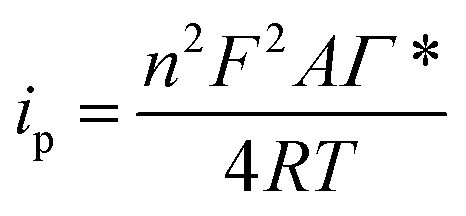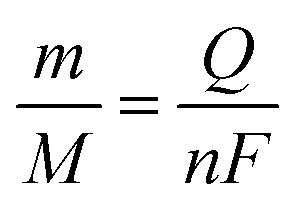DOI:
10.1039/C6RA15578B
(Paper)
RSC Adv., 2016,
6, 83748-83757
Electrodeposition of nickel nanoflowers on screen-printed electrodes and their application to non-enzymatic determination of sugars†
Received
15th June 2016
, Accepted 29th August 2016
First published on 30th August 2016
Abstract
In this work, the electrodeposition of nickel on screen-printed carbon electrodes was carried out. As the main novelty, a galvanostatic electrodeposition methodology (application of a constant current for a specific time) was chosen to perform the electrodeposition from a Ni(II) solution. Interestingly, these conditions were able to generate nickel nanoflowers of 160 nm all over the surface. The nickel nanoflowers showed a great electrocatalytic effect towards the oxidation of reducing sugars. After the characterization of the electrode surface and the optimization of the experimental conditions, the non-enzymatic electrochemical device was employed for the determination of reducing sugars. A linear range of 25–1000 μM was obtained, showing good performance for the determination of sugars at low concentrations. The reproducibility was 5.5% (intraelectrode) and 6.9% (interelectrode), indicating a high precision using the same or different devices. After fabrication, the electrode is stable at least for 35 days, even using the same device to carry out measurements on different days. Real food samples such as honey and orange juice were also evaluated with the nickel nanoflower electrochemical device.
1. Introduction
Glucose determination is a constant concern for the scientific community due to the fact that the diagnosis of diabetes mellitus disease has increased in the last years, and over 552 million patients are estimated to have this disease in 2030.1 Furthermore, the determination of sugars in food, most importantly, glucose and fructose, has a great interest for the food industry in order to evaluate the nutritional information or to control the quality of the production. The most employed techniques for sugar determination are refractometry, densitometry, titration and for speciation analysis, high performance liquid chromatography.2 These techniques have some disadvantages compared to electrochemical detection such as the bulky and more expensive instrumentation, large sample consumption and, generally, trained personnel is necessary to carry out the analyses. Electrochemical sensors have been widely employed for glucose determination, specially using enzyme-modified electrodes.3,4 This kind of sensors have some drawbacks such as the need to control exhaustively the experimental conditions such as pH or temperature. Changes in these conditions may cause a change in the stability of the enzyme and, therefore, in the performance of the sensor. For these reasons, the development of non-enzymatic devices for glucose determination is a relentless subject of study.5,6 Many nanomaterials have shown a strong catalytic effect towards sugars oxidation such as Pt, Cu or Au.6 Currently, these non-enzymatic nanostructured electrodes are widely used in the food industry, particularly in the area of food safety, traceability and quality control, as they provide low detection limits and high stability.7 Nanostructured electrodes have several advantages such as providing a more active surface, enhancing the electron transfer and may show catalytic properties towards different electrochemical reactions. For instance, different nickel nanomaterials have shown a catalytic effect towards the oxidation of reducing sugars. Nickel nanoparticles seems to be the most employed for the non-enzymatic determination of sugars using different strategies such as the addition to a carbon paste electrode,8 embedded in a chitosan membrane9 or electrodeposited on boron-doped diamond electrodes.10 Nickel nanowires have also shown a strong catalytic effect and have been employed for sugar determination coupled to a glassy carbon electrode,11 to disposable electrodes12 or in a nanowire array strategy.13 However, nickel nanoflowers (NiNFs) have not been used as broadly for electroanalytical applications, and instead, they have shown interesting features for batteries14 or supercapacitors.15 However, the high surface area of these flower-like nanoparticles could lead to promising analytical applications. Non-enzymatic glucose electrochemical devices based on nickel oxide or hydroxide nanoflowers have been previously published,16,17 but the complex synthesis of the nanoflowers by a hydrothermal method and also the complex modification of the electrode surface, does not allow the fast generation of simple, small and low-cost electrochemical devices. For instance, Ibupoto et al.16 synthesized NiO nanoflowers after growing Ni(OH)2 on a gold substrate in alkaline media for 4–6 hours at 98 °C and annealing for 2–3 hours at 450 °C. Yang et al.17 synthesized Ni(OH)2 nanoflowers by heating at 45 °C for 2 hours an ammoniacal solution of nickel hexammine. The product was washed and dried for one day. Electrode modification was carried out by drop-casting using a dispersion obtained after mixing the nickel-based product with carbon nanotubes in a solution of Nafion in ethanol by ultrasonication. Therefore, the development of an easier synthesis and modification of nickel nanoflowers on electrodes would be a very interesting methodology to study the performance of this nickel nanomaterial for electroanalytical applications.
