
A computational study of the mechanism of the photocyclization reaction of α-methylamino ketone†
Shih-Hao Sua and
Ming-Der Su*ab
aDepartment of Applied Chemistry, National Chiayi University, Chiayi 60004, Taiwan. E-mail: midesu@mail.ncyu.edu.tw
bDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung 80708, Taiwan
First published on 19th August 2016
Abstract
The mechanism for the photochemical cyclization reaction is studied theoretically using a model system of α-N-methylamidoacetophenone with the M06-L/6-311G(d,p) method. These model studies indicate that the preferred reaction pathway for α-N-methylamino ketone, which leads to the photocyclization product, is as follows: reactant (singlet) → Franck–Condon region (triplet) → local minimum (triplet) → transition state (triplet) → intersystem crossing → intermediate (singlet) → transition state (singlet) → photoproduct (singlet). In other words, the intersystem crossing mechanism plays a decisive role in these photo-rearrangement reactions for α-N-alkylamino ketone systems. These computational observations are in agreement with the available experimental results.
I. Introduction
Carbon dioxide (CO2) is a useful carbon source because of its abundance, low-cost, and non-toxicity. As a result, the use of CO2 as a starting material for the production of chemicals means that it is an excellent chemical feedstock.1 However, CO2 is well known to be difficult to activate because the molecule is stable both thermodynamically and kinetically.2 Sunlight is easily harvested and it is the main carbon-neutral energy source for the future3 so the development of photo-catalyzed CO2 incorporation reactions into organic compounds is of interest to the field of synthetic organic chemistry.4For many years, C–C bond forming carboxylation reactions of organic compounds with CO2 have obtained considerable attention in organic synthesis.5 However, the larger driving force of these carboxylation reactions comes from the chemical reagents. Recently, Murakami and co-workers6 have reported that upon exposure to sunlight the photolysis of α-N-methylamidoacetophenone (1)7 results in a very high yield of the corresponding N-substituted 3-azetidinol (2) to produce a four-membered ring. CO2 is then successfully incorporated into 2, upon exposure of a solution of 2 in N,N-dimethylacetamide (DMA) to gaseous CO2 in the presence of a base. See Scheme 1. The resulting amino-based cyclic carbonate (3) is a valuable building block for pharmaceutical agents, insecticides and fuel additives.8 The first step of this solar-driven reaction is to construct a high-energy four-membered ring by inserting a photo-induced carbonyl fragment into the C–H bond of a CH3 group on a nitrogen atom. In other words, the net molecular circumstance for this photo-assisted reaction requires the insertion of a carbonyl group into a C–H σ bond to form a new carbon–carbon σ bond and a new oxygen–hydrogen σ bond. The carbon–hydrogen σ bond is also broken.9 There have been no experimental or theoretical studies of the photo-formation mechanism for a four-membered azetidinol 2.
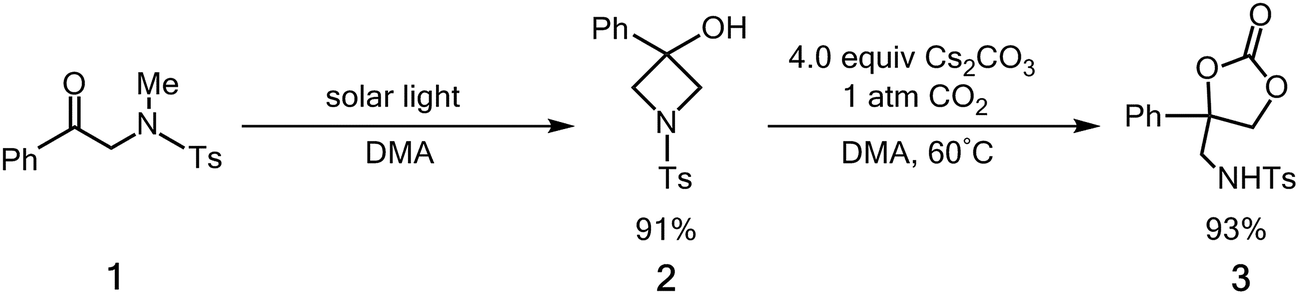 | ||
| Scheme 1 The solar-driven cyclization of 1 and its incorporation of CO2 incorporation. Ts = toluenesulfonyl. | ||
A computational study of the photo-cyclization of α-N-methylamino ketone (1)5 is therefore undertaken in order to gain information about its photochemical behaviour. This work shows that the singlet–triplet crossing plays a prominent role in the photo-rearrangement of α-amino ketone systems. Indeed, sunlight is easily harvested and it is the main carbon-neutral energy source for the future so the development of photo-catalyzed CO2 incorporation reactions into organic compounds is of interest to the field of synthetic organic chemistry. It is hoped this study gives an improved explanation of the energetic aspects of these photo-cyclization reactions and may allow optimization of the design for other related synthetic organic chemistry.
