DOI:
10.1039/C6RA13160C
(Paper)
RSC Adv., 2016,
6, 76632-76641
Amino acid appended cholic acid–azobenzene dyad: an effective & smart phase selective gelator for aromatic solvents†
Received
20th May 2016
, Accepted 8th August 2016
First published on 8th August 2016
Abstract
A series of amino acid appended cholic acid–azobenzene dyads have been synthesized and studied for their gelation behaviour. One of the L-alanine based dyads showed excellent gelation behaviour in a variety of solvents at room temperature with minimum gel concentration in the range 0.8% (w/v)–1.8% (w/v). The morphology of the stable gels indicated the formation of a lamellar or a dense sheet network in different solvents. Variable temperature 1H-NMR and FT-IR studies revealed an evident role of intermolecular hydrogen bonding in the self-assembly process. The photo-isomerization between the trans and cis forms of the azobenzene unit was established by UV-visible spectroscopy, and a comparison of 1H NMR and SEM images of the gel and sol forms. In addition, π → π stacking between phenyl groups of azobenzene might have provided an additional driving force for the formation of a dense three-dimensional network capable of phase selective gelation of aromatic solvents from a water/solvent mixture. This selective gelation of aromatic solvents remains unaffected even in the presence of the common salts usually present in water from different sources. The phase selective gelation ability of the dyad was successfully explored towards the removal of toxic fat-soluble rhodamine dye from water and selectively gelatinizing petrol and crude oil from an oil/water mixture at room temperature. Thus, these smart systems possess the potential to be used effectively in water purification and oil-spill remediation.
Introduction
Immobilization of organic solvents by low-molecular-weight gelators (LMWGs) through a molecular self-assembly process is a well-established strategy for preparing stable gels that find wide-ranging applications as templated materials, drug delivery agents, cosmetics, sensors, and enzyme-immobilization matrices.1–4 These gelators form a three-dimensional (3D) network in organic solvents by self-organization of the monomeric species to higher-order structures, such as fibrous, tubular, or helical,5 usually driven by non-covalent intermolecular interactions such as electrostatic, dipole–dipole, van der Waals, π–π stacking, and hydrogen bonding.6 Although numerous effort has been devoted to establishing a structure–property relationship for the development of low-molecular weight gelators,7 however predicting the gelation ability of a targeted compound and understanding the mechanism of self-assembly process remains a challenging task. In addition, specific effort has been laid towards exploring phase selective gelation ability of novel organic structures. Such phase selective gelators (PSGs) could be used either for separating toxic aromatic dyes/solvents during water purification procedures8 or oil-spill remediation9 which is one of the environmental concern in minimizing marine pollution.10 Also, the use of eco-friendly, abundant and cheap chemical precursors such as amino acids,11 carbohydrates,12 lipids13 for preparing such PSGs provides an additional advantage. Since the development of first amino acid based phase selective gelator by Bhattacharya and Krishnan-Ghosh,14 several other amino acid based PSGs have been developed.15 Das et al. have reported few examples of peptide based organogelators and explored their applications in dye absorption, phase selective gelation and template for nanoparticle synthesis.8 In addition to amino acids, bile acids have also been proven as potential components of supramolecular gels due to their amphiphilic scaffolds, availability of multiple easily derivatizable functional groups, low cost, and adjustable self-assembling characteristics.16
Interestingly, most of the examples reported for phase selective gelation of aromatic solvents requires external stimuli such as heating,8a,14,17 mechanical shaking,18 sonication19 etc. to initiate the gelation process and heating/distillation20 for recovering the aromatic solvent from gelatinized material. Such protocols appear to be highly expensive in the context of large scale water purification or oil-spill remediation processes. Thus, instant gelation of aromatic solvents at room temperature and their recovery by simple filtration are dictating factors for an ideal PSG, which could be addressed by preparing photo-responsive PSGs that could reformed gel back to sol state by photo-irradiation.
In the last decade, emphasis has been laid on the development of photo-responsive gels that have offered promising opportunities for designing useful functional materials such as sensors, actuators, etc.21 The gel–sol transition of such smart materials upon light irradiation could provide a potential solution for recovering aromatic solvents from the recovered gel without heating/distillation. Very few examples on the use of photo-responsive materials for phase selective gelation of aromatic solvents have been reported.22 Ajayaghosh reported the use of N-glucosyl azobenzene as an excellent phase selective gelator for removal of toxic organic solvents from water.23 Very recently, Díaz Díaz reported the application of tetrapeptides bearing a side-chain azobenzene moiety for ultrasound-induced phase selective gelation of organic solvents, water purification by removal of toxic dyes and oil-spill remediation.24 However, the development of bile acid based photo-responsive and efficient room temperature PSGs for the separation of aromatic solvents remains unexplored. In this context, we herein report the first example of amino acid appended cholic acid–azobenzene dyad as an effective and smart phase selective gelator for aromatic solvents at room temperature. The phase selective gelation ability of one of the dyads has been explored towards the practical utility in water purification by removal of toxic fat-soluble dyes and oil-spill remediation.
Results and discussion
Synthesis
Although it is difficult to generalize the criteria for an ideal organogelator, however, hydrogen-bonding enabling functional groups, long alkyl chains that facilitate the van der Waals force interactions, and π-conjugated planar aromatic moieties that promote the π–π are typical of the features found to facilitate the gelation process. In addition, with an aim to prepare smart amino acid appended cholic acid based gelator, we envisaged the hybridization of photo-responsive azobenzene system to cholic acid scaffold via an α-amino acid linker. Thus, (E)-ethyl 4-((4-hydroxyphenyl)diazenyl)benzoate (1) required to initiate the synthesis of targeted system was obtained by the reaction of p-ethylcarboxylate aniline and phenol via standard diazotization-coupling process.26 Alkylation at phenolic-OH in 1 with long chain alkyl bromides (2a–c) using K2CO3 in acetone yielded the corresponding O-alkylatedphenyl diazenyl benzoate derivatives (3a–c), which on reduction with LAH/THF at room temperature yielded the corresponding O-alkylatedphenyl diazenyl methanols (4a–c). Coupling of 4 with Boc protected α-amino acids (5a–b) using EDC HCl/HOBt/DMAP in DCM afforded Boc protected amino acyl O-alkylatedphenyl diazenyl conjugates (6a–f) in good yields. Deprotection of Boc group using HCl/dioxane in 6 followed by coupling with cholic acid (8) using EDC·HCl/HOBt/NEt3 in DCM at room temperature in 6–8 h gave amino acid appended cholic acid–azobenzene dyads (9a–f) in excellent yields (Scheme 1). All the intermediates 3a–c, 4a–c, 6a–f, 7a–f were characterized on the basis of 1H & 13C NMR, IR data and the final compounds 9a–f were characterized on the basis of 1H, 13C NMR, IR and mass spectroscopic studies.
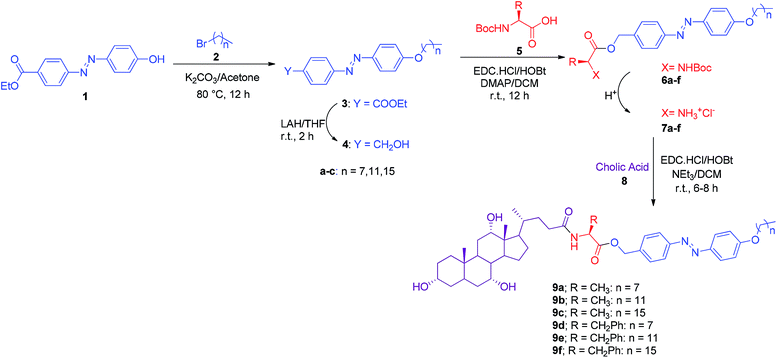 |
| | Scheme 1 Synthesis of amino acid appended cholic acid–azobenzene dyads. | |
Gelation
The gelation behaviour of 9a–f was examined in various non-polar and polar solvents using inverted test tube method25 and the results are summarized in Table 1. In general, all the compounds gelatinized most of the solvents at room temperature forming either stable or weak gels.
