
Specific and highly sensitive spectrofluorimetric method for determination of febuxostat in its tablets and real human plasma. Application to stability studies†
Mahmoud A. Omar*a,
Abdel-Maaboud I. Mohamedb,
Sayed M. Derayeaa,
Mohamed A. Hammada and
Abobakr A. Mohameda
aAnalytical Chemistry Department, Faculty of Pharmacy, Minia University, Minia, Egypt. E-mail: momar1971g@yahoo.com
bPharmaceutical Analytical Chemistry Department, Faculty of Pharmacy, Assiut University, Assiut, Egypt
First published on 27th July 2016
Abstract
A simple, rapid, specific and highly sensitive spectrofluorimetric method has been developed for determination of febuxostat (FEB) in its tablets and real human plasma. The proposed method is based on the investigation of the fluorescence spectral behavior of FEB in an aqueous acidic system. The fluorescence intensity was measured at 400 nm after excitation at 335 nm. The fluorescence–concentration plot was rectilinear over the range 0.5–20.0 ng ml−1, with a limit of detection of 38.9 pg ml−1 and limit of quantification of 117.3 pg ml−1. The high sensitivity of the proposed method permits its application for the determination of FEB in real human plasma with good percentage recovery (81.73 ± 2.61). The application of the proposed method was further extended to stability studies of FEB after exposure to different forced degradation conditions, such as acidic, alkaline and oxidative conditions, according to ICH guidelines. A proposal for the degradation pathways was postulated.
1. Introduction
Febuxostat (FEB), chemically known as 2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-methylthiazole-5-carboxylic acid, is a novel non-purine selective inhibitor of xanthine oxidase, and it is used for the treatment of hyperuricemia in patients with chronic gout.1,2 Gout is a type of arthritis in which uric acid, a naturally occurring substance in the body, builds up in the joints and causes sudden attacks of redness, swelling, pain, and heat in one or more joints. At present, it is primarily treated by promoting uric acid excretion and restraining the formation of uric acid. Allopurinol, an inhibitor of xanthine oxidase with a purine-like structure, has long been the main option for gout treatment because of its ability to inhibit uric acid production. However, allopurinol inhibits other enzymes involved in purine and pyrimidine metabolism, and causes rare serious hypersensitivity reactions, such as Stevens–Johnson syndrome or toxic epidermal necrolysis, as well as various drug interactions. Thus, its use is restricted in some patients.3Several analytical techniques were developed for the determination of FEB in biological fluids and pharmaceutical formulations, including HPLC,4–11 thin-layer chromatography10,12 and spectrophotometric13–16 methods. Although HPLC methods are sufficiently sensitive, the limitation with them is that they require a robust extraction procedure as the extracted samples contain a large quantity of phospholipids, further, they are time-consuming due to a long run time. Also, it was noticed that the reported spectrophotometric methods for the determination of FEB have low sensitivity.
To date, two fluorimetric methods17,18 were published concerning the determination of FEB in tablets dosage form with 800 and 400 times less sensitive than the proposed method, respectively, and only one fluorimetric method19 was published concerning the determination of FEB in plasma with limit of quantitation of 50 ng ml−1, which is 420 times less sensitive than the proposed method (limit of quantification 0.117 ng ml−1). In our method, the limit of detection and limit of quantitation, which are indicative of the sensitivity of the method are lower than those of other reported methods, thus the proposed method considered to be the most sensitive method to date. The high sensitivity of the proposed method expands the applicability to bulk drug, dosage form and biological fluids. To the best of our knowledge, up till now nothing has been published concerning stability indicating study for the determination of FEB. Thus, the proposed method can be considered as a simple and fast alternative to the already existing stability-indicating HPLC procedures.