II. Computational methods
Quantum chemistry computations were performed using the Gaussian-09 system of programs.10 All of the stationary points on the potential energy surfaces studied in this work were characterized by calculating vibrational frequencies, using the M06-L functional11 with the 6-311G(d,p) basis set. The reason for using M06-L functional is because it was recommended by Truhlar and co-worker that M06-L is the only local functional (no Hartree–Fock exchange) with better across-the-board average performance than B3LYP. This is quite important because only local functionals are affordable for many demanding applications on very large systems.11 The other reason is based on our computational experiences.12 We found that the M06-L functional can obtain reliable TD-DFT results about the organic and organometallic molecules.The vertical energies of the singlet–triplet excited-state transitions were computed using the time-dependent density functional theory (TD-DFT) method,13 TD-M06-L/6-311G(d,p). It was established that TD-DFT gives good qualitative accuracy for some small molecules (such as H2O and CH4).13d The method is computationally economical because TD-DFT is eminently suited for application to more complicated compounds.13e The TD-DFT/M06-L/6-311G(d,p) computational results are listed in the ESI.† The minimum energy crossing points between the triplet and singlet potential energy surfaces for 1 were computed using the program that was developed by Harvey et al.,14 which is incorporated in the Gaussian 09 package of programs.10 The Cartesian coordinates and the energies that were computed for these stationary points are available as ESI.†
III. Results and discussion