Table 1 Minimum gelation concentration of CA-AA-AZA (9a–f) in different solventsa,b
| Solvent |
9a |
9b |
9c |
9d |
9e |
9f |
| G = gel formed at room temperature (minimum gel concentration (% w/v)); WG = weak gel (unstable above 20 °C); PS = partially soluble (a part of it becomes soluble upon heating but re-precipitation was observed after cooling to room temperature); S = soluble; I = insoluble (was not soluble at all even on heating). DMSO = dimethyl sulfoxide, DMF = N,N-dimethylformamide, DCE = 1,2-dichloroethane, DCM = dichloromethane, tert-bb = tert-butyl benzene, THF = tetrahydrofuran. |
| Ethyl acetate |
S |
S |
S |
S |
S |
S |
| Hexane |
G (1) |
I |
PS |
WG (4) |
I |
PS |
| DCM |
S |
S |
S |
S |
S |
S |
| DCE |
S |
S |
S |
S |
S |
S |
| 1,4-Dioxane |
S |
S |
S |
S |
S |
S |
| THF |
S |
S |
S |
S |
S |
S |
| Cyclohexanol |
PS |
PS |
PS |
PS |
PS |
PS |
| Methanol |
S |
S |
S |
S |
S |
S |
| Ethanol |
S |
S |
S |
S |
S |
S |
| Cyclohexane |
G (1) |
I |
PS |
I |
I |
PS |
| Heptane |
I |
I |
PS |
I |
I |
PS |
| Decane |
I |
I |
PS |
I |
PS |
PS |
| Dodecane |
I |
I |
PS |
I |
G (1) |
PS |
| Tetradecane |
I |
I |
PS |
I |
I |
PS |
| Hexadecane |
I |
I |
PS |
I |
I |
PS |
| Toluene |
G (1.8) |
S |
S |
WG (6) |
S |
S |
| Benzene |
G (1.2) |
S |
S |
WG (5.5) |
S |
S |
| Xylene |
G (0.8) |
WG (4) |
S |
WG (6.5) |
WG (4.3) |
S |
| Mesitylene |
G (0.8) |
WG (6) |
S |
WG (8.1) |
WG (5.5) |
S |
| tert-bb |
G (0.9) |
WG (7.5) |
S |
WG (7.6) |
WG (6.2) |
S |
| DMF |
S |
S |
WG (10) |
S |
S |
WG (11) |
| DMSO |
S |
S |
WG (10.5) |
S |
S |
WG (11.5) |
For instance, compound 9a, an L-alanine linked dyad possessing 8-carbon alkyl chain at the phenolic-OH showed remarkable gelation ability in seven solvents, namely hexane, cyclohexane, toluene, xylene, benzene, mesitylene and t-butyl benzene. It is worth mentioning that no external stimuli such as heating/sonication/mechanical shaking was required for gelation process and the gel was formed within 3–4 minutes upon gentle swirling of organic solvents with powdered LMWGs. The minimum gel concentration of 9a was found to be 0.8% (w/v) in xylene (mixture of all isomers) and mesitylene, and it ranged from 1 to 1.8% (w/v) in other solvents. Compound 9e, another L-alanine linked dyad possessing 16-carbon alkyl chain at the phenolic-OH gelates dodecane at MGC of 1% (w/v). The remaining compounds (9b–d, 9f) could form weak gels which were not stable above 20 °C (Table 1).
All the stable gels were found to be thermo-reversible i.e. all the gels can be melted at higher temperature and reformed into gel upon cooling. Interestingly, the gels were transformed to solution state under UV irradiation within few minutes and got reformed under visible light. The gelation abilities of 9a remains unchanged after repetitions of several gel–sol cycles. In addition, gels of 9a were found to be quite stable at room temperature over a period of 2–3 months without any phase separation thus indicating their stability. Surprisingly, L-phenylalanine linked dyads (9d–f) possessing different alkyl chains at the phenolic-OH were found to be incapable in generating stable gels in most solvents above 20 °C, thus indicating a crucial role of sterics in gelation process.
Morphology of self-assembled gels
It is well documented27 that organogelation represents a physical state formed via non-covalent hydrophilic and hydrophobic interactions that leads to the variation in the morphologies of the resultant gels, including fibers, nanosheets, fibrils, helical ribbons and other 3-D structures. To get an insight into the microscopic morphology of the stable gels obtained, Scanning Electron Microscopy (SEM) of the xerogels were recorded. The SEM image of the gels of 9a obtained in toluene and xylene showed a dense lamellar sheet network consisting of bundle of fibres entangled with each other (Fig. 1a and b). The gel of 9a in cyclohexane, hexane, benzene, tert-butyl benzene and mesitylene also showed similar lamellar sheet type structures consisting of dense fibre network (Fig. S1a–e, ESI† file). In addition, the gel of compound 9e in dodecane showed a beautiful flowery lamellar dense sheet network (Fig. S1f, ESI† file).
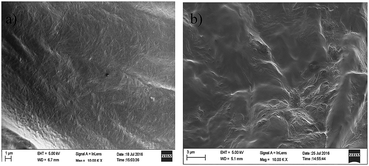 |
| | Fig. 1 SEM images of xerogels of 9a in (a) xylene (b) toluene. | |
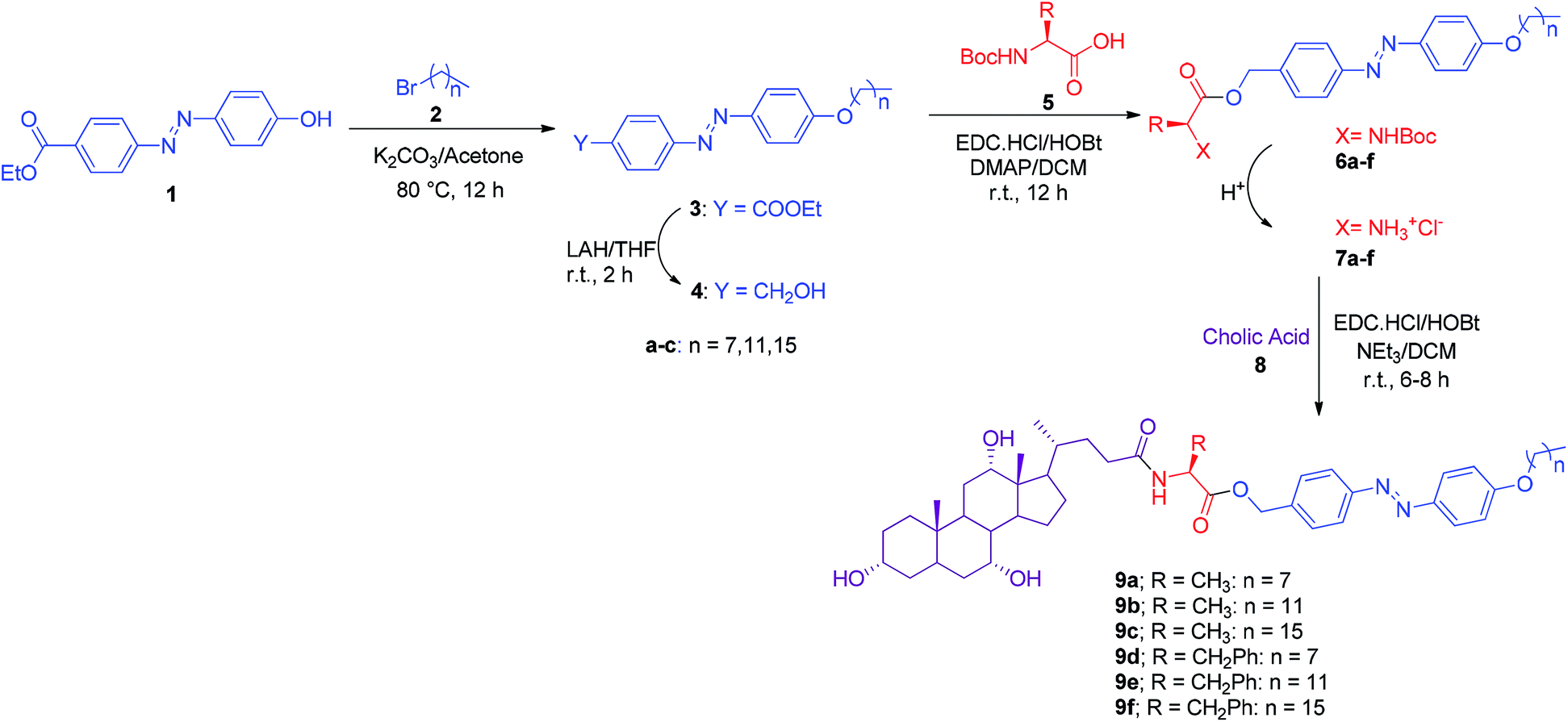
The Transmission Electron Microscopy (TEM) images of the xerogel of 9a in xylene and toluene further supported the lamellar sheet type packing stacked over each other (Fig. 2).
