications such as catalysis, sensors, drug delivery, biological labeling as well as antimicrobial agents. Several studies have been performed on the formation of metal nanoparticle–hydrogel composites through in situ reduction of the metal ions by the gelator molecule.11,13 For instance, Nandi and co-workers have used the interaction of riboflavin and melamine together with the reducing ability of riboflavin moiety for the formation of Ag nanoparticles incorporated hydrogel.13 In case of nucleotides, the sugar moiety is known to induce reduction resulting in the formation of metal nanoparticles. However there are no reports of pure nucleobases acting as reducing agents leading to metal nanoparticles from metal salts. In the present case, in situ generation of Ag nanoparticles was observed in Ag–C, Ag–T and Ag–U hydrogels (Fig. 3a) on incubating the hydrogels for 48 hours at room temperature. The gels were not disrupted during the formation of Ag nanoparticles, suggesting that excess Ag+ ions present in the hydrogel matrix were reduced without disturbing the fibrous network. Ag nanoparticles exhibited their characteristic surface plasmon resonance band with the peak maxima at 460 nm in case of Ag–T and Ag–U hydrogels and 484 nm in Ag–C (Fig. 3b). The powder XRD spectrum of the hydrogels (Fig. S15†) exhibited distinctive reflections at 2θ values of 38.2° and 44.4° corresponding to the (111) and (200) planes of Ag14 in addition to diffraction peaks owing to crystalline structure of the gels. TEM images of the xerogels showed small and nearly spherical Ag nanoparticles having an average size of 4 ± 1.2 nm, decorated along the fibers of the hydrogels (Fig. 3c–e). Under similar conditions, no nanoparticle formation was observed in the Ag–A gel (Fig. S30†), suggesting the probable involvement of the carbonyl groups in cytosine, thymine or uracil in the reduction of the metal salt to metal nanoparticles.
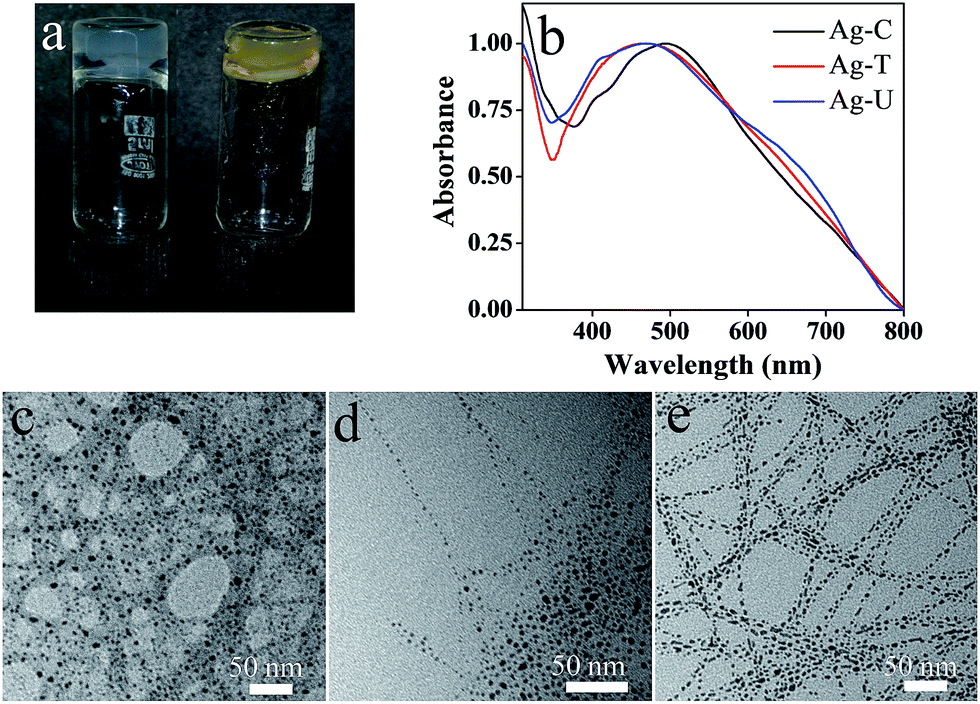 | ||
| Fig. 3 (a) Digital image of freshly prepared Ag–T hydrogel (white) and after 120 hours (yellow). (b) Solid state UV-visible spectrum of Ag–C, Ag–T and Ag–U hydrogels. (c), (d) and (e) TEM images of the Ag nanoparticles formed within the Ag–C, Ag–T and Ag–U hydrogels respectively. | ||