Screen-printed electrodes are devices comprising a 3-electrode electrochemical cell on a small card, which can be mass produced reducing the fabrication cost and generating a reproducible disposable surface. The low-cost, small size and the integrated electrochemical cell are ideal characteristics of these electrodes, which result in a very convenient platform for sensing devices. Nickel-based screen-printed electrodes (SPEs) have also been employed for sugars determination. For instance, García et al.12 employed SPEs modified by drop-casting with nickel nanowires and the fabricated devices were used for the determination of total carbohydrates in several food samples. The good stability of the modification allowed them to use the devices in a flow-injection analysis system for semi-automatic detection. In a similar work, the same authors evaluated the possibility to use nickel–copper nanowires, but they found that the nickel nanowires were most suitable as the fabrication was simpler.18 Several nickel–carbon composites have been reported as useful materials for the modification of the SPE surface in order to determine glucose in different samples. A Ni/nanoporous carbon composite19 has been employed for the modification of the working screen-printed electrode. The nanoporous carbon material has a high surface area, which increases significantly the available electrode area. In other work, a composite formed by graphene oxide, chitosan and Ni(II) was simultaneously electrodeposited by multiple cathodic cyclic voltammetry on the SPE surface generating an interesting structure composed by reduced graphene oxide, chitosan and nickel nanoparticles.20 Nickel paste have also been mixed with carbon ink in order to obtain a material appropriate for screen-printing the working electrode on a ITO substrate.21 This way, nickel-based devices are fabricated directly and the modification of the electrode surface is not necessary, although the electrochemical activation of the working electrode is still required. A hybrid Ni–Co hydroxide material was simultaneously deposited on the surface of SPEs as reported in Lien et al. work.22 The addition of Co seems to decrease the potential needed for the detection of glucose, although severe interferences by other species is found. An interesting device is reported by Niu et al.23 Electrodeposition of nickel is performed in severe conditions (0.2 M Ni(II), 1 M H2SO4) applying a high current to the electrode (0.1 A for 30 s). In these conditions, nickel is electrodeposited on the electrode surface while a great amount of hydrogen bubbles is generated and a three-dimensional porous nickel structure is created on the electrode surface. Although, promising analytical characteristics are found, only the working electrode is a small, portable screen-printed electrode, and the system uses a conventional electrochemical cell with conventional auxiliary and reference electrodes, decreasing its usefulness for in situ analysis. As SPEs typically have a solid quasireference electrode, in certain experimental conditions, applying a constant potential or a potential sweep (potentiostatic/potentiodynamic methods) may not be the best choice for the electrodeposition of nanomaterials. Electrochemical reactions occurring in the electrode–solution interface are processes strongly affected by the electrode surface, and a small change of the electrode surface may cause a big change in the electrochemical reactivity. A good alternative is the electrodeposition by a galvanostatic method (application of a constant current), since, in this case, the electrodeposition is controlled by the current flowing between the working and counter electrodes. This methodology has already been successfully applied for gold nanoparticles electrodeposition with a high control of size and density or for the reduction of graphene oxides on SPEs.24,25 As far as we know, studies about the electrodeposition of nickel nanoflowers on screen-printed electrodes using a galvanostatic method have not been reported.
In this work, we carried out the electrodeposition of nickel on the surface of screen-printed carbon electrodes. As a novelty, a galvanostatic electrodeposition method was employed, which allows a fast and simple generation of nickel nanoflowers on screen-printed electrodes. The screen-printed electrodes modified with nickel nanoflowers (NiNFSPEs) were employed for the non-enzymatic determination of reducing sugars.
2. Methods and materials
2.1. Apparatus and electrodes
Electrochemical measurements were performed with an Autolab PGSTAT10 potentiostat/galvanostat controlled by GPES 4.9 software. All measurements were carried out at room temperature. Screen-printed carbon cards (ref. DRP-110) were purchased from DropSens (Spain). Each card is formed by a 3-electrode electrochemical cell with carbon-based working and counter electrodes, whereas quasireference electrode and electric contacts are fabricated in silver. The diameter of the working electrode was 4 mm. Screen-printed electrodes were connected to the potentiostat through a specific connector, DRP-DSC. A JEOL 6610LV scanning electron microscope was used to characterize the electrodes modified with the nickel nanoflowers. X-ray photoelectron measurements were performed on a SPCEs Phoibos 150/MCD-5 spectrometer, using monochromatic Al Kα excitation source with an energy of 1486.74 eV. The survey and high-resolution spectra were collected with 90 eV and 30 eV of pass energy and 1 eV and 0.1 eV of step energy, respectively.
2.2. Reagents and solutions
Glucose, absolute ethanol, sodium hydroxide and sodium chloride were purchased from Merck. Fructose, xylose, arabinose, mannose, galactose, glycerol, ascorbic acid, lactic acid, citric acid and nickel(II) sulfate hexahydrate were purchased from Sigma-Aldrich. Ultrapure water obtained with a Millipore Direct Q5 purification system from Millipore Ibérica was used throughout this work. All other reagents were of analytical grade. Working solutions of Ni(II) were prepared in 0.1 M NaCl. Working solutions of sugars and interfering species were prepared in 0.1 M NaOH.
2.3. Electrode modification with nickel nanoflowers
Nickel nanoflowers were electrodeposited on screen-printed carbon electrodes by a galvanostatic method. 40 μL of a 10 mM Ni(II) solution was dropped in the electrochemical cell and a constant current of −25 μA was applied for 60 s. Ni(II) is reduced to Ni(0), which is deposited on the carbon surface. In contact with air, the surface of the deposited Ni(0) is spontaneously oxidized (passivated) to Ni(II) oxides and hydroxides. In order to stabilize the nickel-modified surface, a pretreatment applying 50 cycles of cyclic voltammetry from +0.2 V to +0.7 V in a 0.1 M NaOH solution was performed (100 mV s−1).
2.4. Chronoamperometric measurements
Chronoamperometric measurements for the determination of sugars were carried out by applying a constant potential of +0.6 V for 100 s. The current measured at 100 s was chosen as the analytical signal.
2.5. Sample preparation
For the orange juice samples, 1 μL of sample was diluted in 1 mL of 0.1 M NaOH solution. Several samples with different added amounts of glucose were prepared in order to carry out the determination by the standard additions method.
For the honey samples, 1.0020 ± 0.0001 g of honey is diluted in 50 mL of H2O. Then, 100 μL of this solution is diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :400 in 0.1 M NaOH. Several samples with different added amounts of glucose were prepared in order to carry out the determination by the standard additions method.
:400 in 0.1 M NaOH. Several samples with different added amounts of glucose were prepared in order to carry out the determination by the standard additions method.