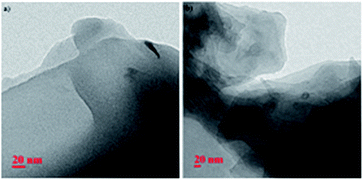 |
| | Fig. 2 TEM images of xerogels of 9a in (a) xylene (b) toluene. | |
Mesomorphic nature of the gelators
To study the mesomorphic behaviour of the gelator, Polarising Optical Microscopy (POM) was recorded. For this purpose, a small amount of gel of 9a at minimum gel concentration in xylene, and toluene was placed on a microscopic glass slide and was visualized under the optical microscope at 10× magnification using visible light. A strong birefringent texture, characteristic of the mesoscopic alignment of the chromophores during gelation was observed (Fig. 3a and b).4g
 |
| | Fig. 3 Polarising Optical Microscopy (POM) images of 9a in (a) xylene (b) toluene under visible light between crossed-polarizers. | |
Fluorescence optical microscopic study
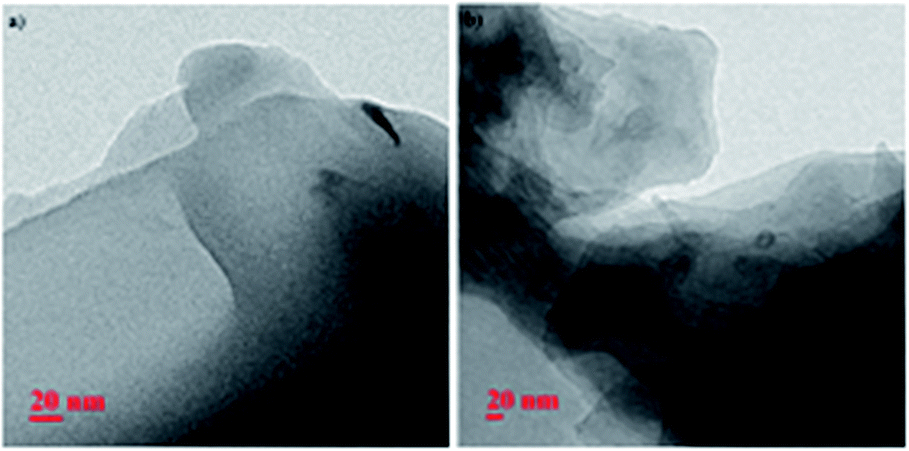
A fibrillar structure of the wet gel of 9a in xylene and toluene respectively was observed by fluorescence optical microscopy (Fig. 4a and b).
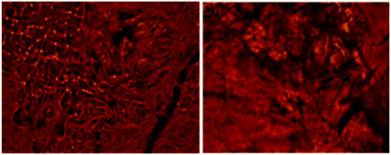 |
| | Fig. 4 Fluorescence optical microscopic images of wet gel 9a in (a) xylene (b) toluene. | |
X-ray diffraction pattern
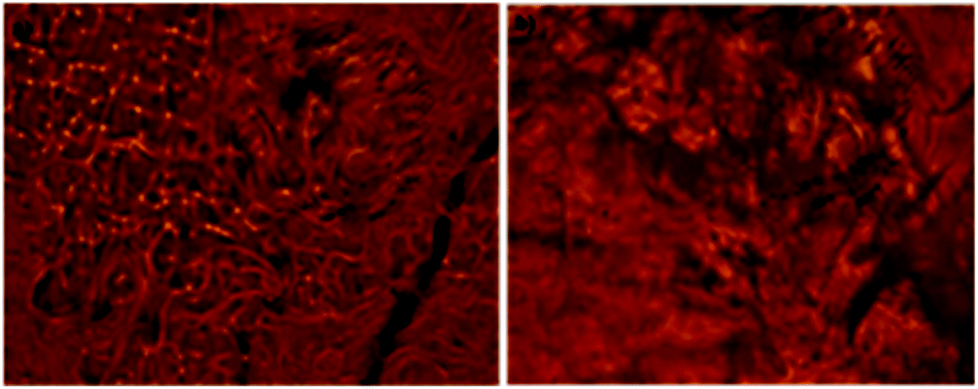
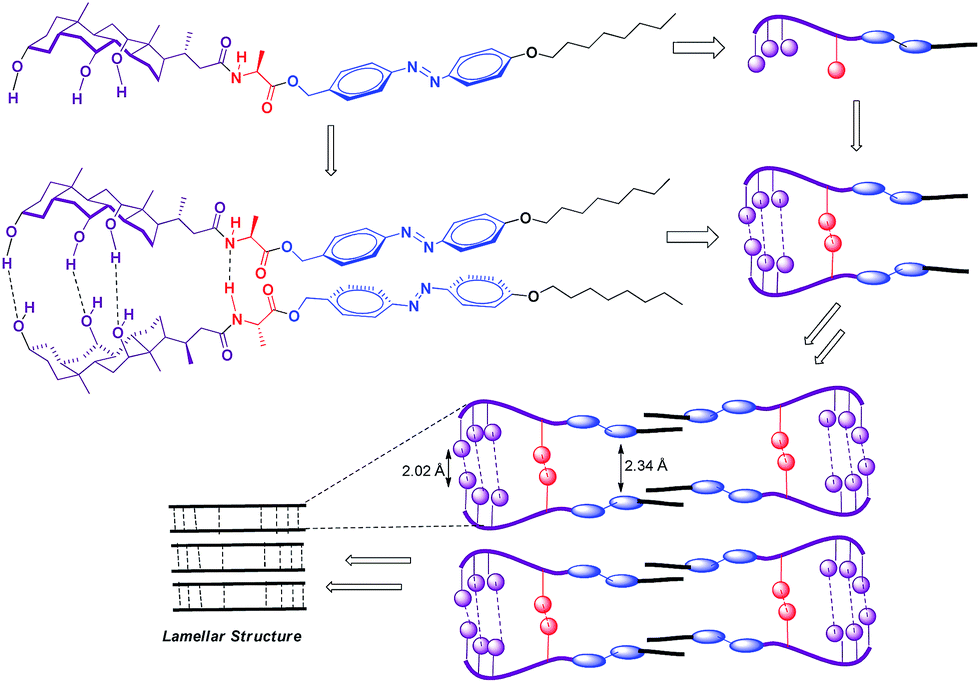
To gain more insight into molecular packing in the gel state, XRD analysis were performed on the xerogel of xylene (Fig. 5). The WAXRD displayed well-resolved diffraction pattern indicating the formation of well-defined arrangement of the molecules during self-assembly process. The xerogel showed reflection peaks (2θ) 38.399, 44.623, 64.914, 77.976 corresponding to spacing (d) 2.34, 2.02, 1.43, 1.22 Å respectively. The sharp peak corresponding to (d) spacing 2.34 Å could be attributed to the inter-layer distance between the azobenzene unit, while the (d) spacing corresponding to 2.02 Å could be the H-bonding distance between the cholic acid and the amino acid units.
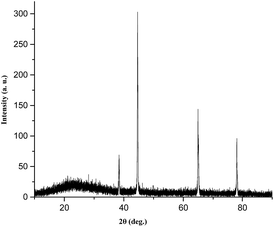 |
| | Fig. 5 X-ray powdered diffraction pattern of xerogel of 9a in xylene. | |
FT-IR analysis
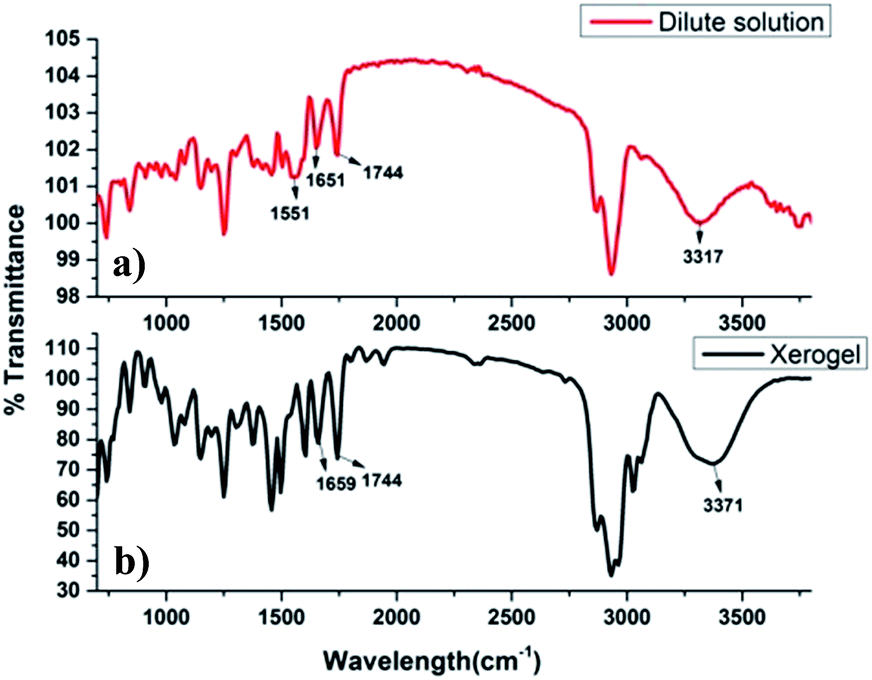
In order to anticipate the effective role of H-bonding in self-assembly process due to presence of amino acid and cholic acid moieties, FT-IR spectra of dilute solution of 9a and its xerogel (in xylene) were recorded and compared (Fig. 6). The dilute solution of compound 9a showed a number of peaks in the range of 3680–3317 cm−1 (O–H/N–H stretching), 1744 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of ester), 1651 cm−1 (CO stretching of amide bond) and 1551 cm−1 (N–H bending), while IR of xerogel showed a prominent band at 3371 cm−1 and no band above it, indicating the possibility of strong involvement of O–H of cholic acid/N–H of amidic bond in the hydrogen bonding in the gel state. A blue shift was observed in the amidic carbonyl from 1651 cm−1 to 1659 cm−1 indicated the role of amide bond in hydrogen bonding.