Generation of single crystals of the Ag–nucleobase metallogels for understanding the exact nature of coordination environment was not successful, therefore density functional theory (DFT) studies were pursued to access the nature of bonding in these materials. Based on the binding energy of Ag(I) at available binding sites in deprotonated nucleobases (Table S3†), the most stable nucleobase–Ag monomeric units were configured as depicted in Fig. 4a, b and S31.† Ag+ ions bind to adenine, cytosine, thymine and uracil through their most preferred N-atoms (N9 of adenine, N1 of cytosine and N3 of thymine and uracil). The second nitrogen atom of the nucleobase is therefore free to bind to another Ag, thus propagating the polymeric network. Again, N3, O2, O4 and O4 sites for adenine, cytosine, uracil and thymine respectively remained free for H-bonding with water which assists in hydrogel formation. In contrast, Ag is most stable in the bridge position of N7 and O6 sites in case of deprotonated guanine. Therefore, second nucleobase unit can bind to Ag only through the bridge position which prevents the formation of polymeric network beyond a dimer. Since the O6 is also involved in bonding with Ag+ ions, so the formation of polymeric network via H-bonding is not feasible in this case. The most energetically stable nucleobase–Ag dimeric configurations in alkaline conditions (Fig. 4c, d and S32†) suggested that whereas Ag–G complex cannot extend beyond a dimeric unit, the other four Ag–nucleobase complexes could be extended further to give polymeric chains. Based on the DFT calculations and experimental studies, we propose the following structure for the Ag–A hydrogel under alkaline condition (Fig. 4e). The polymer chain is extended by the coordination of Ag+ ions to adenine units at the N9 and N7 positions and the N1 position of each adenine unit is hydrogen bonded with water molecules. The structure is further stabilized through intermolecular hydrogen bonding between hydrogen of the NH2 group in one adenine unit with N3 of another adenine. Similarly the nature of bonding in Ag–guanine metallogels in acidic environment can be predicted using DFT studies (Fig. S33 and S34†).