In order to understand the photo-cyclization mechanisms of 1, its reaction profiles, which are summarized in Fig. 1, form the basis for discussion. In this figure, the relative energies of the crucial points with respect to the ground-state minimum 1 are given. Fig. 2 (path I) and 3 (path II) contain all of the stationary points and some specifically optimized geometrical parameters.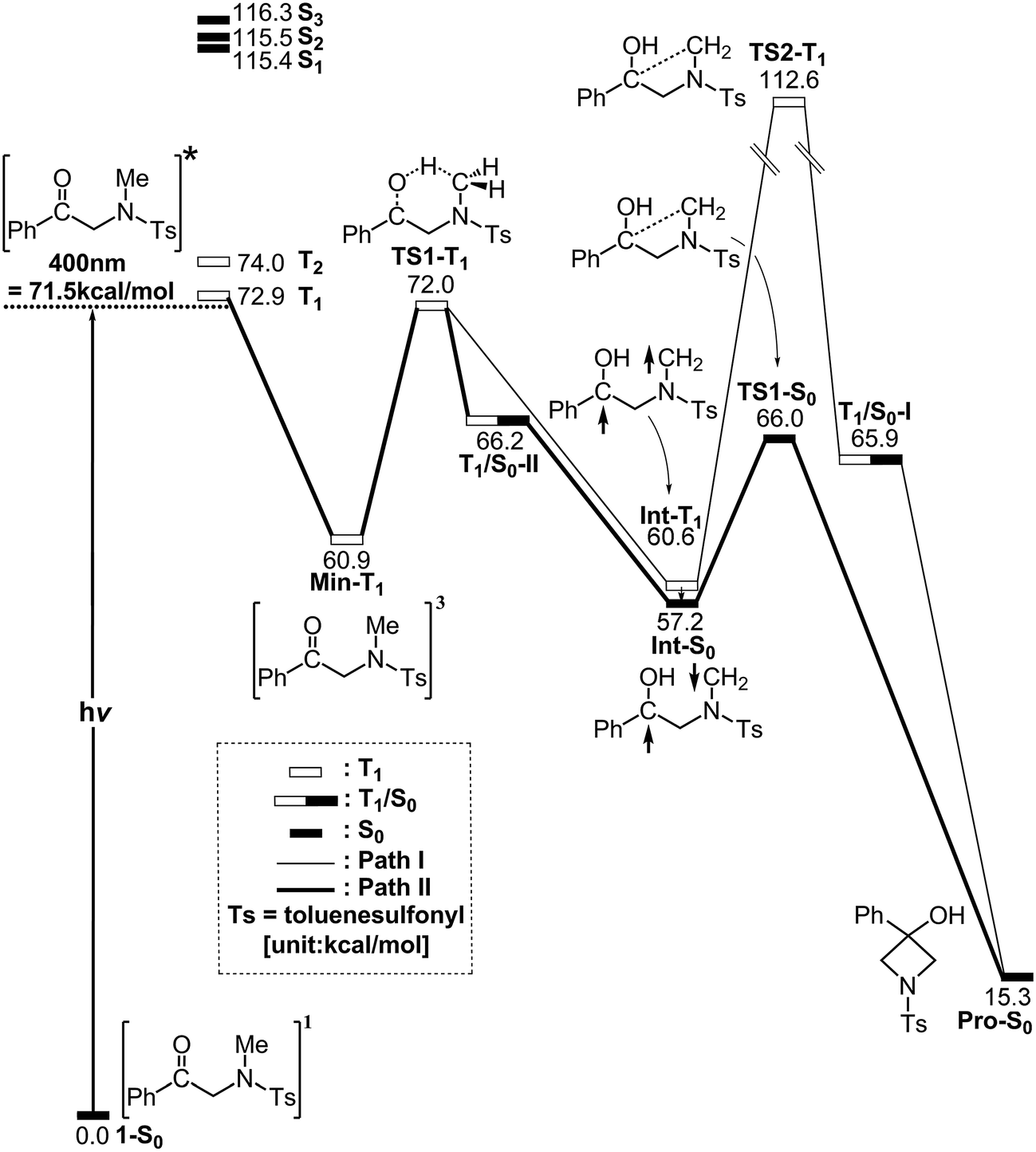 | ||
| Fig. 1 The energy profiles for the photochemical cyclization mode for 1. The abbreviations, FC, TS, Int and T1/S0 respectively represent Frank–Condon, transition state, intermediate and intersystem crossing. The relative energies are calculated at the M06-L/6-311G(d,p) level of theory. For more information see the text. | ||
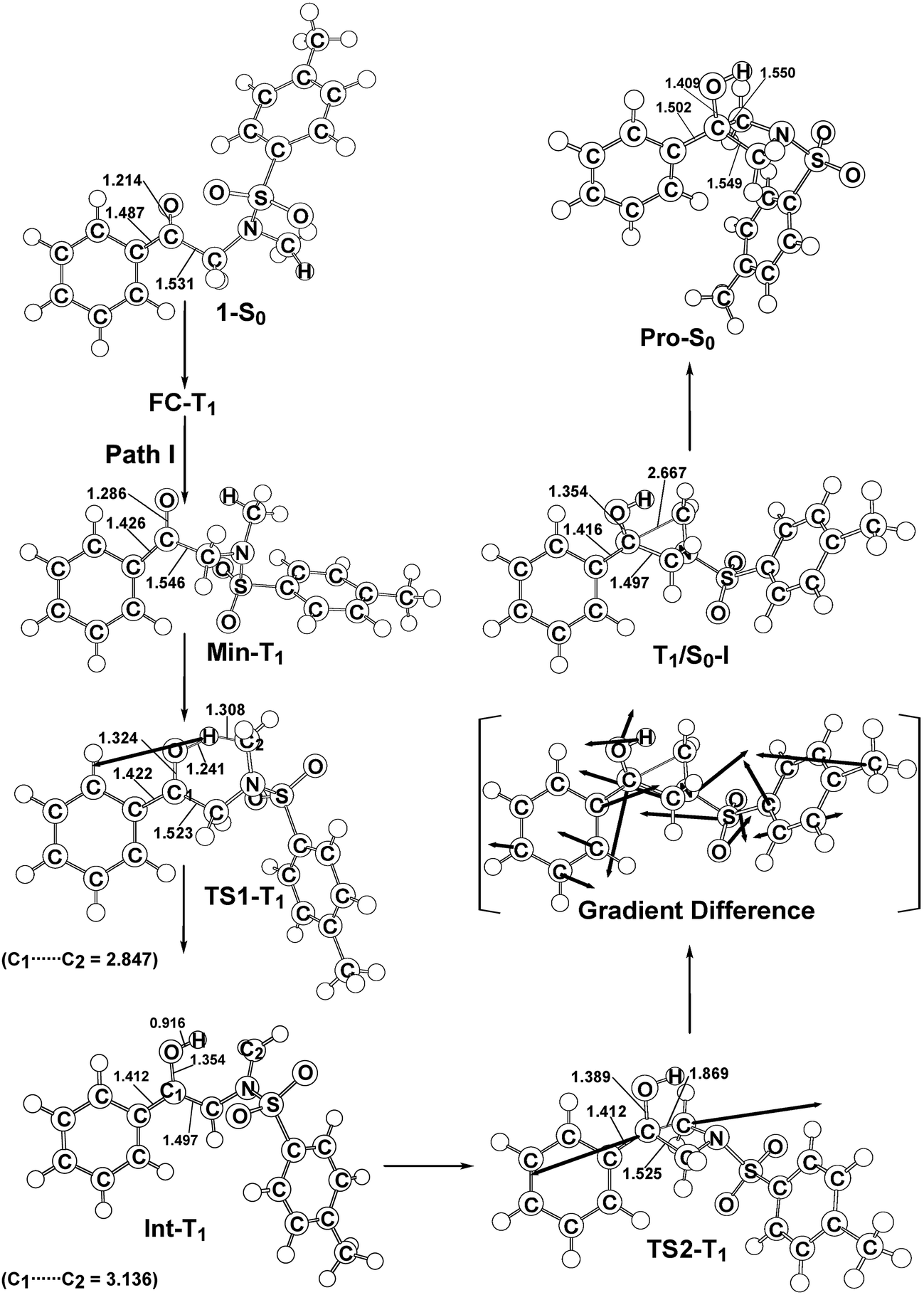 | ||
| Fig. 2 The M06-L/6-311G(d,p) geometries (in Å and deg) for the reactant (1-S0), Frank–Condon (FC-T1), minimum (Min-T1), transition state (TS1-T1), intermediate (Int-T1), TS2-T1, intersystem crossing (T1/S0-I), and photoproduct (Pro-S0). The heavy arrows in TS indicate the main atomic motions in the transition state eigenvector. The gradient difference vector of T1/S0-I, which is computed using M06-L, is shown in the square bracket. Hydrogen atoms are omitted for clarity. For more details see the ESI.† | ||
Gray et al.15 showed that TD-DFT results largely depend on the choice of the functional and basis sets so a multi reference treatment might prevent these problems. However, the limitations of disk and memory space render an exhaustive study unfeasible so the TD-DFT/M06-L/6-311G(d,p) level of theory is used for this paper. The TD-DFT/M06-L/6-311G(d,p) calculations are initially performed for molecule 1, which gives the vertical excitation energies and oscillator strengths for the fifteen lowest energy triplet (Table S1†) and singlet (Table S2†) excited states. These TD-DFT calculations show that several electronic levels that are near to the energy of a 400 nm photon: two triplet states, T1 (72.9 kcal mol−1) and T2 (74.0 kcal mol−1), and three singlet states, S1 (115.4 kcal mol−1), S2 (115.5 kcal mol−1) and S3 (116.3 kcal mol−1). In other words, the TD-DFT results demonstrate that the lowest-lying excited state for molecule 1 is the first excited triplet state (T1). These theoretical calculations show that direct population of the singlet excited states is unlikely, so this study focuses only on the triplet state.16
After photo-irradiation, molecule 1 is promoted to its lowest-lying triplet state by a vertical excitation, which is denoted by FC-T1 (Fig. 1). Although this Frank–Condon point is on the triplet surface, it still has the ground-state geometry (i.e., T1 (S0 geom)). The vertical triplet excitation energy for molecule 1 was computed to be 77 kcal mol−1 (S0 → T1 (S0 geom)) at the M06-L/6-311G(d,p) level of theory. According to the experimental results reported by Murakami and co-workers,1 light at wavelengths of less than 400 nm (=72 kcal mol−1 in energy) is required for this photochemical reaction. Therefore, this comparison shows that these provide a reasonable estimate of the relative energies for the α-N-methylamino ketone system.
From FC-T1, molecule 1 relaxes to a local minimum on the triplet surface, which is denoted by Min-T1. This is calculated to be about 61 kcal mol−1 above the ground-state minimum (1-S0). With reference to Fig. 2, a comparison of the Min-T1 geometry with that of its corresponding 1-S0 readily shows that the triplet state has a significantly greater C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond distance than its singlet ground state.