O stretching of ester), 1651 cm−1 (CO stretching of amide bond) and 1551 cm−1 (N–H bending), while IR of xerogel showed a prominent band at 3371 cm−1 and no band above it, indicating the possibility of strong involvement of O–H of cholic acid/N–H of amidic bond in the hydrogen bonding in the gel state. A blue shift was observed in the amidic carbonyl from 1651 cm−1 to 1659 cm−1 indicated the role of amide bond in hydrogen bonding.
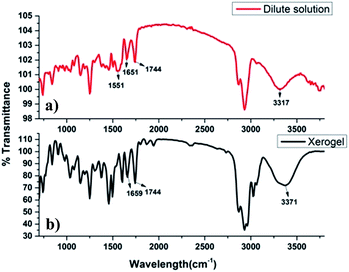 |
| | Fig. 6 (a) FT-IR of the dilute solution of 9a, (b) FT-IR of the xerogel of 9a (in xylene) at minimum gel concentration. | |
Thus, it can be concluded that hydrogen bonding plays a crucial role in self-assembly process of 9a in different solvents. Not much change in the frequency and intensity of carbonyl ester was observed upon gelation, indicating its non-involvement in the H-bonding.
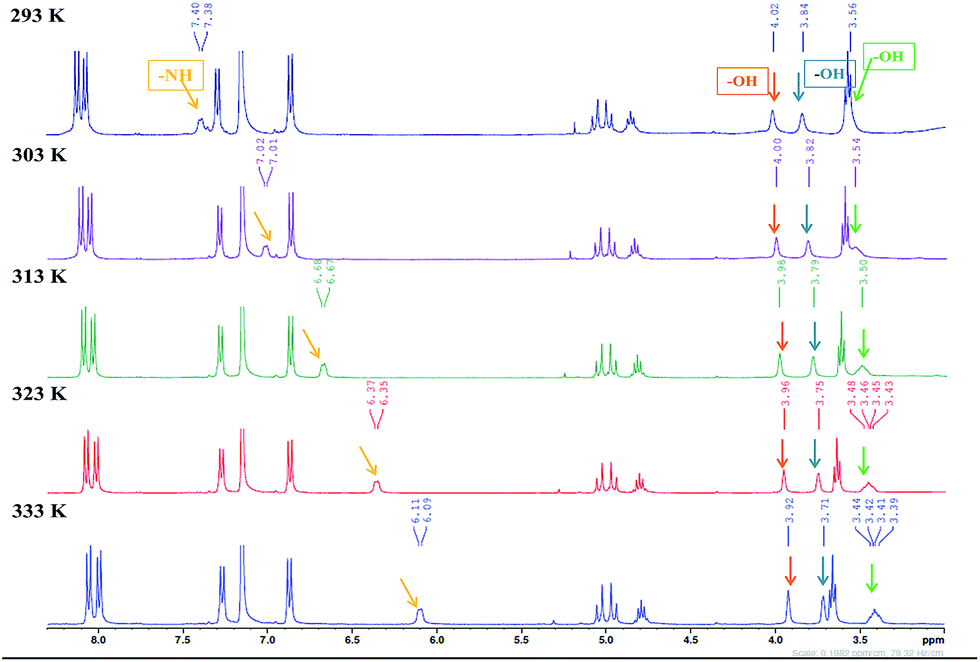
Variable temperature-dependent 1H NMR studies
The SEM images of most xerogels of 9a suggested the existence of dense sheet or lamellar network. The IR studies also clued the existence of strong intermolecular hydrogen bonding in the self-assemble process. To further understand the role of hydrogen bonding in the self-assembly process, variable temperature 1H NMR experiments were conducted at a minimum gelation concentration of 9a in gelation solvent benzene-d6 over a temperature range of 293 K to 333 K. Generally, temperature dependent shift of –NH and –OH peaks have been widely used to understand the role of hydrogen bonding in self-assembly process.28 As evident from the temperature dependent NMR of 9a, a consistent shift in the –NH and –OH proton peaks was observed as the temperature was increased (Fig. 7). The broad multiplet of –NH proton shifted from 7.4 to 6.11 ppm, while the three –OH groups of cholic acid also showed a small upfield shift (Fig. 7). The dependence of chemical shift on temperature (Δδ/ΔT) provides a handy tool to study the role of hydrogen bonding in the self-assembly process in the gelling solvent.
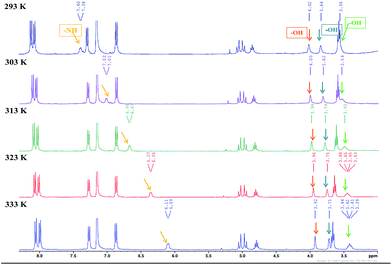 |
| | Fig. 7 Variable temperature 1H-NMR study of 9a in gelling solvent benzene (d6) at 293 K, 303 K, 313 K, 323 K and 333 K showing a change in the chemical shifts of NH and OH protons. | |
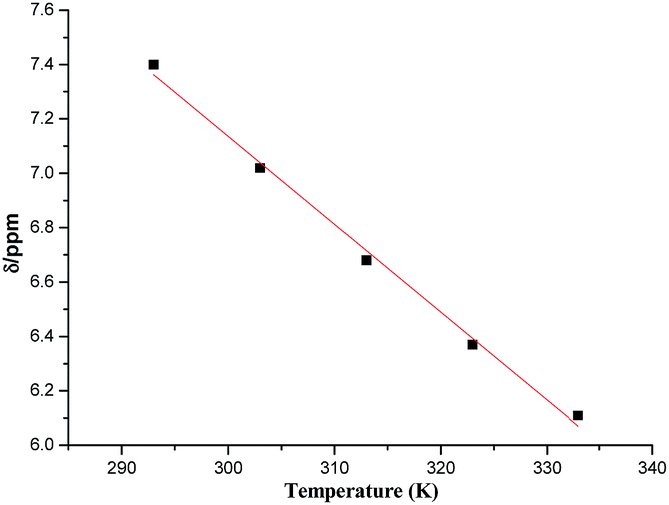
Earlier reports have suggested that higher the value of temperature coefficient (Δδ/ΔT), greater is the role of NH in intermolecular hydrogen bonding.29 In the present case, a relatively large temperature coefficient value for –NH (32.3 ppb K−1, calculated from slope of the graph between change in chemical shift and temperature) as compared to earlier reports29 (Δδ/ΔT of the order of 15.46 ppb K−1) confirmed the formation of strong intermolecular hydrogen bonding using –NH bond (Fig. 8).