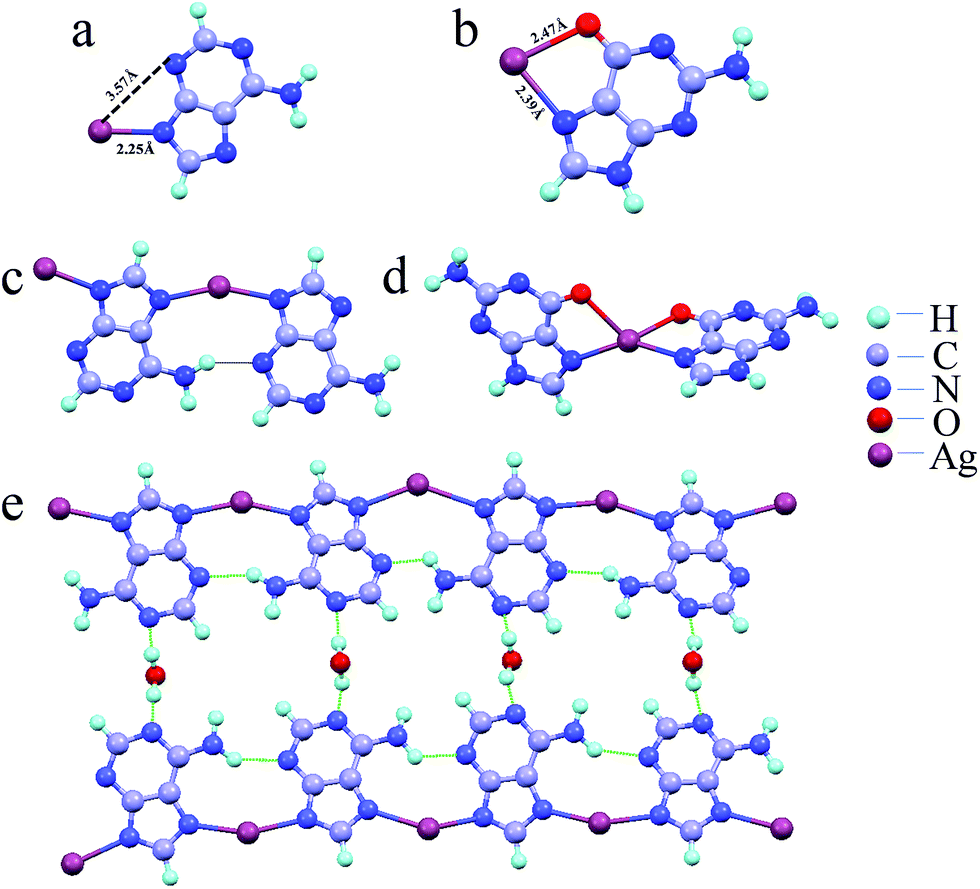 | ||
| Fig. 4 Most stable monomeric units of (a) Ag–A and (b) Ag–G. (c) Ag–A dimeric unit that could be extended to form infinite coordination polymer. (d) Ag–G dimeric unit showing no further extension possible for one-dimensional growth. (e) Proposed structure of the polymeric Ag–A hydrogel. | ||
Since ancient times, silver and its compounds are known to exhibit broad spectrum bactericidal activity and have been used to treat a variety of infections such as burn wounds, arthroplasty as well as to prevent bacterial colonization on prostheses, catheters, dental materials etc.15 Therefore, it was imperative for us to study the antimicrobial activity of the synthesized hydrogels. The antimicrobial activity of the Ag–nucleobase hydrogels was tested against the Gram negative, rod shaped bacteria Escherichia coli (E. coli) and the Gram positive bacterium, Staphylococcus aureus (S. aureus), using the cylinder-plate or cup-plate method. It was observed that for both the microbes, the hydrogels showed excellent antimicrobial activity that prevented their growth in a particular region, known as zone of inhibition (Fig. 5). As observed from the diameter of the zone of inhibition it was found that the Ag–uracil gel was the most effective of the four hydrogels, whereas the Ag–adenine hydrogel was the least effective. The average diameter of the zone of inhibition for E. coli was 12 ± 0.5, 14 ± 0.7, 15 ± 0.6 and 18 ± 1.0 mm, respectively for the Ag–adenine, Ag–cytosine, Ag–thymine and Ag–uracil gels, when 40 μL of the gel was used. On the other hand, the zone of inhibition for S. aureus was 13 ± 0.6, 17 ± 0.8, 18 ± 1.1 and 21 ± 1.2 mm, for the Ag–adenine, Ag–cytosine, Ag–thymine and Ag–uracil gels respectively, when the same amount of gel was used (40 μL). The effect of the amount of the hydrogels on the growth of the microbes was studied by varying the amount of the hydrogels and the results suggested that the antimicrobial activity of the hydrogels was directly dependent on the amount of the hydrogels (Tables S4 and S5†).