3. Results and discussion
3.1. Characterization of nickel nanoflowers-modified screen-printed electrodes
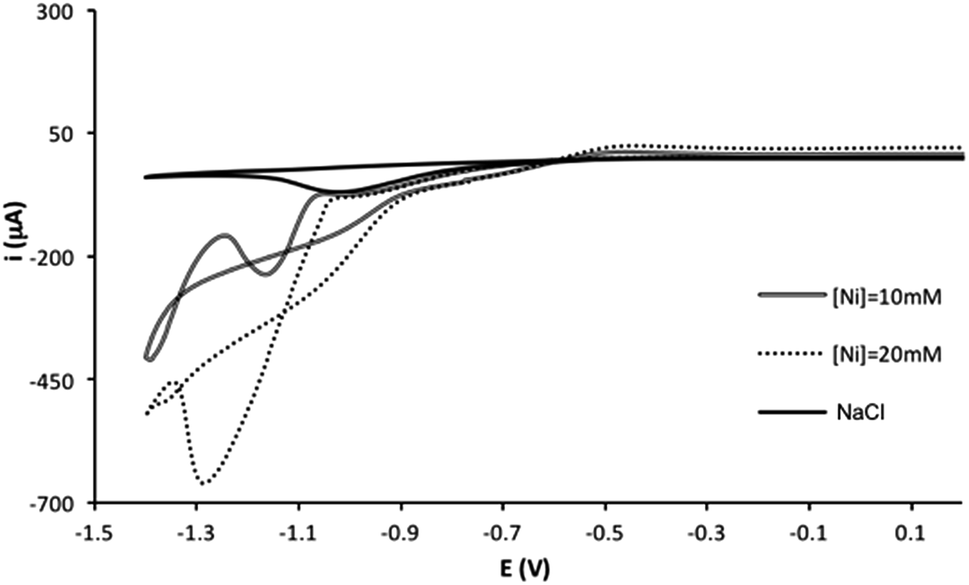
The electrodeposition of nickel on screen-printed carbon electrodes was studied using cyclic voltammetry. Fig. 1 shows the voltammograms for different concentration of Ni(II) (0, 10 and 20 mM) in a 0.1 M NaCl solution. For all cases, two cathodic processes are observed, a process with a peak potential at about −1.0 V, which it is attributed to the oxygen reduction (also observed in the blank solution), and a process with a peak potential at −1.25 V, attributed to the Ni(II) to Ni(0) reduction. The backward curve crossed the forward curve as it is typically found for an electrodeposition process. Therefore, the nickel electrodeposition can be carried out under these experimental conditions using screen-printed electrodes. However, as mentioned in the introduction, the screen-printed cards have a silver quasireference electrode, and the applied potential could be slightly different for different electrodes (or it could shift in the same measurement). Surface processes such as electrodeposition can be very sensitive to these potential changes, and therefore, different amounts/density of metal may be electrodeposited using a potentiostatic/potentiodynamic method. In order to minimize these issues, a galvanostatic method was chosen for the electrodeposition of nickel on the screen-printed electrode surface. A comparison between the galvanostatic and the potentiostatic methods is presented in the ESI,† under similar electrodeposition conditions. Although a comparable electrocatalytic effect observed, a slightly better reproducibility is found for blank solution using the galvanostatic method, even carrying out the electrodeposition in mild conditions for the silver quasireference electrode (0.1 M NaCl aqueous solution).
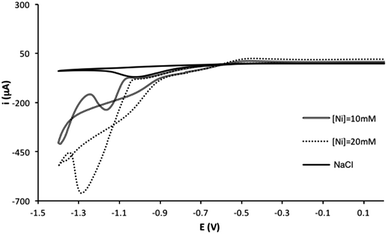 |
| | Fig. 1 Cyclic voltammograms of several concentrations of Ni(II) and blank in a 0.1 M NaCl solution. | |
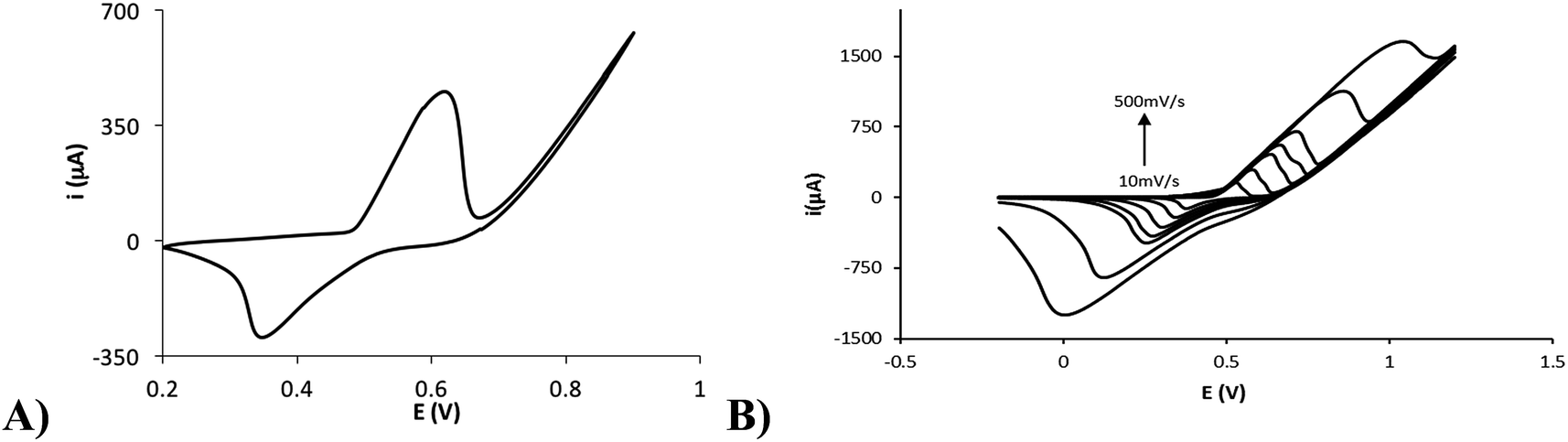
Therefore, the nickel electrodeposition was performed by applying a constant current of −25 μA for 60 s on a 10 mM Ni(II) solution in 0.1 M NaCl, and the modified electrode was characterized. The chronopotentiogram obtained for the galvanostatic electrodeposition is shown in the Fig. S1.† The potential taken by the electrode at the beginning is about −0.75 V. This potential is not kept constant and varies quickly until a value of −0.94 V at 10 s. As described previously, the first process occurring could be due to the oxygen reduction. However, the nickel reduction could also be produced at these potentials. When the oxygen concentration decreases, the potential reaches −0.94 V, which is likely due only to the reduction of Ni(II) to Ni(0). From 10 s to the end of the current application, the potential remains practically constant (varying only from −0.94 to −0.91 V), suggesting that not all the Ni(II) in solution is electrodeposited under these conditions. If this were the case, a new decrement in the potential should happen. As the Fig. 2 shows, the cyclic voltammetry of the modified electrode in 0.1 M NaOH showed an anodic process at +0.60 V and a cathodic process at +0.34 V (Fig. 2A). NaOH is an electrolytic medium widely employed for the non-enzymatic electrochemical detection of sugars because it has been demonstrated that OH− ions in the solution play a crucial role in the reaction.26 The observed processes are assigned to the oxidation of Ni(II) to Ni(III) and its correspondent reduction (eqn (1)). The generation of Ni(II) on the electrode surface from the electrodeposited Ni(0) could happen by two ways: on the one hand, the application of positive potentials (by the cyclic voltammetry) in a NaOH medium could easily oxidize Ni(0) to oxygenated Ni(II), and on the other hand, the spontaneous oxidation of Ni(0) to Ni(II) by atmospheric oxygen has been proposed previously.27,28
| | |
Ni(OH)2 + OH− ⇄ NiO(OH) + H2O + e−
| (1) |
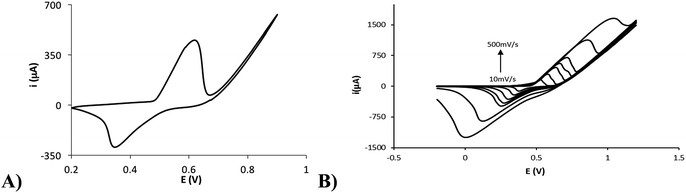 |
| | Fig. 2 (A) Cyclic voltammogram of the nickel-modified electrode in 0.1 M NaOH. (B) Cyclic voltammograms of the nickel-modified electrode in 0.1 M NaOH at several scan rates (10, 25, 50, 75, 100, 250, 500 mV s−1). | |