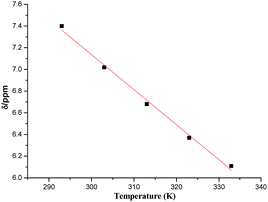 |
| | Fig. 8 The temperature coefficient for –NH calculated from slope (32.3 ppb K−1). A relatively large temperature coefficient value confirmed the formation of strong intermolecular hydrogen bonding using –NH bond. | |
Sol–Gel transformation of CA-AA-AZA (9a) by light stimuli
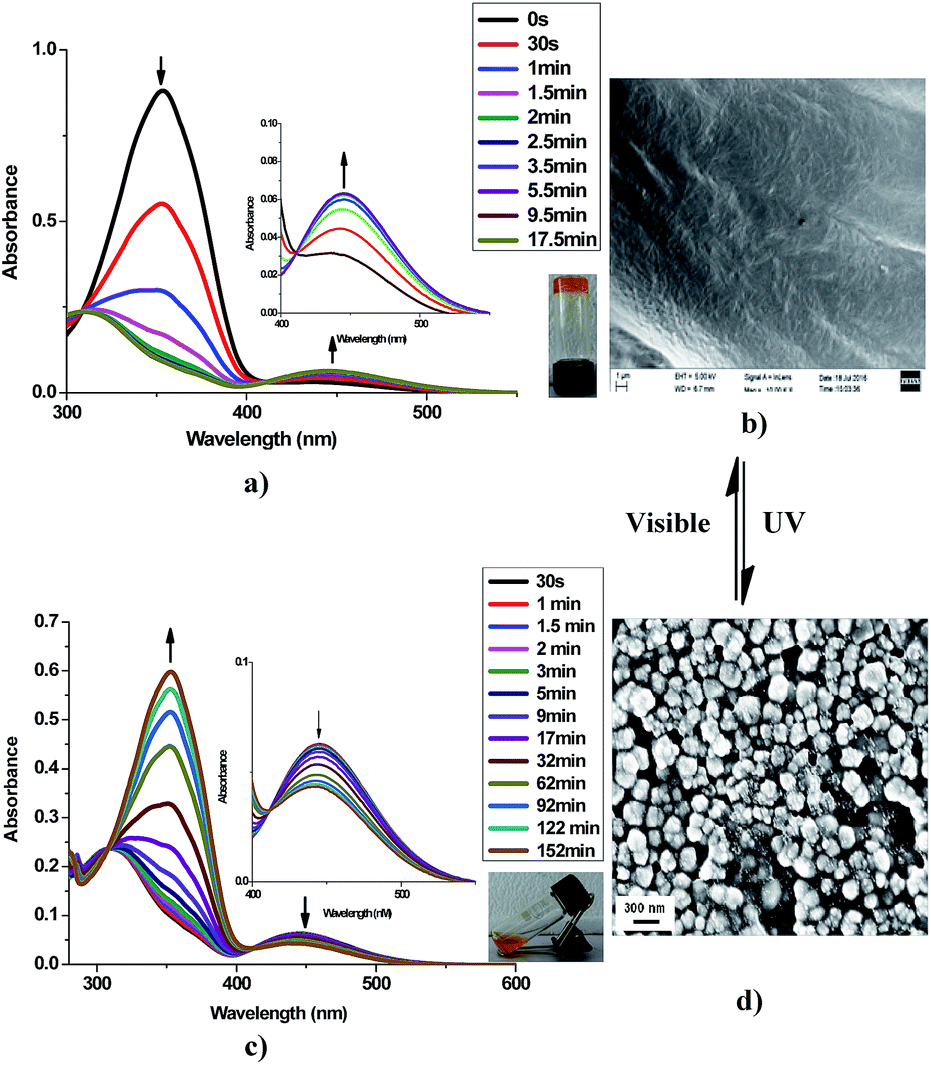
Azobenzene is a photo-responsive chromophore unit which undergoes trans–cis isomerization by exposure to UV light and vice versa by exposure to visible light respectively. It has been well documented30 that trans form of the azobenzene favours self-assembly process by long range networking which could be an important parameter for the selective gelation of solvents in water–solvent mixture. As earlier stated23 that trans form of azobenzene unit favours self-assembly process that favours long range entangled network capable of forming gels with solvent molecules (aromatic) via π–π interaction. It was observed that the gel of 9a in xylene turned into sol on exposure to UV light within few minutes. Subsequently, the regeneration of the gel was observed upon exposure to visible light at room temperature within few minutes. This photo-responsive behaviour (gel ⇄ sol) was found to be reversible and could be repeated many times. This configurable change in the azobenzene scaffold on irradiation with UV light was further studied and confirmed by UV-Visible spectroscopy. With an increase in the UV exposure time (under hand held UV lamp at 365 nm) of xerogel of 9a, a gradual decrease in the intensity of the absorption band at 352 nm (π–π* transition) and a concomitant increase in the intensity of a band at 455 nm (n–π* transition) was observed (Fig. 9a). Subsequently an opposite phenomena was observed when the same sample was irradiated with visible light (>460 nm under a normal bulb) (Fig. 9c). These spectral changes can account for the change in the photo-responsive behaviour of the gel at the macroscopic state. The sol formed after the exposure of UV light on the gel, when analyzed through SEM showed small aggregates, while the subsequent gel obtained from this sol in visible light showed dense sheets when analyzed through SEM images. Thus, it could be concluded that the UV irradiation of gel disturbed the molecular packing of 9a in xylene and thus the network of gel converted to sol (Fig. 9).
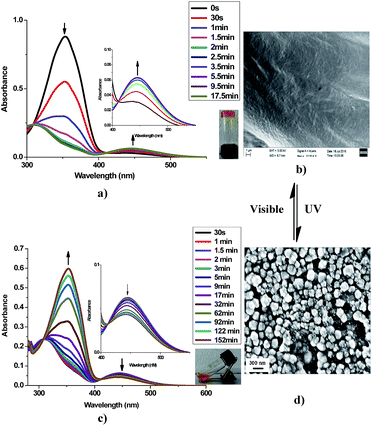 |
| | Fig. 9 (a) Absorption spectra of 9a under UV-light irradiation (365 nm) at different time scale (inverted vial showing gel of xylene (2% w/v) after exposing to visible light). (b) SEM image of the xylene gel showing a lamellar network. (c) Absorption spectra of 9a upon exposure to visible light (>460 nm) at different time scale (tilted vial showing sol form after exposure to UV-light of xylene). (d) SEM image of the sol form showing aggregates. | |
Comparative 1H NMR studies for cis–trans isomerization
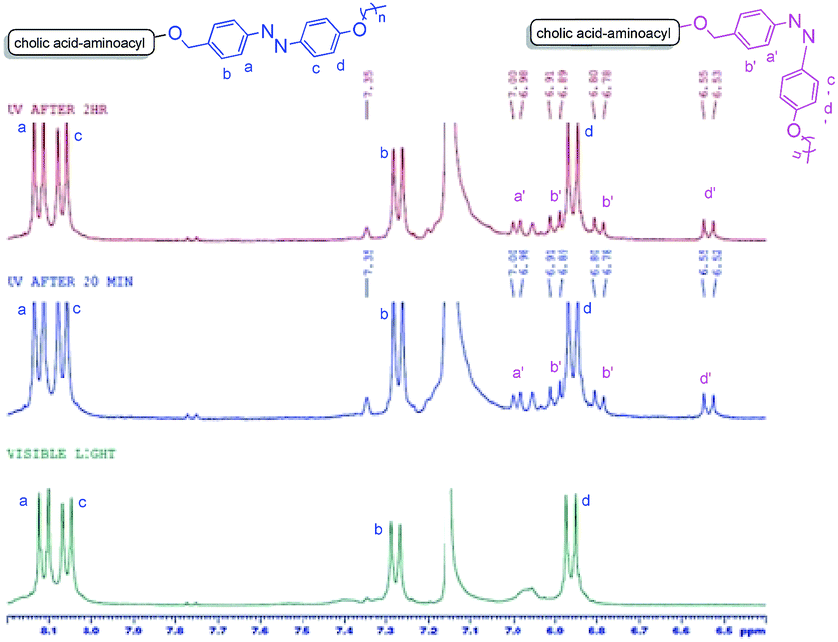
The trans to cis photo-isomerization phenomena further got support by recording the 1H NMR of a sample of 9a (in benzene-d6) after exposing it to UV light at different intervals of time (Fig. 10). It became evident from these 1H-NMR experiments that extra peaks of cis-isomer in the aromatic region appeared after irradiating the sample with UV light after 20 min and 120 min respectively. Cis isomer protons are consistently more shielded than their trans isomer counterparts. The relative shifts in the position of cis protons (a′–d′) as compared to trans protons (a–d) are in accordance with earlier reports.31 As cis–trans photo-isomerization is a reversible phenomenon and trans form is more is more stable than the cis-form, thus relatively small amount of cis-form was found even after exposure for a longer time (Fig. 10).
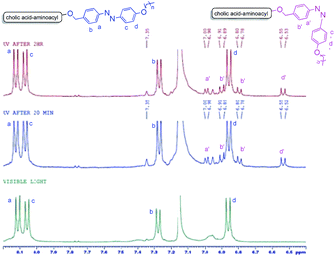 |
| | Fig. 10 Comparison of 1H-NMR of 9a (in benzene-d6) in visible light (>460 nm) and after irradiating UV light (365 nm) for 20 min and 120 min. Appearance of new peaks (a′–d′) after UV irradiation confirms cis–trans isomerism of the azobenzene unit. | |
Mechanism of self-assembly process
Based on the variable temperature 1H NMR and IR studies, it can be concluded that the involvement of N–H of amide bond and O–H groups of cholic acid in strong intermolecular hydrogen bonding, might be a crucial parameter in self-assembly process. In addition, trans geometry of the azobenzene unit could be another important parameter that favors the face to face orientation/stacking of molecules that favors gelation. The driving force in the remaining scaffold could be face to face packing of amphibilic cholic acid units that gets stabilized due to inter-molecular H-bonding between three hydroxyl groups, which further got support from intermolecular H-bonding between and amide bonds in the extended amino acid skeleton, which remain undisturbed upon irradiation by UV light. Complete inter-conversion of trans to cis form is chemically a slow phenomena due to large steric repulsions, however photo-isomerization of few molecules from trans to cis might be enough to perturb the self-assembled three-dimensional network and thus physically the gel got transformed into sol. Moreover, re-gelation of sol within few minutes under visible light because of back-isomerization indicates that the remaining scaffold of the gelator favors aggregation process in trans form. Similar, visual observations could be inferred by gel to sol reversible conversion that occurs upon cooling and heating process respectively. As evident from variable temperature 1H NMR studies that heating of the gel breaks the strong intermolecular hydrogen bonding between N–H bonds of amide bond linker or O–H bonds of two consecutive possible parallel stacked cholic acid units, thereby distorting the self-assembled aggregates temporarily, which regains their original format upon cooling. However, no change in the chemical shift of the azobenzene protons or appearance of no extra peaks in the aromatic region (corresponding to cis isomer) indicated that the proposed π → π stacking between trans-azobenzene units remain unaffected upon heating. Thus, based on the above observations and literature report,23 it could be inferred that CA-AA-AZA (9a) type of scaffold might have formed by face to face packing stabilized by strong intermolecular hydrogen bonding between N–H of amide bonds of the amino acid linker and three O–H of the cholic acid motif leading to formation of lamellar sheet networks containing bundle of fibers entangled into each other which are also evident from SEM, TEM and XRD analysis. Such dense packing facilitates the entrapping aromatic solvents easily and this facilitates the phase selective gelation behavior of gelator (Fig. 11).