O bond distance than its singlet ground state.
From Min-T1, isomerization to the final photoproduct (Pro-S0) involves two possible reaction routes (path I and path II), as shown in Fig. 1. On path I, Min-T1 undergoes a 1,2-hydrogen shift (TS1-T1) to give an intermediate on a triplet surface, Int-T1, which subsequently reaches the second transition state, TS2-T1. Vibrational frequency calculations indicate that TS1-T1 and TS2-T1 respectively involve breaking a carbon–hydrogen bond (1687i cm−1) and the formation of a carbon–carbon bond (868i cm−1). The computed transition vectors for these processes are shown in Fig. 2. Fig. 1 also shows that the TS2-T1 transition state connects the triplet local minimum Int-T1 to the corresponding intersystem crossing point (T1/S0-I). From T1/S0-I, this system then arrives at the final photoproduct on the ground singlet surface (Pro-S0). The M06-L calculations for this study suggest that the relative energies of TS1-T1, Int-T1, and T1/S0-I are above those of the corresponding reactant 1-S0 by 72, 61 and 66 kcal mol−1, respectively. In particular, the energy of TS2-T1 is 41 kcal mol−1 higher than that of its corresponding FC-T1, which represents a significant difference. This finding strongly suggests that the triplet-excited energy of 1 is insufficient to yield a photo-cyclization product Pro-S0 via path I. The process for path I is represented as:
| Path I: 1-S0 + hν → FC-T1 → Min-T1 → TS1-T1 → Int-T1 → TS2-T1 → T1/S0-I → Pro-S0 |
Similarly to path I (Fig. 1), on path II, from the FC-T1 point, reactant 1 relaxes to an excited triplet state minimum, Min-T1. This triplet Min-T1 then experiences a 1,2-hydrogen migration to reach a triplet transition state, TS1-T1, followed by an intersystem crossing, T1/S0-II. From this crossing point, the system then goes to the local minimum on the singlet potential energy surface, Int-S0. The M06-L computations suggest that the Int-T1 point also relaxes to the Int-S0 intermediate via a very small release of energy, the energy difference between them being only 3.4 kcal mol−1, with the Int-T1 lying above the Int-S0 species, as shown in Fig. 1. The fully optimized structures for the triplet (Int-T1) and singlet (Int-S0) states are respectively shown in Fig. 2 and 3. Both Fig. 2 (path I) and 3 (path II) show that the Int-T1 and Int-S0 intermediate points have a five-center-staggered structure, wherein two unpaired electrons are located on the C1 and C2 atoms. These two unpaired electrons are sufficiently distant from each other that the triplet Int-T1 (3.1 Å) and the singlet Int-S0 (2.7 Å) states are almost degenerate. In other words, the theoretical findings suggest that the short-lived singlet Int-S0 intermediate can be produced via either the triplet Int-T1 species after a very small release of energy (path I) or via a fast T1/S0-II crossover point (path II). Both pathways generate a large amount of the Int-S0 species during these photochemical cyclization reactions. Finally, after crossing a