The Ni(II)/Ni(III) redox process in 0.1 M NaOH was studied at different scan rates. Fig. 2B shows the electrochemical response with increasing scan rates (10, 25, 50, 75, 100, 250, 500 mV s−1). An increment in the peak potential difference is observed with increasing scan rates as expected for a quasireversible electrochemical process. Furthermore, a linear relationship between the peak currents and the scan rate up to 50 mV s−1 is found (Fig. S2†), indicating a surface-confined process for the reaction of adsorbed Ni(OH)2/NiO(OH). However, a linear relationship between the peak currents and the square root of the scan rate is found at higher scan rates (Fig. S3†). This fact could indicate that the reaction at these scan rates is controlled by the diffusion of OH− (involved in the reaction as indicated in eqn (1)) to/from the electrode surface, as has been previously found by other authors.8 At lower scan rates, the flow of OH− to the surface is high enough to observe the characteristics of the adsorbed nickel film. Therefore, with the data obtained at low scan rates, the adsorbed concentration of nickel was estimated using the following eqn (2):
| |
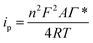 | (2) |
where
ip is the peak current,
n is the number of electrons exchanged in the electrochemical reaction,
A is the electrode area,
Γ* is the surface concentration,
R is the gas constant,
T is the absolute temperature and
v is the scan rate of the cyclic voltammetry. The adsorbed amount of nickel on the screen-printed surface was 7.9 × 10
−9 moles and considering the geometric area of the electrode the surface concentration was 6.6 × 10
−7 mol cm
−2. The expected nickel amount deposited in the electrodeposition step can be estimated considering the transferred charge (
Q =
it), which in these conditions (−25 μA for 60 s) was 1.5 mC. Then, the amount of nickel can be calculated by the Faraday laws using
eqn (3), where
m is the amount of nickel (g),
M is the molar mass (g mol
−1),
Q is the charge transferred (C),
n is the number of electrons exchanged and
F is the Faraday constant (96
480 C mol
−1). The estimated amount of nickel electrodeposited was of 7.8 × 10
−9 moles. This value is very close to the estimated with the voltammetric peak, suggesting that all the applied current is employed for the electrodeposition of nickel on the electrode surface and, besides, the electrodeposited nickel is stable and stick to the electrode surface. The surface concentration found in our device for the optimal conditions (660 nmol cm
−2) seems a higher value than other published works where this concentration was estimated. For instance, Hutton
et al.10 reported a boron-doped diamond electrode modified with 20 nmol cm
−2 of Ni(OH)
2 nanoparticles as the optimal surface concentration or Sharifi
et al.29 reported a glassy carbon electrode modified with about 40 nmol cm
−2 of NiO nanoparticles. In our case, a higher amount of nickel is necessary to obtain the best electrocatalytic effect than for these different electrodes.
| |
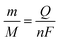 | (3) |
Scanning electron microscopy micrographs were obtained in order to study the morphological and structural aspects of the electrodeposited nickel. Fig. 3A shows different micrographs for the nickel-modified screen-printed electrode. Ni was electrodeposited applying a constant current of −25 μA for 60 s on a solution of 10 mM of Ni(II) in 0.1 M NaCl. In these conditions, as can be seen in the micrographs, the electrode surface is completely coated with flower-shaped nickel nanoparticles. The approximate size of the nanoparticles was 160 ± 24 nm and an uniform distribution is observed across the electrode surface. Furthermore, some porous structure is observed between the nanoflowers, which could lead to larger surface area for the electrochemical reactions. Fig. 3B shows a SEM image of the NiNFSPEs after performing the electrochemical activation (explained in the following sections). It seems that the size and morphology of the nanoflowers did not change after the electrochemical activation, and therefore, the improvement observed in the measurements is probably due to some surface process, as could be the generation of Ni(OH)2 from NiO. In a previous unpublished study, we performed the electrodeposition of Ni(II) under the same conditions but using a 0.1 M H3BO3/NaCl solution (see ESI†). In that case, non-flower shaped spherical nickel nanoparticles can be found with a size of around 130 nm (Fig. 3C). Therefore, the solution in which the electrodeposition is carried out is crucial to determine the shape of the generated nanoparticles.