The current study was aimed to develop and validate a simple, rapid, specific and highly sensitive spectrofluorimetric method for determination of FEB in tablets and human plasma; utilizing the native fluorescence of FEB in aqueous acidic solution. The present study is simpler and more time saving with superior sensitivity in comparison with the reported methods for the analysis of FEB. The proposed method was fully validated according to ICH guidelines.20 Furthermore, the proposed method was extended to establish the inherent stability of the studied drug under different stress conditions such as alkaline, acidic, oxidative and photolytic conditions. Therefore, the proposed method is superior to the previously reported methods for determination of febuxostat in terms of its higher sensitivity and stability-indication capability.
2. Experimental
2.1. Apparatus
A Perkin Elmer LS 45 luminescence spectrometer (United Kingdom) equipped with 150 watt xenon arc lamp and 1 cm quartz cell was used. Slit width for both monochromators were set at 10 nm. The spectrometer is connected to a PC computer loaded with the FL WINLAB™ software. Spectronic™ Genesys™ 2 PC ultraviolet/visible spectrophotometer (Milton Roy Co, USA) with matched 1 cm quartz cell was connected to PC loaded with Winspec™ application software, Laboratory centrifuge 4000 c/s (Bremsen ECCO, Germany), MLW type thermostatically controlled water bath (Memmert GmbH, Schwabach, Germany) was used for heating purposes and a Hanna pH Meter (Romania) was used for pH adjustments.2.2. Material and chemicals
All chemicals used were of analytical grade. Febuxostat pure sample (FEB, 99.8%, Batch No. R 0191-02) was kindly provided by Hikma Pharma Co., Egypt.Sodium dodecyl sulfate (SDS), Tween-80, methanol, acetone, dimethyl formamide, acetic acid, sodium hydroxide, hydrochloric acid and sulfuric acid, all were purchased from (El-Nasr chemical Co., Cairo, Egypt), beta-cyclodextrin (β-CD, Sigma, St. Louis, MO, USA), cetyl trimethyl ammonium bromide (CTAB, Danochemo a subsidiary of Ferrosan, manufacturing chemists, Copenhagen, Denmark), acetonitrile (Merck, Germany), dimethyl sulfoxide (Riedel-deHäen, Germany). The authors got the permission for using plasma sample of human volunteers from Minia University Hospital, Minia, Egypt, according to institutional guidelines. In all cases, informed written consent was obtained from all participants. Human plasma samples were kept frozen at −20 °C until assay after gentle thawing.
The tablets were obtained from local pharmacies: Feburic® tablets labeled to contain 80 mg febuxostat per tablet (batch #1083001), product of Hikma Pharma Co., Egypt.
2.3. Standard solutions
An accurately weighed amount (10 mg) of FEB was quantitatively transferred into a 100 ml calibrated flask, dissolved in 20 ml methanol with the aid of an ultrasonic bath, and then completed to volume with the same solvent to obtain a stock solution of 0.1 mg ml−1. The stock solution was further diluted with methanol to obtain working solutions in the concentration range of 0.5–20 ng ml−1. All solutions were stored in a refrigerator and found to be stable for at least 10 days without alteration.2.4. General procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 659 g-force for 10 min. The clear supernatant was carefully transferred into a clean tube and further processed according to the general procedure. A blank experiment was carried out simultaneously omitting the drug. The relative fluorescence intensity was plotted against the final concentration of the drug. Subsequently, the corresponding regression equation was derived.659 g-force for 10 min. Then 1.0 ml of plasma obtained was quantitatively transferred to a 100 ml calibrated flask and completed to volume with methanol, then the solution was mixed by vortex for 5 min and centrifuged at 18659 g-force for 10 min. The clear supernatant was carefully transferred into a clean tube. Then the general procedure was followed. This procedure was repeated three times to obtain intraday and interday assay.