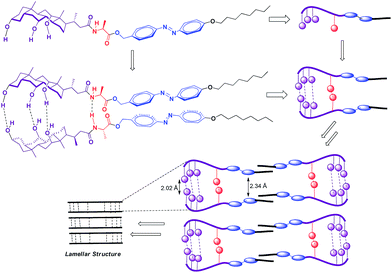 |
| | Fig. 11 Schematic representation of plausible mode of aggregation in the self-assembly process via non-covalent interactions resulting in the formation of lamellar sheet networks containing bundle of fibers entangled into each other. | |
Application of 9a for phase selective gelation. Contrary to other compounds, CA-AA-AZA (9a) showed selective gelation of aromatic solvents from a solvent–water mixture. The phase selective gelation of aromatic solvents could be attributed to the entrapping of aromatic solvent molecules between long range parallel dense network of amino acid appended cholic acid azobenzene molecule stacked one over another via H-bonding and π–π interaction. As model examples, the phase selective gelation of xylene, toluene and benzene were carried by mixing an equiv. volume of these with water (1 mL each) and subsequently shaking with 9a (0.02 g). After few minutes, gelation of the aromatic solvents in their respective vials were observed and dense coloured gels were obtained (Fig. 12). The resultant gels were stable enough to be scooped out with a spatula (Fig. 12e) or via filtration to give pure water. The solvent trapped in gel could be recovered by vacuum distillation and the gelator could be reused.
 |
| | Fig. 12 Phase selective gelation of 9a (0.02 g) in equiv. volume of water with aromatic solvents such as (a) xylene (b) toluene (c) benzene. (d) Tilted vial after phase selective gelation of xylene showing gel stability in water (e) xylene gel scooped out with a spatula. | |
The phase selective nature of the gelator was also tested in presence of various metal salts. To our delight, the phase selective nature of the gelator was unaffected in presence of various salts such as saturated solutions of NaCl, CaCl2, CuSO4, NiCl2, FeCl3, and CdCl2, (Fig. 13), which provided us a handy tool for the use of gelator in real samples such as sea water, tap water etc.
 |
| | Fig. 13 Phase selective gelation of 9a (0.02 g) from the mixture of xylene and water (1 mL each) in the presence of saturated solution of common salts present in water such as (a) NaCl (b) CuSO4 (c) Cd(NO3)2 (d) Ni(NO3)2 (e) CaCl2 (f) FeCl3. | |
Oil-spill remediation
Oil-spill has emerged as one of the major issues in the ever growing environmental problem.32 An oil-spill not only causes damage to marine ecosystems, but also imparts a disastrous health impact on humans due to the consumption of oil polluted sea foods.33 Due to scale-up challenges and a number of other drawbacks associated with the various oil-spill cleaning techniques including bioremediation,34 dispersion,35 absorption,36 and solidification,37 phase selective gelation of oil from a biphasic mixture of water and oil, by low-molecular-weight gelators has provided a potential and effective solution to this problem.38 To overcome this, phase selective gelation has emerged as one of the effective remedy.
To explore the real application of phase selective gelation behaviour of our synthesized dyad, 9a in oil-spill remediation process, we attempted phase selective gelation of petrol and crude oil. Addition of 25 mg of 9a to a mixture of petrol–water (1 mL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5 mL) and crude oil–water (1 mL:2.5 mL) followed by gentle swirling gelatinizes petrol and crude oil effectively in few minutes (4–5 min). The stable gels formed were comfortably scooped out with a spatula as depicted in the Fig. 14.
:2.5 mL) and crude oil–water (1 mL:2.5 mL) followed by gentle swirling gelatinizes petrol and crude oil effectively in few minutes (4–5 min). The stable gels formed were comfortably scooped out with a spatula as depicted in the Fig. 14.
 |
| | Fig. 14 Real-time example of oil-spill remediation by phase selective gelation of petrol and crude oil using 9a from a biphasic mixture of petrol/crude oil and water. (a) Vial showing mixture of crude oil (2 mL, left) and petrol (2 mL right) with water (5 mL) (b) tilted vials showing phase selective gelation of crude oil (left) and petrol (right) (c) crude oil gel (left) and petrol gel (right) scooped out from the vial and the vials after crude oil and petrol removal. | |
Water purification
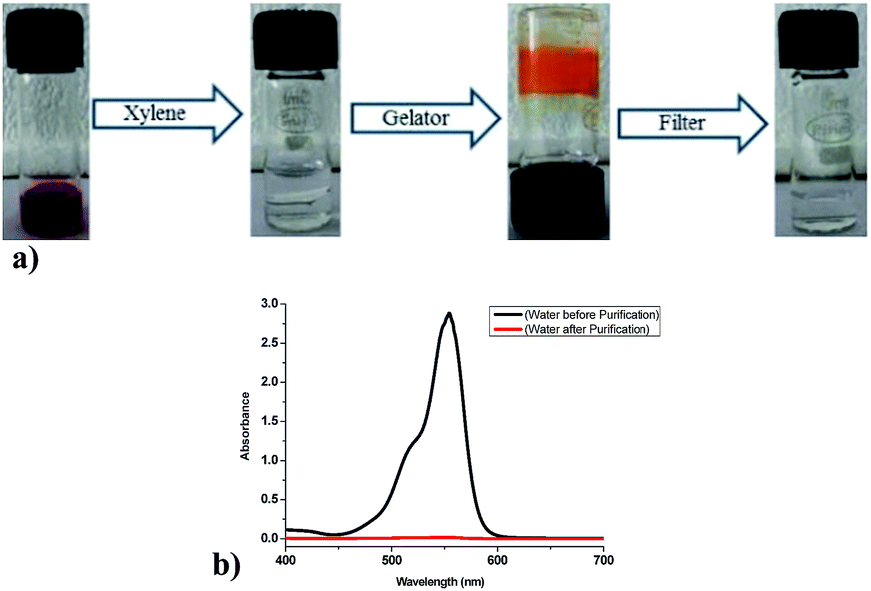
Removal of toxic dyes from waste water is one of the important task in environmental remediation, as it is known that fat-soluble dyes usually gets extracted in organic solvents. Thus, phase selective nature of organogelators could be used as a handy tool for water purification. In order to explore the practical applicability of 9a towards phase selective gelation of xylene from water–xylene mixture, a 0.05 mM aqueous solution of rhodamine B dye was prepared that showed red colour (Fig. 15). Rhodamine B was selected due to its good fat-soluble properties. Upon addition of xylene (1 mL) to the coloured aqueous layer (1 mL) contaminated with rhodamine B and subsequent shaking, the overall solution becomes colourless indicating the solubilization of dye in xylene layer. This xylene layer got gelatinized quickly upon addition of powdered gelator (12 mg) in it, which upon filtration or simple decanting yielded the pure water. Thus, the rhodamine B dye got trapped in three-dimensional (3D) network of gel formed and gets immobilized along with xylene molecules. The water obtained by filtering the gel was analyzed by recording its UV-Visible absorption spectrum and comparing it with the UV-Visible of the water containing rhodamine B dye. The purification efficiency was found to be around 99.5% (Fig. 15b). Thus, phase selective behaviour of 9a could be efficiently used in waste water purification by removal of similar fat-soluble dyes from it.