 |
| | Fig. 3 (A) SEM micrograph of the nickel-modified electrode. (B) SEM micrograph of the nickel-modified electrode after the activation with 50 CV cycles. (C) SEM micrograph of the nickel-modified electrode using a H3BO3/NaCl solution. | |
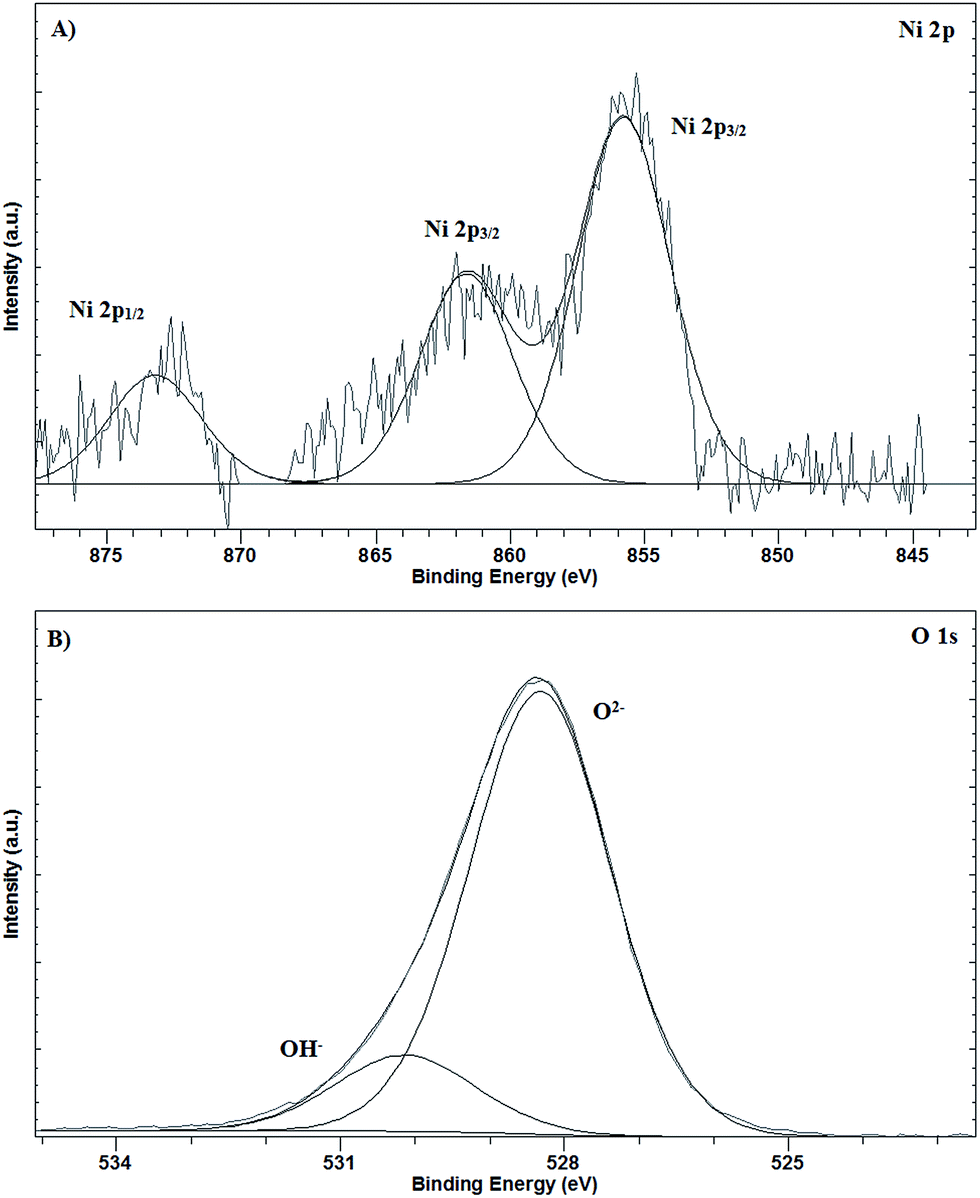
The oxidation state of the electrodeposited nickel was studied by XPS. Fig. 4 shows the Ni 2p and O 1s regions of the spectrum. The peaks with binding energies of 873.2 eV and 855.8 eV can be tentatively assigned to Ni 2p1/2 and Ni 2p3/2 of Ni(II), respectively, and characteristic of Ni(OH)2.30 The peak with binding energy of 861.6 eV could be assigned to a multielectron excitation of Ni2+, but difficult to be assigned to NiO or Ni(OH2).31 The spectrum for O 1s shows peaks with binding energies of 528.3 and 530.2 eV, which can be tentatively assigned to O2− (NiO) and OH− (Ni(OH)2), respectively.30,32 These results confirm the presence of NiO and Ni(OH)2 species on the electrode surface. Comparing the O 1s spectra against the obtained previously for the bare electrode,33 significant differences can be observed. Two XPS peaks appear for the O 1s spectra for the bare electrode at around 534.2 eV (smaller intensity) and 531.8 eV (higher intensity), assigned to different C–O bonds. For the nickel-modified electrodes, no peaks appeared at these binding energies, suggesting that the response is completely due to the different Ni–O bonds.
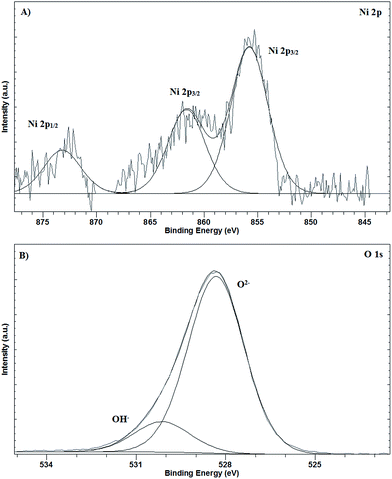 |
| | Fig. 4 XPS spectrum of the NiNFSPE electrode: (A) Ni 2p region and (B) O 1s region. | |
3.2. Electrocatalytic effect of nickel nanoflowers towards sugars oxidation
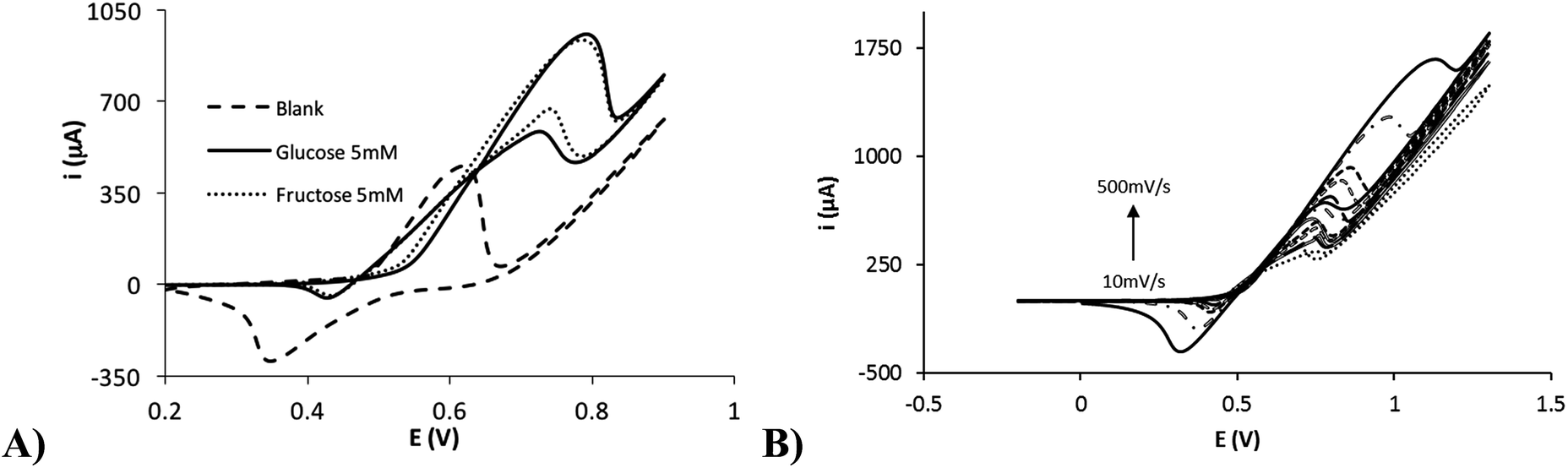
The electrocatalytic effect of nickel nanoflowers towards the oxidation of reducing sugars was studied by cyclic voltammetry. Fig. 5A shows the electrochemical response of NiNFSPEs in presence of 5 mM glucose, 5 mM fructose and a 0.1 M NaOH solution (blank). Cyclic voltammetries for other reducing sugars such as arabinose, galactose, mannose and xylose are shown in the Fig. S4.† It is easily detected as the Ni(II) to Ni(III) oxidation process is enhanced in presence of the reducing sugar in the solution. However, for the Ni(III) to Ni(II) reduction process a significant decrease in the peak current is observed. The catalytic oxidation of sugars is attributed to the following reactions34 (specified for the glucose case):| | |
Ni(OH)2 + OH− → NiO(OH) + H2O + e−
| (4) |
| | |
NiO(OH) + glucose → Ni(OH)2 + gluconolactone
| (5) |
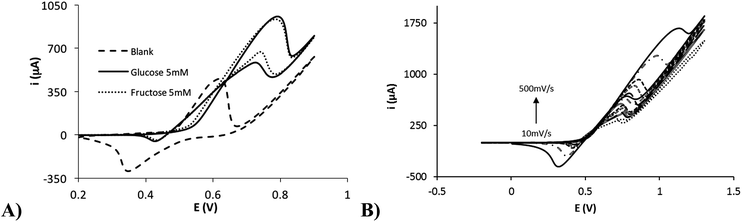 |
| | Fig. 5 (A) Cyclic voltammograms of the NINFSPE device in presence of 5 mM of glucose (solid line), 5 mM of fructose (dotted line) and blank (dashed line). (B) Cyclic voltammograms of the NiNFSPE device in presence of 5 mM of glucose at several scan rates (10, 25, 50, 75, 100, 250 and 500 mV s−1). | |
This mechanism is consistent with the observed electrochemical response. The NiO(OH) species reacts chemically with the sugar, and, therefore, the electrochemical oxidation of Ni(OH)2 to NiO(OH) will increase to regenerate the NiO(OH) consumed by the coupled chemical reaction. For this reason, an increased current flow is expected to carry out the oxidation of Ni(II) to Ni(III). For the reduction process (Ni(III) to Ni(II)) and due to that the sugar oxidation regenerates the Ni(II) chemically, the intensity of the electrochemical process decreases as less Ni(III) will be available. Although in some works where nickel-modified electrodes are used, the catalytic effect is only observed for glucose,23,35 in our case, the electrodeposited nickel nanoflowers catalyze the oxidation of several reducing sugars. This is also observed in other nickel-based electrodes previously described in the literature.12,22,36 Studies are scarce explaining why