659 g-force for 10 min. The clear supernatant was carefully transferred into a clean tube and further processed according to the general procedure. A blank experiment was carried out simultaneously omitting the drug. The relative fluorescence intensity was plotted against the final concentration of the drug. Subsequently, the corresponding regression equation was derived.659 g-force for 10 min. Then 1.0 ml of plasma obtained was quantitatively transferred to a 100 ml calibrated flask and completed to volume with methanol, then the solution was mixed by vortex for 5 min and centrifuged at 18659 g-force for 10 min. The clear supernatant was carefully transferred into a clean tube. Then the general procedure was followed. This procedure was repeated three times to obtain intraday and interday assay.2.4.5.1. Procedure for alkaline and acidic degradation. Aliquots of methanolic FEB stock solutions (equivalent to 1.0 μg of the drug) were transferred into a series of small tubes; 1.0 ml of either 2 M NaOH or 2 M HCl were added. The solutions were heated in thermostatically controlled water bath at 60 °C for different time intervals (15–90 min or 15–60 min for acid or base hydrolysis, respectively). The forced degradation in either acidic or basic media was performed in the dark to exclude the possible degradative effect of light. At the specified time, the contents of each tube were cooled, neutralized to pH 7.0 and transferred into a series of 10.0 ml volumetric flasks. The volumes were completed with distilled water to achieve a concentration of 100 ng ml−1 of FEB. 1.0 ml of each solution was then transferred into 10.0 ml volumetric flask and the “General analytical procedure” was performed. The drug concentration was derived either from the previously plotted calibration graph, or using the regression equation.
2.4.5.2. Procedure for oxidative degradation. Aliquots of the methanolic FEB stock solutions (equivalent to 1.0 μg of the drug), were transferred into a series of 10 ml volumetric flasks; different volumes (0.5, 1.0, 2.0, 3.0 and 4.0 ml) of 30% H2O2 were added, and then kept in the dark for 30 min at room temperature. The solutions were then heated on a boiling water bath for 5 min to get rid of excess hydrogen peroxide, and then cooled and completed to the volume with distilled water. 1.0 ml of each solution was transferred into 10 ml volumetric flasks and the “General analytical procedure” were performed.
2.4.5.3. Procedure for photolytic degradation. Aliquots of appropriate methanolic FEB standard solutions (equivalent to 1.0 μg of the drug) were transferred into a series of 10 ml volumetric flasks and completed to volume with methanol. The volumetric flasks were exposed to direct artificial day light for different time intervals (0.5–120 h). 1.0 ml of each solution was transferred into 10.0 ml volumetric flask and the “General analytical procedure” was performed.
3. Results and discussion