small energy barrier (8.8 kcal mol−1) for the singlet transition state (TS1-S0) with one imaginary frequency (229i cm−1), the Int-S0 intermediate leads to a photoproduct with a four-membered-ring, Pro-S0, as shown in Fig. 1 and 2. These computational results show that the energies of T1/S0-II, Int-S0 and TS1-S0 lie above that of the corresponding reactant, 1-S0, by 66, 57, and 66 kcal mol−1, respectively. However, these relative energies are all smaller than the vertical triplet excitation energy (72 kcal mol−1). These model studies therefore demonstrate that, when sunlight irradiates α-N-methylamino ketone (1), molecule 1 results in the final photoproduct, Pro-S0, without any difficulty. These theoretical results indicate that the photo-cyclization reaction for path II should proceed as follows:
| Path II: 1-S0 + hν → FC-T1 → Min-T1 → TS1-T1 → T1/S0-II → Int-S0 → TS1-S0 → Pro-S0 |
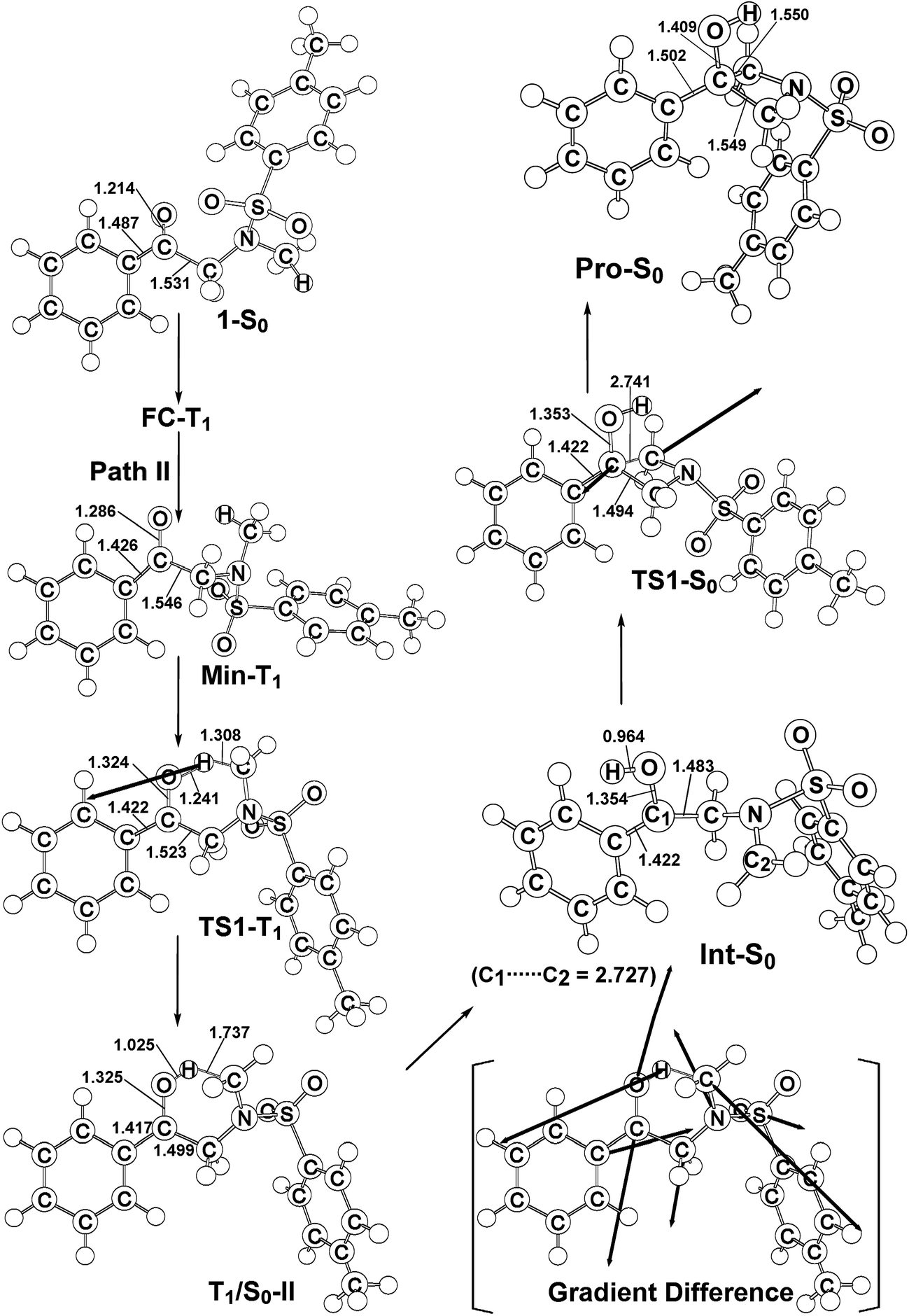 | ||
| Fig. 3 The M06-L/6-311G(d,p) geometries (in Å and deg) for the reactant (1-S0), Frank–Condon (FC-T1), minimum (Min-T1), transition state (TS1-T1), intersystem crossing (T1/S0-II), intermediate (Int-S0), TS1-S0, and photoproduct (Pro-S0). The heavy arrows in TS indicate the main atomic motions in the transition state eigenvector. The gradient difference vector of T1/S0-II, which is computed using M06-L, is shown in the square bracket. Hydrogen atoms are omitted for clarity. For more details see the ESI.† | ||
Moreover, from the above theoretical analysis (Fig. 1), it would have the third photo-cyclization reaction pathway (path III), which is represented as follows:
| Path III: 1-S0 + hν → FC-T1 → Min-T1 → TS1-T1 → Int-T1 → Int-S0 → TS1-S0 → Pro-S0 |
IV. Conclusion
The photo-rearrangement mechanisms for α-N-methylamino ketone (1) are theoretically studied using density functional theory (M06-L/6-311G(d,p)). These model studies demonstrate that upon absorption of sunlight, molecule 1 is excited vertically to the triplet state (FC-T1). From this point, the system can return to the singlet ground state via three radiation-less decay pathways (path I, path II, and path III). The M06-L studies point to the fact that path I is quite inefficient for this photo-cyclization reaction because the process has a high activation barrier. However, the theoretical observations strongly suggest that either path II or path III is more favorable, both energetically and kinetically.The photochemical cyclization reaction for 1 originates from the excited triplet surface and eventually advances along the singlet ground-state path. Therefore, the spin intersystem crossing between the triplet (T1) and singlet (S0) surfaces must play a prominent role in characterizing the mechanistic photo-induced reactions for these α-N-alkylamino ketone species because the spin crossover influences their fundamental photochemistry. The mechanism that uses the intersystem crossing gives a better understanding of the photochemical reactions for α-N-alkylamino ketone molecules and allows experimental observations.6,17