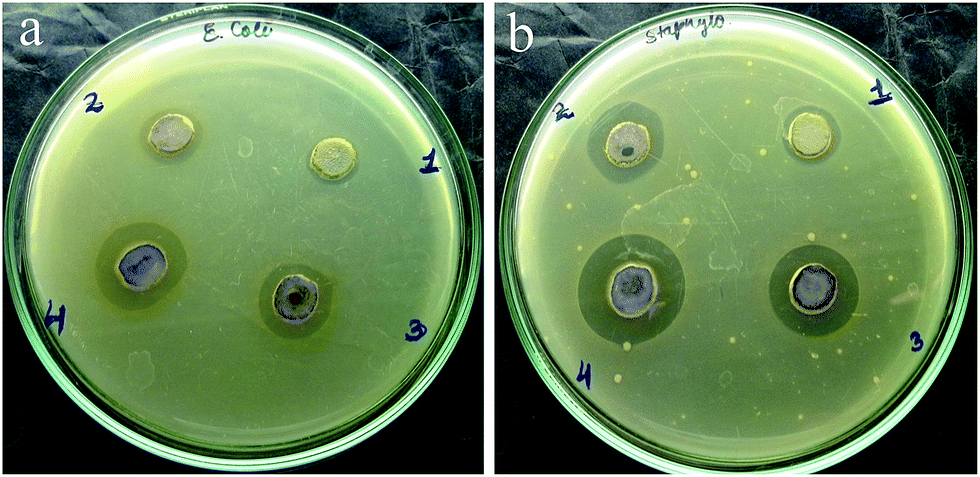 | ||
| Fig. 5 Antimicrobial activity of the four hydrogels against (a) E. coli and (b) Staphyllococcus aureus. (1) – Ag–A gel, (2) – Ag–C gel, (3) – Ag–T gel and (4) – Ag–U gel. | ||
The release of the antimicrobial agent, i.e. Ag+ ions from the hydrogels was studied in physiological buffered saline using inductively coupled plasma (ICP) method to understand the trends in the antimicrobial activity of the hydrogels. From the ICP analysis, it was found that the concentration of the released silver species in case of Ag–A hydrogel was 11.2 μg mL−1 and 20.6 μg mL−1 respectively after one and six hours. On the other hand the concentration of the released silver species in case of Ag–U gel was 18.1 μg mL−1 and 31.3 μg mL−1 after one and six hours respectively. These results clearly indicated that the better activity of the Ag–U gel was due to higher concentration of the antimicrobial species released from the Ag–U hydrogel in solution. A relatively lower concentration of Ag+ ions was released from the Ag–A gel, resulting in a poorer antimicrobial activity. The presence of Ag nanoparticles in the Ag–U gel might contribute towards the higher release of silver ions in the Ag–U gel as compared to the Ag–A gel.
The exact mechanism of antimicrobial activity of the silver based hydrogels is not very clear and is largely debated.15b,16 However, according to the most widely accepted mechanism, silver ions are released from the hydrogels which can interact with the thiol groups of many vital enzymes present in the cell wall of the microbes and inactivate them.16 The bacterial cells in contact with silver take up the silver ions, thus inhibiting several functions of the cells. Then reactive oxygen species are generated, possibly through the inhibition of a respiratory enzyme by silver ions, which attack the bacterial cells. Further, silver being a soft acid can react with soft bases such as phosphorous, a major component of DNA in the microbes and cause damage to the DNA, thereby inhibiting the growth of the microorganisms.
Conclusions
In summary, the possibility of using unsubstituted nucleobases as a low molecular weight gelator for metallogels through spontaneous coordination with Ag(I) adds to the repertoire of complex molecular architectural design using basic biological building blocks. The simple biomediated metal-coordinate gels will provide inspiration for design of soft materials for recognition events, porous materials as well as catalysis. Further the in situ generation of metal nanoparticles in the gel matrix opens up broad applications in catalysis and nanoelectronics.17 The exhibition of excellent antimicrobial activity by hydrogels generated using basic biological building units indicate their potential for use in biomedical field. Further applications of these metallogels for (photo)catalytic organic transformations and encapsulation of biomolecules for biological applications are in progress in our laboratory.Acknowledgements
The authors are grateful to Dr Deepa Dey and Dr Anand Nighojkar for helpful scientific inputs. This work was supported by research funding from SERB, Department of Science and Technology, India (SR/S1/PC-32/2010). B. S. and S. M. acknowledge research fellowships from UGC, India and CSIR, India respectively. The authors thank IIT Indore for lab and computation facilities, SIC, IIT Indore for instrumentation facilities, SAIF, NEHU, Shillong and Department of Physics, Savitribai Phule Pune University for TEM facilities.Notes and references
- (a) A. M. Spokoyny, D. Kim, A. Sumrein and C. A. Mirkin, Chem. Soc. Rev., 2009, 38, 1218–1227 RSC; (b) A. Carné, C. Carbonell, I. Imaz and D. Maspoch, Chem. Soc. Rev., 2011, 40, 291–305 RSC; (c) C. He, D. Liu and W. Lin, Chem. Rev., 2015, 115, 11079–11108 CrossRef CAS PubMed; (d) I. W. Hamley, Angew. Chem., Int. Ed., 2003, 42, 1692–1712 CrossRef CAS PubMed.