Febuxostat containing a thiazole moiety which imparts a weak native fluorescence in aqueous solution measured at 400 nm after an excitation at 335 nm. In acidic aqueous solution, the fluorescence intensity of FEB was enhanced nearly 20-fold in comparison with its native fluorescence intensity in aqueous medium, (Fig. 1). FEB exhibits two excitation wavelengths of 315 and 335 nm. A wavelength of 335 nm was selected as the optimum excitation wavelength since it gave emission spectrum with best reproducibility and linearity and nearly the same sensitivity. Practically, when trying to make calibration of FEB using 315 nm as the excitation wavelength, it was found that the results were not reproducible and the calibration plot was not rectilinear over the used concentration range. This was due to high blank reading (5.0 M HCl per dist. water) at 400 nm by excitation at 315 nm.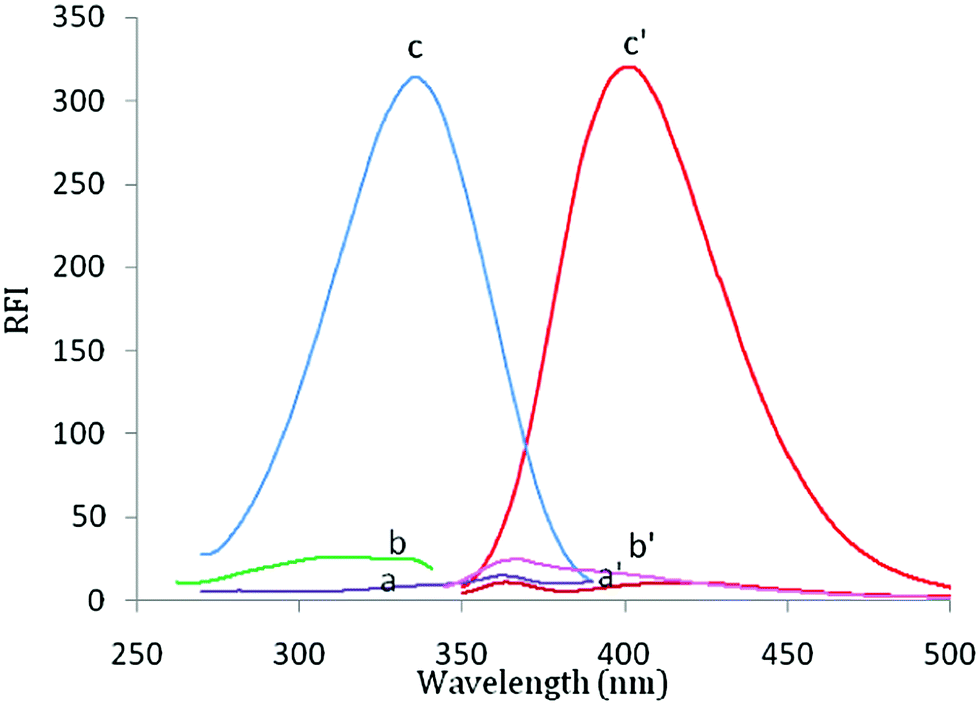 | ||
| Fig. 1 Fluorescence spectra of FEB (20 ng ml−1) in; (c and c′) aqueous acidic system, (b and b′) aqueous solution and (a and a′) blank of aqueous acidic system, where: (a–c) are the excitation spectra and (a′–c′) are the emission spectra. | ||
FEB has a pKa value of 3.3, this may explain the reason for the maximum RFI observed in acidic solution, at which the studied drug is mostly present in its protonated form, and also, thus make the carboxylic group presents in its un dissociated form, and consequently, its electrons capable to conjugate with the thiazole ring, resulting in increase fluorescence intensity. As, it is reported that the fluorescence intensity for protonated species is always higher than that for neutral species.21,22 Therefore, its intrinsic fluorescence intensity was enhanced (20-fold) in the presence of acidic solution.23,24
As a consequence, we aimed to utilize this emission band of FEB, in order to explore a new methodology for the analysis of FEB in tablets and human plasma. Different experimental parameters affecting the fluorescence intensities of FEB were carefully studied and optimized. Such factors were changed individually, where others kept constant. Furthermore, the developed method was applied to establish the inherent stability of FEB under different stress conditions.
3.1. Optimization of experimental conditions
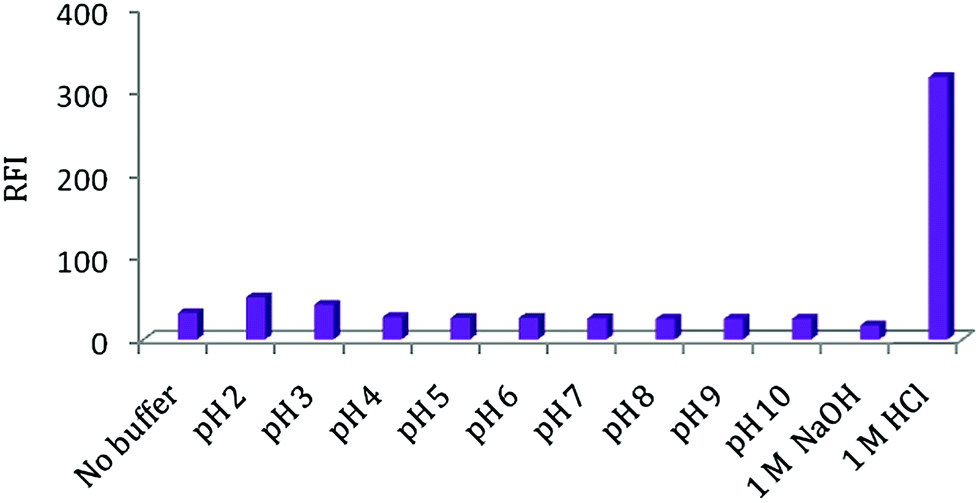 | ||
| Fig. 2 Effect of pH, 1 M NaOH and 1 M HCl on RFI of FEB (20 ng ml−1). | ||
| Acid | RFI |
|---|---|
| Hydrochloric | 313 |
| Sulphuric | 282 |
| Perchloric | 298 |
| Phosphoric | 190 |
| Acetic | 53 |
3.2. Validation of the proposed method
The developed methods were validated according to ICH guidelines of the validation of analytical methods20 regarding linearity, limits of detection (LOD), limits of quantification (LOQ), accuracy, precision and robustness.| Parameter | FEB |
|---|---|
| λex (nm) | 335 |
| λem (nm) | 400 |
| Concentration range (ng ml−1) | 0.5–20.0 |
| LOD (pg ml−1) | 38.90 |