certain nickel-based electrode materials show a catalytic effect only for glucose and in other cases, it occurs for several reducing sugars. Oxidation of sugars is produced as a dehydrogenation reaction, although in all reducing sugars the group involved in the reaction is a hemiacetal group. Compton et al.37 suggested that the electrocatalysis process is generally observed to occur via the adsorption of the analyte to the electrode surface, probably involving d-electrons and empty d-orbitals of the metallic substrate (in this case, the Ni(III) species). It is, therefore, probable that the adsorption of the sugars on the catalyst is crucial in order to achieve the oxidation reaction. Although they have similar structures, there are some differences in the reducing sugars such as the number of carbons in pentoses and hexoses, or especially, the configurational differences in hydroxide groups. Therefore, it is likely that these small differences in the structure play a significant role in the adsorption of the sugars over the electrode materials, process which appears to be highly important for the catalysis. For this reason, it seems that in some materials only glucose adsorption would occur, being selective to this sugar instead of other also oxidizable sugars. This is a possible explanation for the different selectivity of the various materials described in the literature, however, it seems clear that more studies are needed to clarify these processes and the influence of the nickel structures, although this is not within the scope of this article.
The catalytic process of the glucose oxidation (500 μM) was studied at different scan rates (10–500 mV s−1). Fig. 5B shows the electrochemical response with increasing scan rates. The anodic peak current obtained is linearly proportional to the root of the scan rate (Fig. S5†), indicating that the limiting step of the electrochemical reaction is the diffusion of the glucose to the electrode surface.
3.3. Analytical performance of NiNFSPEs for sugar determination
The optimization of the different parameters affecting the analytical signal was carried out. Firstly, the optimization of the experimental conditions for the nickel electrodeposition such as the electrodeposition current and time or the nickel concentration was performed. Several conditions were used for the Ni(II) concentration (0.1, 0.5, 1, 5, 10 and 15 mM), for the electrodeposition current (−5, −25, −75, −100 μA) and for the electrodeposition time (30, 60, 90 and 120 s). The highest signal/background ratio was obtained by applying −25 μA for 60 s to a Ni(II) solution of 10 mM.
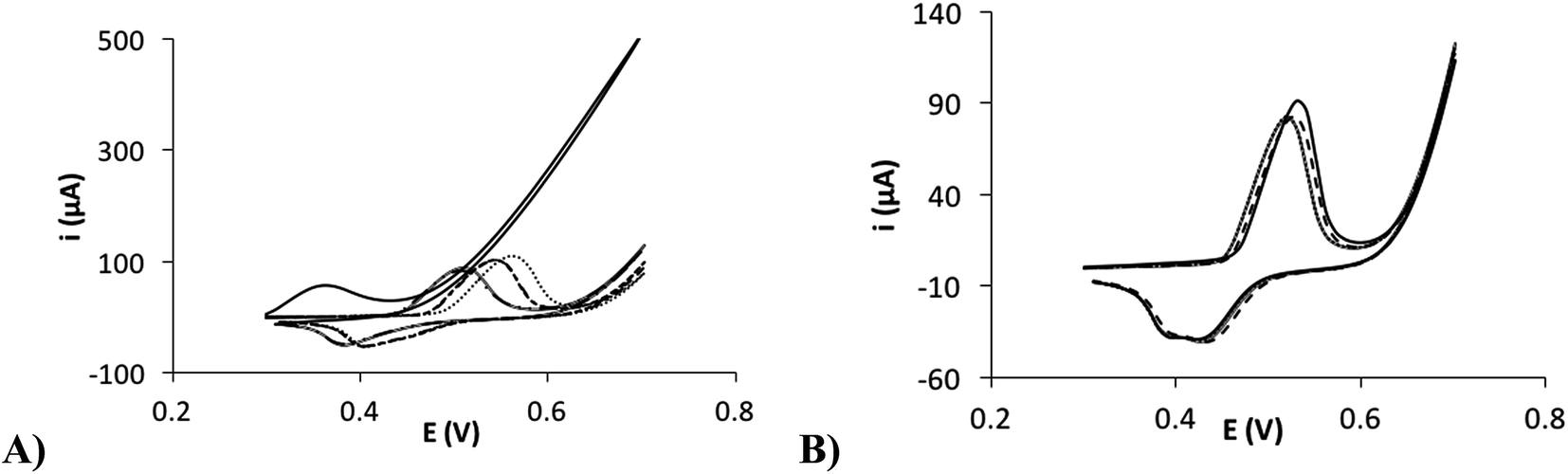
Secondly, it was necessary to perform an activation to the nickel electrodeposited surface in order to improve the stability of the formed film, as shown in the Fig. 6A. In this figure, the electrochemical response of several cycles of cyclic voltammetry using a non-activated electrode is shown. In Fig. 6B, a higher stability on the response is observed after performing an electrochemical activation to the electrode surface. This fact could be due to that different Ni(II) species are presented in the surface, which shows a different electrochemical behavior. After the activation in a NaOH solution, it is probably that the most stable Ni(OH)2 species are generated all over the surface, preventing Ni(II) mixed processes and improving the stability.38 It is possible to apply a wide number of activation methods in order to improve the surface stability. In our case, we chose to carry out several cycles of cyclic voltammetry from +0.2 to +0.7 V (100 mV s−1). The number of cycles was optimized with the aim to improve the stability of the surface and enhance the signal/background ratio. It was achieved after the application of 50 cycles.