- (a) S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC; (b) I. Imaz, M. Rubio-Martínez, J. An, I. Solé-Font, N. L. Rosi and D. Maspoch, Chem. Commun., 2011, 47, 7287–7302 RSC; (c) A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540–1567 RSC; (d) I. Imaz, M. Rubio-Martínez, W. J. Saletra, D. B. Amabilino and D. Maspoch, J. Am. Chem. Soc., 2009, 131, 18222–18223 CrossRef CAS PubMed; (e) S. Saha, G. Das, J. Thote and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 14845–14851 CrossRef CAS PubMed; (f) W. Lin, W. J. Rieter and K. M. L. Taylor, Angew. Chem., Int. Ed., 2009, 48, 650–658 CrossRef CAS PubMed; (g) M. Cametti, M. Cetina and Z. Džolić, Dalton Trans., 2015, 44, 7223–7229 RSC; (h) J. Schiller, J. V. Alegre-Requena, E. Marqués-López, R. P. Herrera, J. Casanovas, C. Aléman and D. Díaz Díaz, Soft Matter, 2016, 12, 4361–4374 RSC; (i) T. Feldner, M. Häring, S. Saha, J. Esquena, R. Banerjee and D. Díaz Díaz, Chem. Mater., 2016, 28, 3210–3217 CrossRef CAS.
- (a) J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982–986 CrossRef CAS PubMed; (b) B. Xing, M. F. Choi and B. Xu, Chem.–Eur. J., 2002, 8, 5028–5032 CrossRef CAS PubMed; (c) S. Sarkar, S. Dutta, S. Chakraborti, P. Bairi and T. Pal, ACS Appl. Mater. Interfaces, 2014, 14, 6308–6316 CrossRef PubMed; (d) O. R. Evans and W. Lin, Acc. Chem. Res., 2002, 35, 511–522 CrossRef CAS PubMed; (e) P. Chen, Q. Li, S. Grindy and N. H. Andersen, J. Am. Chem. Soc., 2015, 137, 11590–11593 CrossRef CAS PubMed.
- (a) N. V. Hud, Nucleic Acid–Metal Ion Interactions, RSC Publishing, Cambridge, 2009 Search PubMed; (b) A. Sigel, H. Siegel and R. K. O. Sigel, Interplay Between Metal Ions and Nucleic Acids, Springer, Dordrecht, 2012 CrossRef; (c) G. H. Clever, C. Kaul and T. Carrel, Angew. Chem., Int. Ed., 2007, 46, 6226–6236 CrossRef CAS PubMed.
- (a) L. Berti and G. A. Burley, Nat. Nanotechnol., 2008, 3, 81–87 CrossRef CAS PubMed; (b) S. Verma, A. K. Mishra and J. Kumar, Acc. Chem. Res., 2010, 43, 79–91 CrossRef CAS PubMed; (c) G. Beobide, O. Castillo, A. Luque and S. Pérez-Yáñez, CrystEngComm, 2015, 17, 3051–3059 RSC; (d) J. An, O. K. Farha, J. T. Hupp, E. Pohl, J. I. Yeh and N. L. Rosi, Nat. Commun., 2012, 3, 604 CrossRef PubMed; (e) J. An, C. M. Shade, D. A. Chengelis-Czegan, S. Petoud and N. L. Rosi, J. Am. Chem. Soc., 2011, 133, 1220–1223 CrossRef CAS PubMed; (f) R. Nishiyabu, N. Hashimoto, T. Cho, K. Watanabe, T. Yasunaga, A. Endo, K. Kaneko, T. Niidome, M. Murata, C. Adachi, Y. Katayama, M. Hashizume and N. Kimizuka, J. Am. Chem. Soc., 2009, 131, 2151–2158 CrossRef CAS PubMed; (g) H. Wei, B. Li, Y. Du, S. Dong and E. Wang, Chem. Mater., 2007, 19, 2987–2993 CrossRef CAS; (h) S. Sivakova and S. J. Rowan, Chem. Soc. Rev., 2005, 34, 9–21 RSC; (i) H. Liang, Z. Zhang, Q. Yuan and J. Liu, Chem. Commun., 2015, 51, 15196–15199 RSC; (j) P. Amo-Ochoa and F. Zamora, Coord. Chem. Rev., 2014, 276, 34–58 CrossRef CAS.