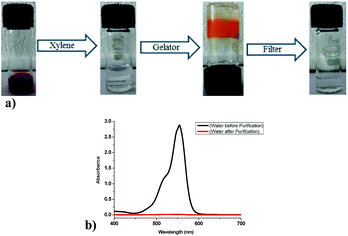 |
| | Fig. 15 (a) Removal of toxic rhodamine B dye from water by phase selective gelation of xylene using 9a and recovery of pure water by simple filtration. (b) UV-visible spectra of water before and after purification depicting the efficacy of process. | |
Conclusions
In summary, we have designed and synthesized a series of amino acid appended cholic acid–azobenzene dyads as photo-responsive low-molecular-weight gelators. Among these, L-alanine based dyad possessing O-octyl c hain proved to be excellent phase selective gelator of a variety of solvents such as hexane, cyclohexane, toluene, xylene, benzene, mesitylene and t-butyl benzene at room temperature. The minimum gel concentration of 9a was found to be 0.8% (w/v)–1.8% (w/v) in these solvents. The SEM images of the stable gels of 9a showed a lamellar or a dense sheet network in different solvents. The self-assembly process of such morphologies was explained by variable temperature 1H-NMR studies, FT-IR that revealed a strong role of intermolecular hydrogen bonding. In addition, UV-visible spectroscopy and a comparison of SEM images of the gel and sol forms gave an insight into the existence of photo-isomerization between the trans and cis form of azobenzene unit and π → π stacking between phenyl groups of trans azobenzene. This regular stacking and long range networking stabilized by H-bonding make them suitable for phase selective gelation of aromatic solvents from a water–solvent mixture and which remains unaffected in presence of common salts usually present in water from different sources. The phase selective gelation ability of 9a was successfully explored towards the removal of toxic fat-soluble rhodamine dye from waste-water and selectively gelatinizing petrol and crude oil from oil/water mixture at room temperature. Thus, these smart systems possess the potential to be used effectively in water purification and oil-spill remediation.
Acknowledgements
The authors acknowledge Department of Science & Technology (DST), New Delhi for providing research grant (SB/FT/CS-033/2012) for the work. We also thank BITS Pilani for providing additional research grant for the work. We would also like to thank Jitendra Kumar and Rajnish Singh, for providing POM images and Fluorescence Optical Microscopic images respectively. We would also like to thank Dr Subhashish Gangopadhyay, Dr Anshuman Dalvi and Sumita choudhary for the XRD analysis and DST FIST Project (SR/FST/PSI-150/2010) for facilitating XRD facility at Department of physics, BITS Pilani. The authors would also like to thank Materials Research Centre (MRC), MNIT, Jaipur for TEM analysis. The author DA thank DST, New Delhi, India for providing JRF fellowship. The author, NG thanks IASST, Guwahati for the characterization facility and CSIR, New Delhi for the fellowship.
Notes and references
-
(a) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133 CrossRef CAS PubMed;
(b) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821 RSC;
(c) A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644 CrossRef CAS PubMed;
(d) A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109 RSC;
(e) J. H. Jung, M. Park and S. Shinkai, Chem. Soc. Rev., 2010, 39, 4286 RSC;
(f) S. S. Babu, K. K. Kartha and A. Ajayaghosh, J. Phys. Chem. Lett., 2010, 1, 3413 CrossRef CAS;
(g) A. Dawn, T. Shiraki, S. Haraguchi, S. Tamaru and S. Shinkai, Chem.–Asian J., 2011, 6, 266 CrossRef CAS PubMed;
(h) V. K. Praveen, C. Ranjith, E. Bandini, A. Ajayaghosh and N. Armaroli, Chem. Soc. Rev., 2014, 43, 4222 RSC;
(i) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973 CrossRef CAS PubMed.
-
(a) M. Suzuki, Y. Sakakibara, S. Kobayashi, M. Kimura, H. Shirai and K. Hanabusa, Polym. J., 2002, 34, 474 CrossRef CAS;
(b) R. J. H. Hafkamp, B. P. A. Kokke, I. M. Danke, H. P. M. Geurts, A. E. Rowan, M. C. Feiters and R. J. M. Nolte, Chem. Commun., 1997, 545 RSC;
(c) Y. Ono, K. Nakashima, M. Sano, Y. Kanekiyo, K. Inoue, J. Hojo and S. Shinkai, Chem. Commun., 1998, 1477 RSC;
(d) J. H. Jung, H. Kobayashi, M. Masuda, T. Shimizu and S. Shinkai, J. Am. Chem. Soc., 2001, 123, 8785 CrossRef CAS PubMed;
(e) E. D. Sone, E. R. Zubarev and S. I. Stupp, Angew. Chem., Int. Ed., 2002, 114, 1781 (Angew. Chem., Int. Ed., 2002, 41, 1705) CrossRef.
-
(a) S. Kobayashi, N. Hamasaki, M. Suzuki, M. Kimura, H. Shinkai and K. Hanabusa, J. Am. Chem. Soc., 2002, 124, 6550 CrossRef CAS PubMed;
(b) C. Zhan, J. Wang, J. Yuan, H. Gong, Y. Liu and M. Liu, Langmuir, 2003, 19, 9440 CrossRef CAS;
(c) S. Kobayashi, K. Hanabusa, N. Hamasaki, M. Kimura, H. Shirai and S. Shinkai, Chem. Mater., 2000, 12, 1523 CrossRef CAS;
(d) F. S. Schoonbeek, J. H. van Esch, B. Wagewijs, D. B. A. Rep, M. P. de Haas, T. M. Klapwijk, R. M. Kellog and B. L. Feringa, Angew. Chem., Int. Ed., 1999, 111, 1486 (Angew. Chem., Int. Ed., 1999, 38, 1393) CrossRef;
(e) F. Placin, J.-P. Desvergne and J.-C. Lassegues, Chem. Mater., 2001, 13, 117 CrossRef CAS;
(f) A. Shumburo and M. C. Biewer, Chem. Mater., 2002, 14, 3745 CrossRef CAS;
(g) G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312 CAS.
-
(a) T. Polubesova, S. Nir, D. Zakada, O. Rabinovitz, C. Serban, L. Groisman and B. Rubin, Environ. Sci. Technol., 2005, 39, 2343 CrossRef CAS PubMed;
(b) R. Denoyel and E. S. Rey, Langmuir, 1998, 14, 7321 CrossRef CAS;
(c) A. Sayari, S. Hamoudi and Y. Yang, Chem. Mater., 2005, 17, 212 CrossRef CAS;
(d) M. Arkas, D. Tsiourvas and C. M. Paleos, Chem. Mater., 2005, 17, 3439 CrossRef CAS;
(e) P. Kofinas and D. R. Kioussis, Environ. Sci. Technol., 2003, 37, 423 CrossRef CAS PubMed;
(f) V. Bekiari and P. Lianos, Chem. Mater., 2006, 18, 4142 CrossRef CAS;
(g) S. Ray, A. K. Das and A. Banerjee, Chem. Mater., 2007, 19, 1633 CrossRef CAS;
(h) E. J. Cho, I. Y. Jeong, S. J. Lee, W. S. Han, J. K. Kang and J. H. Jung, Tetrahedron Lett., 2008, 49, 1076 CrossRef CAS.
-
(a) M. Suzuki, T. Sato, A. Kurose, H. Shirai and K. Hanabusa, Tetrahedron Lett., 2005, 46, 2741 CrossRef CAS;
(b) M. Suzuki, S. Owa, M. Kimura, A. Kurose, H. Shiraib and K. Hanabusa, Tetrahedron Lett., 2005, 46, 303 CrossRef CAS.
-
(a) N. Mohmeyer and H. W. Schmidt, Chem.–Eur. J., 2005, 11, 863 CrossRef CAS PubMed;
(b) M. Suzuki, M. Nanbu, M. Yumoto, H. Shiraib and K. Hanabusa, New J. Chem., 2005, 29, 1439 RSC;
(c) M. Suzuki, T. Nigawara, M. Yumoto, M. Kimura, H. Shirai and K. Hanabusa, Org. Biomol. Chem., 2003, 1, 4124 RSC;
(d) H. Yang, T. Yi, Z. Zhou, Y. Zhou, J. Wu, M. Xu, F. Li and C. Huang, Langmuir, 2007, 23, 8224 CrossRef CAS PubMed;
(e) M. Suzuki, T. Sato, H. Shiraib and K. Hanabusa, New J. Chem., 2006, 30, 1184 RSC;
(f) N. Mohmeyer and H. W. Schmidt, Chem.–Eur. J., 2007, 13, 4499 CrossRef CAS PubMed;
(g) M. Suzuki, Y. Nakajima, M. Yumoto, M. Kimura, H. Shirai and K. Hanabusa, Org. Biomol. Chem., 2004, 2, 1155 RSC.
-
(a) S. Roy, A. Dasgupta and P. K. Das, Langmuir, 2007, 23, 11769 CrossRef CAS PubMed;
(b) A. Shome, S. Debnath and P. K. Das, Langmuir, 2008, 24, 4280 CrossRef CAS PubMed.
-
(a) S. Debnath, A. Shome, S. Dutta and P. K. Das, Chem.–Eur. J., 2008, 14, 6870 CrossRef CAS PubMed;
(b) S. Dutta, D. Das, A. Dasgupta and P. K. Das, Chem.–Eur. J., 2010, 16, 1493 CrossRef CAS PubMed;
(c) D. Das, T. Kar and P. K. Das, Soft Matter, 2012, 8, 2348 RSC.
- S. R. Jadhav, P. K. Vemula, R. Kumar, S. R. Raghavan and G. John, Angew. Chem., Int. Ed., 2010, 49, 7695 CrossRef CAS PubMed.
- S. Mukherjee and B. Mukhopadhyay, RSC Adv., 2012, 2, 2270 RSC.