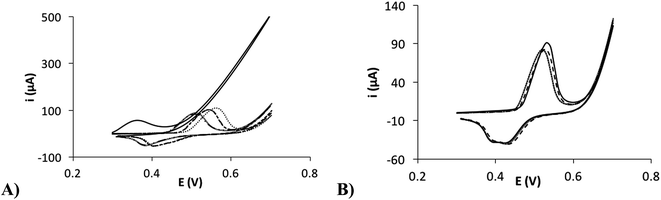 |
| | Fig. 6 (A) Consecutive cyclic voltammograms of the NiNFSPE in 0.1 M NaOH without activation. (B) Consecutive cyclic voltammograms of the NiNFSPE in 0.1 M NaOH after the activation. | |
As chronoamperometry was used to carry out the determination of sugars, the effect of the applied potential was also evaluated. In order to perform a measurement, 40 μL of the glucose solution in 0.1 M NaOH was added to the device and a potential able to oxidize the Ni(II) film is applied for a certain time. The chronoamperometric current obtained at 100 s was considered as the analytical signal. Therefore, several potentials were tested in order to improve the signal/noise ratio using a 250 μM glucose solution. 0.6 V was chosen as the most appropriate potential to carry out the detection of sugars.
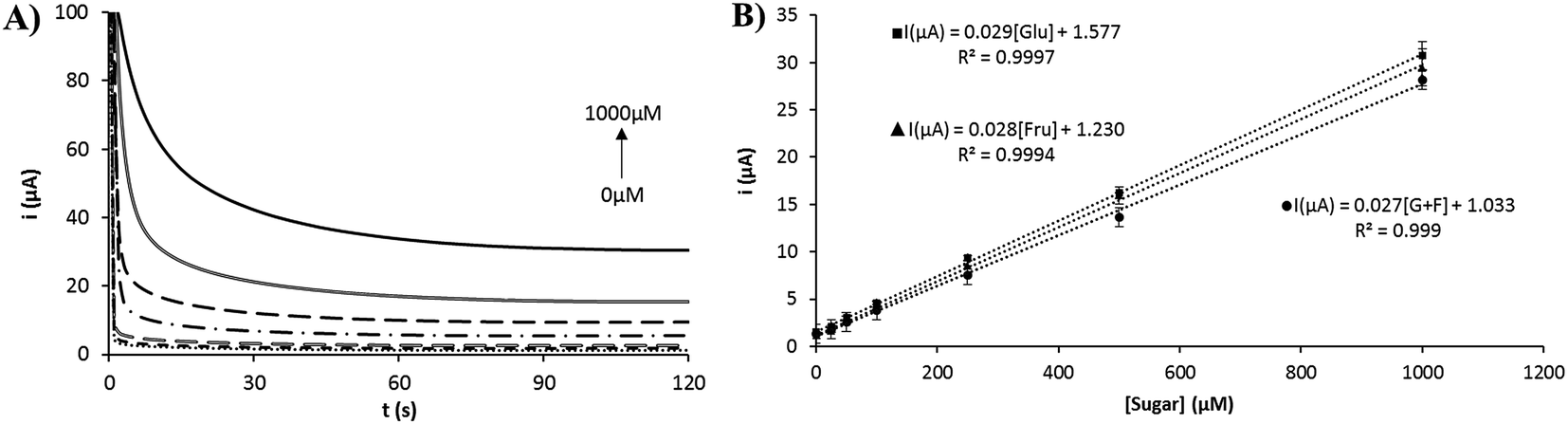
The electrochemical response for different concentrations of glucose, fructose and a 1:1 mixture in 0.1 M NaOH was evaluated. Fig. 7 shows the chronoamperograms for different concentrations of glucose (Fig. 7A) and the calibration plots obtained with a linear range from 25 to 1000 μM for all cases (Fig. 7B). Similar results were found for other reducing sugars such as arabinose, galactose, mannose and xylose (see Fig. S6†). The slope of the calibration plots was similar for all the reducing sugars evaluated. The reproducibility obtained for the slopes of the calibration plots was in all cases under 8% (RSD, n = 3). The sensitivity obtained was between 0.21 and 0.23 μA μM−1 cm−2 and a detection limit between 8 and 20 μM was estimated. The limit of detection was calculated as the concentration corresponding to three times the standard deviation of the estimate, as proposed by Miller.39 A quantitative comparison of several devices for non-enzymatic detection of sugar using nickel-modified screen-printed electrodes is shown in Table 1. The device fabricated with 3D nickel nanoporous structures stand out over the other devices in terms of linear range and limit of detection. This fact is due to that the screen-printed electrode is used in a stirred high-volume conventional cell, with a Pt wire and Ag/AgCl conventional electrodes. A higher volume of the sample and the improved mass transfer due to the stirring of the solution allows to achieve a lower limit of detection. It could be interesting to evaluate this electrode in a quiescent solution, as generally employed for screen-printed electrodes. Comparing the other nickel-based screen-printed electrodes, our device is highly competitive in terms of the linear range and limit of detection. Furthermore, the high stability shown by the nickel nanoflowers and the simplicity for the fabrication of the nanostructured surface are really interesting characteristics in order to apply this device in a real world application, such as the determination of sugars in food.