- (a) N. Sreenivasachary and J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5938–5943 CrossRef CAS PubMed; (b) J. Dash, A. J. Patil, R. N. Das, F. L. Dowdall and S. Mann, Soft Matter, 2011, 7, 8120–8126 RSC; (c) R. N. Das, Y. P. Kumar, S. Pagoti, A. J. Patil and J. Dash, Chem.–Eur. J., 2012, 18, 6008–6014 CrossRef CAS PubMed; (d) J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS PubMed.
- K. Araki and I. Yoshikawa, Top. Curr. Chem., 2005, 256, 133–165 CrossRef CAS PubMed.
- P. K. Sukul and S. Malik, Soft Matter, 2011, 7, 4234–4241 RSC.
- T. D. Hamilton, D. K. Bučar, J. Baltrusaitis, D. R. Flanagan, Y. Li, S. Ghorai, A. V. Tivanski and L. R. MacGillivray, J. Am. Chem. Soc., 2011, 133, 3365–3371 CrossRef CAS PubMed.
- (a) G. O. Lloyd and J. W. Steed, Nat. Chem., 2011, 1, 437–442 CrossRef PubMed; (b) Q. Liu, Y. Wang, W. Li and L. Wu, Langmuir, 2007, 23, 8217–8223 CrossRef CAS PubMed; (c) H. J. Kim, W. C. Zin and M. Lee, J. Am. Chem. Soc., 2004, 126, 7009–7014 CrossRef CAS PubMed.
- (a) M. Cametti and Z. Džolić, Chem. Commun., 2014, 50, 8273–8286 RSC; (b) P. Thoniyot, M. J. Tan, A. A. Karim, D. J. Young and X. J. Loh, Adv. Sci., 2015, 2, 1400010 CrossRef PubMed.
- R. N. Mitra and P. K. Das, J. Phys. Chem. C, 2008, 112, 8159–8166 CrossRef CAS.
- S. Chaterjee and A. K. Nandi, Chem. Commun., 2011, 47, 11510–11512 RSC.
- T. Jiao, H. Guo, Q. Zhang, Q. Peng, Y. Tang, X. Yan and B. Li, Sci. Rep., 2015, 5, 11873 CrossRef CAS PubMed.
- (a) Y. Liu, W. Ma, W. Liu, C. Li, Y. Liu, X. Jiang and Z. Tang, J. Mater. Chem., 2011, 21, 19214–19218 RSC; (b) W. K. Jung, H. C. Koo, K. W. Kim, S. Shin, S. H. Kim and Y. H. Park, Appl. Environ. Microbiol., 2008, 74, 2171–2178 CrossRef CAS PubMed; (c) C. Lossaso, S. Belluco, V. Cibin, P. Zavagnin, I. Mičtić, F. Gallocchio, M. Zanella, L. Bregoli, G. Biancotto and A. Ricci, Front. Microbiol., 2014, 5, 227 Search PubMed; (d) I. Chakraborthy, T. Udayabhaskararao, G. K. Deepesh and T. Pradeep, J. Mater. Chem. B, 2013, 1, 4059–4064 RSC; (e) F. Xu, H. Padhy, M. Al-Dossari, G. Zhang, A. R. Behzad, U. Stingl and A. Rothenberger, J. Mater. Chem. B, 2014, 2, 6406–6411 RSC.
- (a) S. Prabhu and E. K. Poulose, Int. Nano Lett., 2012, 2, 32 CrossRef; (b) L. Cui, P. Chen, S. Chen, Z. Yuan, C. Yu, B. Ren and K. Zhang, Anal. Chem., 2013, 85, 5436–5443 CrossRef CAS PubMed.
- (a) B. O. Okesola, S. K. Suravaram, A. Parkin and D. K. Smith, Angew. Chem., Int. Ed., 2016, 55, 183–187 CrossRef CAS PubMed; (b) H. B. Aiyappa, S. Saha, P. Wadge, R. Banerjee and S. Kurungot, Chem. Sci., 2015, 6, 603–607 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11137h |
| This journal is © The Royal Society of Chemistry 2016 |