| LOQ (pg ml−1) | 117.30 |
| Determination coefficients (r2) | 0.997 |
| Correlation coefficient (r) | 0.9985 |
| Slope | 15.93 |
| Intercept | 9.75 |
| SD the intercept (Sa) | 0.187 |
| SD of slope (Sb) | 0.24 |
| LOQ = 10Sa/b |
| LOD = 3.3Sa/b |
Accuracy and precision were further examined through statistical analysis27 of the results obtained by the proposed and reported method16 using Student's t-test and variance ratio F-test showed no significant differences between the two methods regarding accuracy and precision, respectively (Table 4). Furthermore, the validity of the suggested method was checked by applying the standard addition technique, which was performed by adding known amounts of the studied drug to a known concentration of the commercial product (Table 5). According to the results revealed in Tables 4 and 5, good accuracy was observed for the proposed methods and there is no interference from the frequently encountered tablet excipients.
3.3. Pharmaceutical applications
The proposed method gave satisfactory results with the studied drug in its marketed product, Feburic® tablets, as its pharmaceutical formulation was subjected to the analysis of its FEB contents by the proposed and the reported methods.16 The label claim percentages were 98.10 ± 0.92% for the proposed fluorimetric method (Table 4). These results were compared with those obtained from the reference method by statistical analysis using Student's t-test and F-test and showed no significant differences at 95% confidence level proving similar accuracy and precision in the determination of FEB by proposed method.3.4. Application to human plasma
The high sensitivity of the proposed method permits the in vitro and in vivo determination of FEB in human plasma without interference from the components of plasma. To ensure that the plasma components do not interfere with the proposed method, a blank experiment was carried out by applying the same procedures on plasma sample free from the studied drug. It was found that plasma sample did not has any significant fluorescence intensity at the specified proposed method conditions (ESI Fig. 4†).For in vitro study the concentration of FEB was computed from its responding regression equations. The obtained mean recovery values were 99.22 ± 5.0 and 100.82 ± 3.50%, for intra and inter-day assay, respectively. For in vivo study; FEB is absorbed after its oral administration tablet (80 mg) producing a mean peak plasma concentration of 2.5 μg ml−1 in 1.5 h.6 The following equation24 was used in calculating the % recovery of the FEB in human plasma samples:
% recovery in vivo = delivery in vivo × % recovery in vitro/delivery in vitro where; % recovery in vivo is % recovery for drug in human sample, delivery in vivo is concentration of the drug in human sample, % recovery in vitro is % recovery for drug in spiked human sample and delivery in vitro is the concentration of the drug in spiked human sample.