-
(a) L. A. Estroff and A. D. Hamilton, Angew. Chem., Int. Ed., 2000, 112, 3589 (Angew. Chem., Int. Ed., 2000, 39, 3447) CrossRef;
(b) K. Hanabusa, A. Itoh, M. Kimura and H. Shirai, Chem. Lett., 1999, 767 CrossRef CAS;
(c) F. M. Menger and K. L. Caran, J. Am. Chem. Soc., 2000, 122, 11679 CrossRef CAS;
(d) K. Hanabusa, H. Nakayama, M. Kimura and H. Shirai, Chem. Lett., 2000, 1070 CrossRef CAS;
(e) H. M. Willemen, T. Vermonden, A. T. M. Marcelis and E. J. R. Sudhçlter, Eur. J. Org. Chem., 2001, 2329 CrossRef CAS;
(f) X. Luo, B. Lin and Y. Liang, Chem. Commun., 2001, 1556 RSC;
(g) A. Dasgupta, R. N. Mitra, S. Roy and P. K. Das, Chem.–Asian J., 2006, 1, 780 CrossRef CAS PubMed;
(h) J. Makarevic, M. Kokic, B. Peric, V. Tomisic, B. Kojic-Prodic and M. Zinic, Chem.–Eur. J., 2001, 7, 3328 CrossRef CAS PubMed;
(i) J. Becerril, M. I. Burguete, B. Escuder, S. V. Luis, J. F. Miravet and M. Querol, Chem. Commun., 2002, 738 RSC;
(j) M. Jokic, J. Makarevic and M. Zinic, J. Chem. Soc., Chem. Commun., 1995, 1723 RSC;
(k) H. T. Stock, N. J. Turner and R. J. McCague, J. Chem. Soc., Chem. Commun., 1995, 2063 RSC;
(l) M. Suzuki, T. Sato, A. Kurose, H. Shirai and K. Hanabusa, Tetrahedron Lett., 2005, 46, 2741 CrossRef CAS;
(m) M. Suzuki, S. Owa, M. Kimura, A. Kurose, H. Shiraib and K. Hanabusa, Tetrahedron Lett., 2005, 46, 303 CrossRef CAS;
(n) S. Roy, D. Das, A. Dasgupta, R. N. Mitra and P. K. Das, Langmuir, 2005, 21, 10398 CrossRef CAS PubMed;
(o) S. Roy and P. K. Das, Biotechnol. Bioeng., 2008, 100, 756 CrossRef CAS PubMed.
-
(a) O. Gronwald and S. Shinkai, Chem.–Eur. J., 2001, 7, 4329 CrossRef CAS;
(b) A. Friggeri, O. Gronwald, K. J. C. van Bommel, S. Shinkai and D. N. Reinhoudt, J. Am. Chem. Soc., 2002, 124, 10754 CrossRef CAS PubMed;
(c) S. Kiyonaka, S. Shinkai and I. Hamachi, Chem.–Eur. J., 2003, 9, 976 CrossRef CAS PubMed.
- T. Nakashima and N. Kimizuka, Adv. Mater., 2002, 14, 1113 CrossRef CAS.
- S. Bhattacharya and Y. K. Ghosh, Chem. Commun., 2001, 185 RSC.
-
(a) T. Kar, S. Debnath, D. Das, A. Shome and P. K. Das, Langmuir, 2009, 25, 8639 CrossRef CAS PubMed;
(b) M. Konda, I. Maity, D. B. Rasale and A. K. Das, ChemPlusChem, 2014, 79, 1482 CrossRef CAS;
(c) M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2009, 38, 967 RSC.
-
(a) H. Svobodova, V. Noponen, E. Kolehmainen and E. Sievanen, RSC Adv., 2012, 2, 4985 RSC;
(b) V. S. Pore, S. G. Agalave, S. G. Pharande, P. A. Patil and A. S. Kotmale, New J. Chem., 2015, 39, 453 RSC.
- Y. Ni, Z. HeLan, X. Z. Yan and F. Yu, Chin. Sci. Bull., 2012, 57, 4310 CrossRef.
- M. Xue, D. Gao, K. Liu, J. Peng and Y. Fang, Tetrahedron, 2009, 65, 3369 CrossRef CAS.
-
(a) X. Yu, L. Chen, M. Zhanga and T. Yi, Chem. Soc. Rev., 2014, 43, 5346 RSC;
(b) D. Trivedi, A. Ballabh and P. Dastidar, Chem. Mater., 2003, 15, 3971 CrossRef CAS.
- S. Mukherjee, C. Shang, X. Chen, X. Chang, K. Liu, C. Yu and Y. Fang, Chem. Commun., 2014, 50, 13940 RSC.
- X. Yang, G. Zhang and D. Zhang, J. Mater. Chem., 2012, 22, 38 RSC.
- Y. Zang, Y. Ma, M. Deng, H. Shang, C. Liang and S. Jiang, Soft Matter, 2015, 11, 5095 RSC.
- R. Rajaganesh, A. Gopal, T. M. Das and A. Ajayaghosh, Org. Lett., 2012, 14, 748 CrossRef CAS PubMed.
- J. Bachl, S. Oehm, J. Mayr, C. Cativiela, J. J. M. Tellado and D. D. Diaz, Int. J. Mol. Sci., 2015, 16, 11766 CrossRef CAS PubMed.
- R. Yang, S. Peng and T. C. Hughes, Soft Matter, 2014, 10, 2188 RSC.
-
(a) A. R. Katritzky, R. Sakhuja, L. Khelashvili and K. Shanab, J. Org. Chem., 2009, 74, 3062 CrossRef CAS PubMed;
(b) P. K. Vemula, J. Li and G. John, J. Am. Chem. Soc., 2006, 128, 8932 CrossRef CAS PubMed.
-
(a) S. Bhattacharya and S. N. G. Acharya, Chem. Mater., 1999, 11, 3504 CrossRef CAS;
(b) O. Gronwald and S. Shinkai, Chem.–Eur. J., 2001, 7, 4328 CrossRef CAS PubMed.
-
(a) A. R. Hirst and D. K. Smith, Org. Biomol. Chem., 2004, 2, 2965 RSC;
(b) A. K. Das, S. Manna, M. G. B. Drew, S. Malik, A. Nandi and A. Banerjee, Supramol. Chem., 2006, 18, 645 CrossRef CAS;
(c) S. K. Maji, S. Malik, M. G. B. Drew, A. Nandi and A. Banerjee, Tetrahedron Lett., 2003, 44, 4103 CrossRef CAS.
- G. Palui and A. Banerjee, J. Phys. Chem. B, 2008, 112, 10107 CrossRef CAS PubMed.
-
(a) S. G. Kumar and D. C. Neckers, Chem. Rev., 1989, 89, 1915 CrossRef;
(b) K. Ichimura, Chem. Rev., 2000, 100, 1847 CrossRef CAS PubMed;
(c) D. E. W. Guang, J. Phys. Chem. A, 2004, 108, 950 CrossRef.
- N. A. Wazzan, P. R. Richardson and A. C. Jones, Photochem. Photobiol. Sci., 2010, 30, 968 Search PubMed.
-
(a) L. Guterman, Science, 2009, 323, 1558 CrossRef CAS PubMed;
(b) Estimated to be B900 000 metric tons per annum. http://www.buzzle.com/articles/ocean-pollution-causes.html.
- A. M. Thayer, Chem. Eng. News, 2011, 89, 13 Search PubMed.
- R. P. J. Swannell, K. Lee and M. Mcdonagh, Microbiol. Rev., 1996, 60, 342 CAS.
- R. R. Lessard and G. Demarco, Spill Sci. Technol. Bull., 2000, 6, 59 CrossRef CAS.
-
(a) Y. I. Matatov-Meytal and M. Sheintuch, Ind. Eng. Chem. Res., 1997, 36, 4374 CrossRef CAS;
(b) M. O. Adebajo, R. L. Frost, J. T. Kloprogge, O. Carmody and S. Kokot, J. Porous Mater., 2003, 10, 159 CrossRef CAS.
- E. Pelletier and R. Siron, Environ. Toxicol. Chem., 1999, 18, 813 CrossRef CAS.
-
(a) C. C. Tsai, Y. T. Cheng, L.-C. Shen, K.-C. Chang, I.-T. Ho, J.-H. Chu and W.-S. Chung, Org. Lett., 2013, 15, 5830 CrossRef CAS PubMed;
(b) N. V. Lakshmi, T. M. Babu and E. Prasad, Chem. Commun., 2016, 52, 617 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data of compounds 3a–c, 4a–c, 6a–f, 7a–f, 9a–f. Original copies of 1H NMR and 13C NMR of 3a–c, 4a–c, 6a–f, 7a–f, 9a–f. SEM images of xerogel of 9a in cyclohexane, hexane, benzene, tert-butylbenzene mesitylene and xerogel of 9e in dodecane. See DOI: 10.1039/c6ra13160c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.