
Defect states and morphological evolution in mechanically processed ZnO + xC nanosystems as studied by EPR and photoluminescence spectroscopy
Abstract
An evolution of electron paramagnetic resonance (EPR) and photoluminescence (PL) spectra of various active states (hydrogen donor DH EPR centers, Zn vacancy-related EPR centers 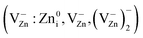 , EPR DS centers from shallow donors (g = 1.9640) in ZnOW, Mn2+ ions in ZnOZB, C EPR centers in carbon nanoparticles, forming the near-band-edge (NBE) PL emission, PL emission typical for Zn-, O- and N-enriched ZnOW particles, as well as oxidized carbon nanodots (OCN)) was observed in the mixtures of ZnO + xC nanoparticles during prolonged high-energy mechanical processing (MP) in a hermetically sealed grinding chamber. The results of EPR and PL spectroscopy, X-ray diffraction analysis, as well as morphological analysis by means of atomic force microscopy (AFM) and laser particle sizer (LPS) measurements show a wide variety of interrelated series–parallel processes in the samples with increasing MP processing time (tMP). These processes include: (a) dramatic reduction in intensity of the DH EPR signal and PL bands at 3.14 (1.57), 2.53 and 2.3 eV during the first minutes of MP, which correlate with sample disaggregation; (b) grinding of ZnO particles and formation of Zn vacancy-related EPR centers in the area of destruction (AD); (c) an increase in sample temperature; (d) annealing of the Zn vacancy-related EPR centers formed; (e) initiation of carbon nanoparticle interaction with oxygen in the grinding chamber; (f) formation and growth of the EPR signal due to carbon nanoparticles; (g) formation of a reducing environment in the grinding chamber; (h) stabilization of donor DS-centers in AD of ZnO nanoparticles; (i) an increase in CO concentration in the grinding chamber; (j) inhibition of DS paramagnetic centers in ZnO; (k) initiation of the EPR signals due to Mn2+ ions in ZnOZB sphalerite phase in the sample with the lowest carbon content; (l) inhibition of nitrogen PL centers in ZnO; (m) the formation of oxidized carbon nanodots (OCN) showing a PL band at 2.8 eV. A detailed analysis for the localization of EPR and PL centers in the MP samples is presented.
, EPR DS centers from shallow donors (g = 1.9640) in ZnOW, Mn2+ ions in ZnOZB, C EPR centers in carbon nanoparticles, forming the near-band-edge (NBE) PL emission, PL emission typical for Zn-, O- and N-enriched ZnOW particles, as well as oxidized carbon nanodots (OCN)) was observed in the mixtures of ZnO + xC nanoparticles during prolonged high-energy mechanical processing (MP) in a hermetically sealed grinding chamber. The results of EPR and PL spectroscopy, X-ray diffraction analysis, as well as morphological analysis by means of atomic force microscopy (AFM) and laser particle sizer (LPS) measurements show a wide variety of interrelated series–parallel processes in the samples with increasing MP processing time (tMP). These processes include: (a) dramatic reduction in intensity of the DH EPR signal and PL bands at 3.14 (1.57), 2.53 and 2.3 eV during the first minutes of MP, which correlate with sample disaggregation; (b) grinding of ZnO particles and formation of Zn vacancy-related EPR centers in the area of destruction (AD); (c) an increase in sample temperature; (d) annealing of the Zn vacancy-related EPR centers formed; (e) initiation of carbon nanoparticle interaction with oxygen in the grinding chamber; (f) formation and growth of the EPR signal due to carbon nanoparticles; (g) formation of a reducing environment in the grinding chamber; (h) stabilization of donor DS-centers in AD of ZnO nanoparticles; (i) an increase in CO concentration in the grinding chamber; (j) inhibition of DS paramagnetic centers in ZnO; (k) initiation of the EPR signals due to Mn2+ ions in ZnOZB sphalerite phase in the sample with the lowest carbon content; (l) inhibition of nitrogen PL centers in ZnO; (m) the formation of oxidized carbon nanodots (OCN) showing a PL band at 2.8 eV. A detailed analysis for the localization of EPR and PL centers in the MP samples is presented.

 Please wait while we load your content...
Please wait while we load your content...