The calculated % recoveries in vivo using the proposed method in human plasma sample by intra and inter-day assay were 81.53 ± 3.76 and 81.93 ± 1.70%, respectively, (Table 6). It can be seen that the proposed method have the suitable sensitivity for the analysis of FEB in real human plasma.
| Assay | Delivery in vitro (ng ml−1) | % recovery in vitroa | Delivery in vivo (ng ml−1) | % recovery in vivoa |
|---|---|---|---|---|
| a Mean of three determinations. | ||||
| Intra-day assay | 2.35 | 93.86 ± 1.90 | 1.93 | 77.2 ± 1.89 |
| 5.20 | 104.00 ± 0.57 | 4.20 | 84.0 ± 1.16 | |
| 9.98 | 99.80 ± 0.30 | 8.34 | 83.4 ± 2.25 | |
| Inter-day assay | 2.44 | 97.60 ± 2.05 | 2.00 | 80.0 ± 2.60 |
| 5.23 | 104.60 ± 1.15 | 4.16 | 83.2 ± 1.08 | |
| 10.02 | 100.26 ± 0.45 | 8.26 | 82.6 ± 0.66 | |
4. Application to stability studies
The proposed spectrofluorimetric method was applied to study the stability of FEB under different stress conditions recommended by ICH guidelines20 including acidic, alkaline, oxidative and photolytic conditions. The results of the forced degradation studies are summarized as follows:4.1. Acidic degradation
Initially 5.0 ml 1 M hydrochloric acid was used at ambient temperature. No degradation was observed for up to 3 h, but upon heating with 2.0 M HCl in water bath for 60 min at 60 °C, about 20.6% of the original drug was degraded.4.2. Alkaline degradation
FEB was found to be insusceptible to alkaline degradation; upon heating the drug sample with 1 ml 2.0 M NaOH for up to 90 min at 60 °C, no marked degradation was observed, proving the stability of FEB under alkaline stress conditions. This observation was coincident with the results that have been previously published.104.3. Oxidative degradation
Degradation of FEB under oxidative stress conditions was also studied, considerable degradation of FEB was observed under oxidative conditions. It was found that treating the drug with H2O2 solution at room temperature, according to the procedure described above, resulted in immediate degradation of FEB. The amounts of degraded drug were dependent on the volume of H2O2 added; 25.2% degradation was observed upon using 4 ml 30% H2O2 solution. The oxidative degradation of FEB is proposed to proceed via oxidation of the nitrogen and sulfur atoms yielding the corresponding N-oxide and sulfoxide derivative, respectively leading to a decrease of the RFI value of FEB. Oxidation of tertiary amino group into the corresponding oxide and sulfur atoms into sulfoxide derivative were previously reported in the literature.23,284.4. Photolytic degradation
Methanolic solution of FEB was stable to sun-light even after exposure for 72 h.This present study gives an indication about the stability of FEB toward alkaline and photo-degradations, but appreciable degradation of FEB was carried out under oxidative and acidic conditions (Table 7).
| Condition | Time (h) | % of degradation products |
|---|---|---|
| 2 M HCl, 60 °C | 1.0 | 20.6 |
| 2 M NaOH, 60 °C | 1.5 | — |
| 4 ml, 30% (w/v) H2O2, room temp. | 0.5 | 25.2 |
| Photodegradation (artificial day light) | 120.0 | — |
A proposal of the possible degradation pathways of FEB was postulated (Fig. 3).