Acknowledgements
The authors are grateful to the National Center for High-Performance Computing of Taiwan for generous gift of computational time. They also thank the National Science Council of Taiwan for the financial support. One of the author (M.-D. Su) also wishes to thank Professor Michael A. Robb, Dr Michael J. Bearpark, (University of London, UK), Professor Massimo Olivucci (Universita degli Sstudi di Siena, Italy), and Professor Fernando Bernardi (University of Bologna, Italy) for their encouragement and support during his stay in London. Special thanks are also due to reviewers 1 and 2 for very help suggestions and comments.References
- For instance: (a) A. Behr, Carbon Dioxide Activation by Metal Complexes, VCH Publisher, Weinheim, Germany, 1988 Search PubMed; (b) T. Ito and A. Yamamoto, Organometallic Reactions of Carbon Dioxide, Organic and Bioorganic Chemistry of Carbon Dioxide, ed. S. Inoue and N. Yamazaki, Kodansha Ltd., Tokyo, Japan, 1982, ch. 3 Search PubMed.
- D. H. Gibson, Chem. Rev., 1996, 96, 2063 CrossRef CAS PubMed.
- N. Armaroli and V. Balzani, Energy for a Sustainable World, From the Oil Age to a Sun-Powered Future, Wiley-VCH, Weinheim, 2011 Search PubMed.
- For instance: (a) D. B. Dell'Amico, F. Calderazzo, L. Labella, F. Marchetti and G. Pampaloni, Chem. Rev., 2003, 103, 3857 CrossRef PubMed; (b) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed; (c) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388 CrossRef CAS PubMed; (d) Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, Weinheim, 2010 Search PubMed; (e) S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347 RSC; (f) A. Behr and G. Henze, Green Chem., 2011, 13, 25 RSC; (g) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Khn, Angew. Chem., Int. Ed., 2011, 50, 8510 CrossRef CAS PubMed; (h) I. Omae, Coord. Chem. Rev., 2012, 256, 1384 CrossRef CAS.
- For a general review: (a) A. S. Lindsey and H. Jeskey, Chem. Rev., 1957, 57, 583 CrossRef CAS; (b) P. Braunstein, D. Matt and D. Nobel, Chem. Rev., 1988, 88, 747 CrossRef CAS; (c) X. Yin and J. R. Moss, Coord. Chem. Rev., 1999, 181, 27 CrossRef CAS; (d) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed; (e) Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, Weinheim, 2010 Search PubMed; (f) S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347 RSC; (g) R. Martin and A. W. Kleij, ChemSusChem, 2011, 4, 1259 CrossRef CAS PubMed; (h) K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435 RSC; (i) I. I. F. Boogaerts and S. P. Nolan, Chem. Commun., 2011, 47, 3021 RSC; (j) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510 CrossRef CAS PubMed; (k) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956 RSC; (l) L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395 RSC.
- N. Ishida, Y. Shimamoto and M. Murakami, Angew. Chem., Int. Ed., 2012, 51, 11750 CrossRef CAS PubMed.
- The full chemical name of molecule 1 is N,4-dimethyl-N-(2-oxo-2-phenylethyl)benzenesulfonamide.
- For instance: (a) B. M. Trost and S. R. Angle, J. Am. Chem. Soc., 1985, 107, 6123 CrossRef CAS; (b) K. Iritani, N. Yanagihara and K. Uchimoto, J. Org. Chem., 1986, 51, 5499 CrossRef CAS; (c) A. G. Myers and K. L. Widdowson, Tetrahedron Lett., 1988, 29, 6389 CrossRef CAS; (d) J. Fournier, C. Bruneau and P. H. Dixneuf, Tetrahedron Lett., 1989, 30, 3981 CrossRef CAS; (e) Y. Kayaki, M. Yamamoto and T. Ikariya, Angew. Chem., Int. Ed., 2009, 48, 4194 CrossRef CAS PubMed; (f) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC; (g) S. Minakata, I. Sasaki and T. Ide, Angew. Chem., Int. Ed., 2010, 49, 1309 CrossRef CAS PubMed; (h) S. Yoshida, K. Fukui, S. Kikuchi and T. Yamada, J. Am. Chem. Soc., 2010, 132, 4072 CrossRef CAS PubMed.
- (a) E. H. Gold, J. Am. Chem. Soc., 1971, 93, 2793 CrossRef CAS; (b) K. L. Allworth, A. A. El-Hamamy, M. M. Hesabi and J. Hill, J. Chem. Soc., Perkin Trans. 1, 1980, 1671 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian, Inc., Wallingford, CT, 2013.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS PubMed.
- (a) S.-H. Su and M.-D. Su, RSC Adv., 2016, 6, 50825 RSC; (b) M.-D. Su, ChemistrySelect, 2016, 1, 1588 CrossRef; (c) S.-H. Su and M.-D. Su, Phys. Chem. Chem. Phys., 2016, 18, 16396 RSC.
- (a) R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454 CrossRef CAS; (b) M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS; (c) R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef CAS; (d) M. Stener, G. Fronzoni, D. Toffoli and P. Decleva, Chem. Phys., 2002, 282, 337 CrossRef CAS; (e) J. E. Monat, J. H. Rodriguez and J. K. McCusker, J. Phys. Chem. A, 2002, 106, 7399 CrossRef CAS.
- (a) J. N. Harvey, M. Aschi, H. Schwarz and W. Koch, Theor. Chem. Acc., 1998, 99, 95 CrossRef CAS; (b) J. N. Harvey and M. Aschi, Phys. Chem. Chem. Phys., 1999, 1, 5555 RSC; (c) J. N. Harvey and M. Aschi, Faraday Discuss., 2003, 124, 129 RSC.
- P. Hummel, J. Oxgaard, W. A. Goddard III and H. B. Gray, Inorg. Chem., 2005, 44, 2454 CrossRef CAS PubMed.
- In fact, it has been shown by several groups that the lowest lying excited state of α-alkylphenyl ketones is the triplet state. This implies that the intersystem-crossing mechanism plays a central role in their photochemical reactions. For instance, see: (a) P. J. Wagner, M. Sobczak and B.-S. Park, J. Am. Chem. Soc., 1998, 120, 2488 CrossRef CAS; (b) N. C. Baird, J. Am. Chem. Soc., 1972, 94, 4941 CrossRef CAS; (c) C. R. Flynn and J. Michl, J. Am. Chem. Soc., 1974, 96, 3280 CrossRef CAS; (d) A. Sevin, B. Bigot and M. Pfau, Helv. Chim. Acta, 1979, 62, 699 CrossRef CAS; (e) J. J. Dannenberg and J. C. Rayes, J. Org. Chem., 1983, 48, 4723 CrossRef CAS.
- As suggested by one reviewer, we also consider the solvent effect to study the correction for solvation with N,N-dimethylacetamide (DMA), whose dielectric constant (ε) is 37.8. We used the self-consistent reaction field (SCRF) method (SCRF = PCM) in Gaussian 09, whose computational results are given in Fig. S1 (ESI†). As seen in Fig. S1,† the relative energies computed with two methods, i.e., M06-L/6–311G(d,p) and M06-L/6–311G(d,p) + PCM (solvent = DMA)//M06-L/6–311G(d,p), are quite similar to each other. Therefore, we are confident that the present theoretical findings from the gas-phase study should be trustworthy for the same system on the solvent effects.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra15248a |
| This journal is © The Royal Society of Chemistry 2016 |