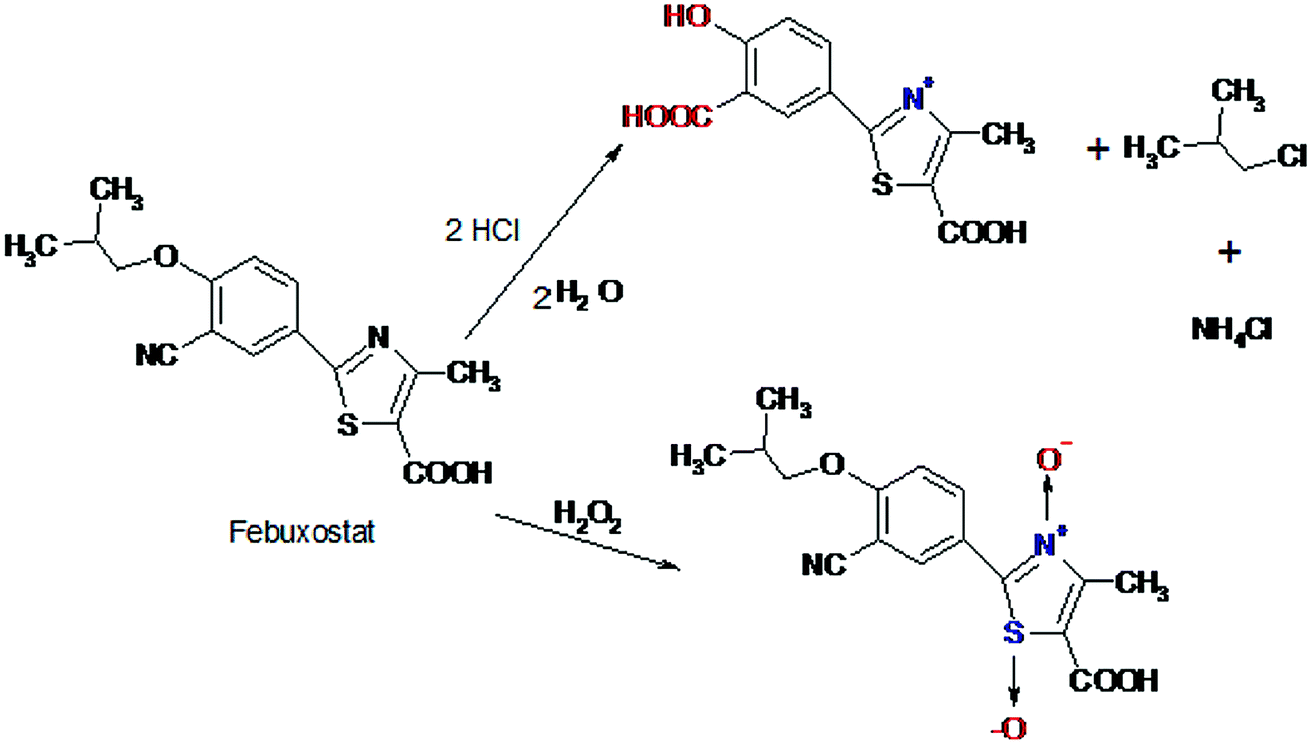 | ||
| Fig. 3 Proposed pathway of acidic and oxidative degradation of FEB. | ||
5. Conclusion
A simple, fast, specific and highly sensitive spectrofluorimetric method has been developed for the determination of FEB in real human plasma. The developed method offered a number of features including enhanced sensitivity, high recovery, simple operation process and short analysis time as well as low cost and eco-friendly than the reported methods. Taking into account, the proposed spectrofluorimetric method is the most sensitive method for the determination of FEB in real human plasma up till now. Therefore, the proposed method not only enabled the determination of FEB in real human plasma, but also with higher sensitivity than the reported methods. Additionally, the developed method has been successfully applied, for stability studies of FEB as a novel stability-indicating method. Finally, the method could be useful for determination of FEB in biological samples and could have clinical applications for patients receiving this drug.References
- M. A. Becker, H. R. Schumacher, Jr, R. L. Wortmann, P. A. MacDonald, D. Eustace, W. A. Palo, J. Streit and N. Joseph-Ridge, N. Engl. J. Med., 2005, 353, 2450–2461 CrossRef CAS PubMed.
- S. C. Sweetman, Martindale: the complete drug reference, Pharmaceutical Press, London, 38th edn, 2016 Search PubMed.
- Y. Takano, K. Hase-Aoki, H. Horiuchi, L. Zhao, Y. Kasahara, S. Kondo and M. A. Becker, Life Sci., 2005, 76, 1835–1847 CrossRef CAS PubMed.
- N. Cooper, R. Khosravan, C. Erdmann, J. Fiene and J. W. Lee, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2006, 837, 1–10 CrossRef PubMed.
- G. Ashwini, N. Aravindsai, T. Karnaker Reddy and A. Anand Kumar, J. Pharma Res., 2012, 5, 1224–1227 Search PubMed.
- B. R. Chandu, K. Kanala, N. T. Hwisa, P. Katakam and M. Khagga, SpringerPlus, 2013, 2, 194 CrossRef PubMed.
- O. Lukram, S. Parmar and A. Hande, Drug Test. Anal., 2013, 5, 492–499 CrossRef CAS PubMed.
- H. Wang, P. Deng, X. Chen, L. Guo and D. Zhong, Biomed. Chromatogr., 2013, 27, 34–38 CrossRef PubMed.
- T. Zhang, Y. Sun, P. Zhang, J. Gao, S. Wang and Z. He, Biomed. Chromatogr., 2013, 27, 137–141 CrossRef CAS PubMed.
- P. G. Sunitha and K. Ilango, Eur. J. Chem., 2014, 5, 545–549 CrossRef.
- U. P. Panigrahy and A. S. K. Reddy, Int. J. Pharma Sci. Res., 2015, 6, 798–807 CAS.
- I. A. Ramallo, S. A. Zacchino and R. L. Furlan, Phytochem. Anal., 2006, 17, 15–19 CrossRef CAS PubMed.
- P. Bagga, M. Salman, H. Siddiqui, A. M. Ansari, T. Mehmood and K. Singh, Int. J. Pharma Sci. Res., 2011, 2, 2655–2659 CAS.
- H. L. Sameer and M. R. Bhalekar, J. Pharma Res., 2011, 4, 3122–3123 Search PubMed.
- M. Sheth, S. Joshi and M. Patel, Int. J. Pharma Sci. Res., 2012, 3, 1621–1624 CAS.
- R. Spandana, E. Pushpa Latha and J. Susmitha, Int. J. Pharm. Pharm. Res., 2015, 3, 38–46 Search PubMed.
- N. Modi, S. Patel, A. Patel, R. Parmar, V. Shah and D. Shah, Inventi Impact: Pharm Anal. Qual. Assur., 2013, 1013–1015 Search PubMed.
- S. Thakar, B. Marolia, S. Sahah and D. Shah, Inventi Impact: Pharm Anal. Qual. Assur., 2013, 863–876 Search PubMed.
- H. R. Kanani, Y. M. Desai, D. M. Rathod, R. R. Parmar and D. A. Shah, Inventi Impact: Pharm Anal. Qual. Assur., 2013, 82–95 Search PubMed.
- ICH Harmonized Tripartite Guideline, Validation of Analytical Procedures: Text and Methodology, Q2(R1), Current Step 4 Version, Parent Guidelines on Methodology Dated November 6 1996, Incorporated in November 2005, http://www.ich.org/LOB/media/MEDIA417.pdf, accessed 15 February 2008.
- L. Ohannesian, Handbook of pharmaceutical analysis, Marcel Dekker, New York, Basel, 2002, electronic version Search PubMed.
- M. I. Walash, F. Belal, N. El-Enany, M. Eid and R. N. El-Shaheny, Luminescence, 2011, 26, 670–679 CrossRef CAS PubMed.
- R. N. El-Shaheny, J. Fluoresc., 2012, 22, 1587–1594 CrossRef CAS PubMed.
- S. M. Derayea, M. A. Omar, I. M. Mostafa and M. A. Hammad, RSC Adv., 2015, 5, 78920–78926 RSC.
- M. Pesez and J. Bartos, Colorimetric and fluorimetric analysis of organic compounds and drugs, Marcel Dekker Inc., New York, 1974 Search PubMed.
- D. A. Skoog, D. M. West, F. J. Holler and S. R. Crouch, Fundamentals of analytical chemistry, Saunders College Publishing, Philadelphia, PA, 8th edn, 2004 Search PubMed.
- J. Miller and J. Miller, Statistics and chemometrics for analytical chemistry, Pearson Education, Harlow, UK, 5th edn, 2005 Search PubMed.
- M. I. Walash, F. Belal, N. El-Enany, M. Eid and R. N. El-Shaheny, J. Fluoresc., 2011, 21, 1659–1667 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11246c |
| This journal is © The Royal Society of Chemistry 2016 |