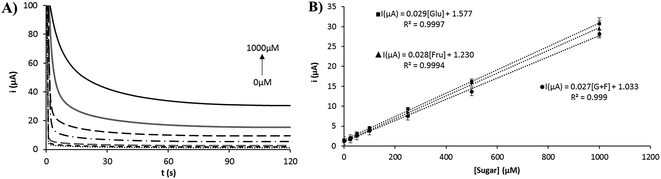 |
| | Fig. 7 (A) Chronoamperometric response for increasing concentrations of glucose. (B) Calibration plots for glucose, fructose and a 1:1 mixture of glucose and fructose. | |
Table 1 Analytical characteristics of different nickel-modified screen-printed electrodes for the determination of sugars
| Electrode material |
Linear range (μM) |
Detection limit (μM) |
Reference |
| Ni nanoflowers |
25–1000 |
8 |
This work |
| NiCu nanowires |
50–1000 |
40 |
18 |
| Ni nanowires |
50–1000 |
— |
12 |
| NiCo |
25–3700 |
— |
22 |
| Ni-Doped nanoporous carbon |
20–240 |
10 |
19 |
| 3D-porous Ni nanostructures |
0.5–4000 |
0.07 |
23 |
| NiNP–chitosan–rGO |
200–9000 |
4.1 |
20 |
| Ni/C composite |
1000–10000 |
400 |
21 |
3.4. Stability and precision studies
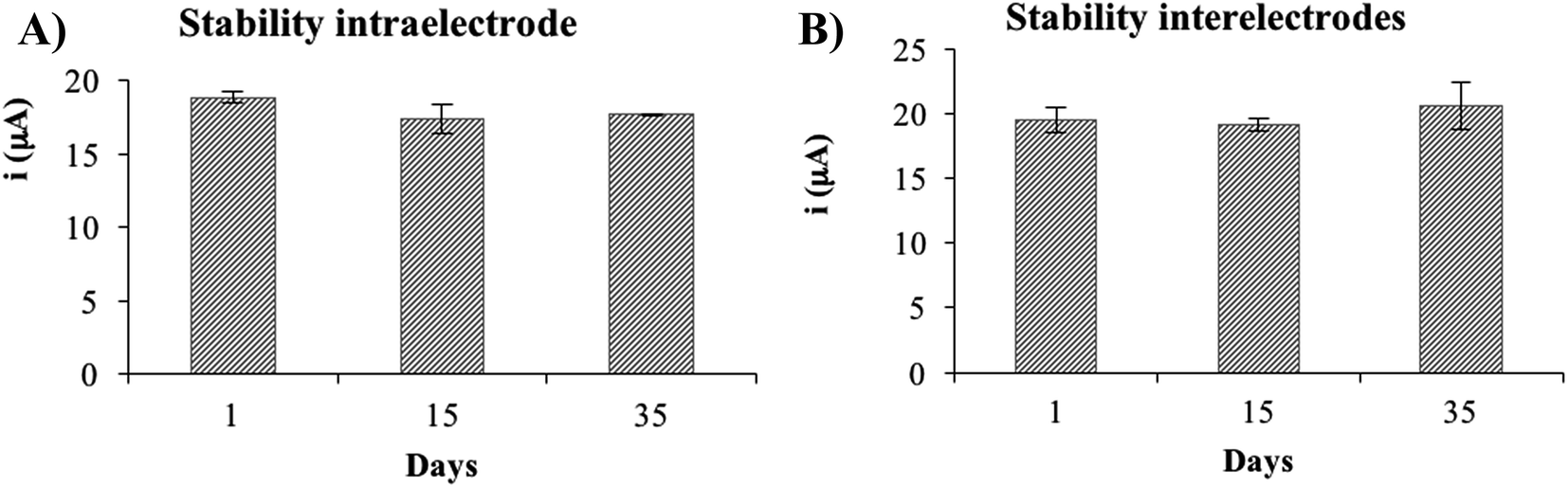
Besides the estimation of the precision of the non-enzymatic electrode by evaluating the residual standard deviation of the calibration slopes as described in the previous paragraph, a study of intra- and interelectrode precision was performed. In order to do so, a solution of 500 μM of glucose was measured using the optimized experimental conditions. The RSD obtained for intraelectrode precision was 5.5% (n = 10), showing that the device is very precise even reusing the same electrode, which can be extremely useful to save costs in an industrial environment. The RSD obtained for the interelectrode precision study was 6.9% (n = 3), showing a high precision using different devices.
Several devices were fabricated and activated (50 voltammetric cycles) on the same day and were stored at room temperature until the day of use. The results show that the NiNFSPE device is stable at least up to 35 days (Fig. 8), considering the signals obtained for different electrodes (interelectrode stability) as for signals obtained using the same electrode (intraelectrode stability). In all cases, the electrode surface was rinsed with ultrapure water before the measurements. No re-activation of the electrode surface was necessary in order to obtain a reproducible response. The fact that the same device can be used in different days and keep its electrochemical response is a great advantage compared to other previously published devices.
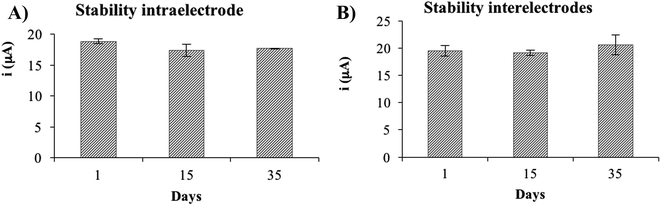 |
| | Fig. 8 (A) Electrochemical response of the NiNFSPE device using the same device in different days (intraelectrode). (B) Electrochemical response of the NiNFSPE device using different devices in different days (interelectrode). | |
3.5. Selectivity study
The effect of different species that could interfere with the determination of sugars due to the proximity of its oxidation processes was evaluated. The interfering species studied were ascorbic acid, citric acid, lactic acid, ethanol and glycerol, as they are species that could be found in food samples, typically at lower concentrations than sugars. Solutions of these species were prepared at different increasing concentrations (from 0.1 mM to 10 mM) and a constant glucose concentration of 1 mM was used. These solutions were measured by chronoamperometry with the optimized experimental conditions. Table 2 shows the lower concentration of the interfering species that influenced the analytical signal and the variation in this signal. No significant interference effect was found for citric and lactic acids up to 10 mM, showing that the device does not respond to these species even at high concentrations. A concentration of 1 mM of ascorbic acid increased the analytical signal by 23% (in order to know the influence of ascorbic acid, it is worth to mention that typical concentrations of sugars/ascorbic acid in orange juice could be at least in a 100:1 ratio). An ethanol concentration of 5 mM increased the analytical signal by 13%, suggesting that the device could have issues to determine the sugar content in high-concentration alcoholic beverages. The higher interfering effect was found for glycerol as a concentration of 0.25 mM was enough to increase the analytical signal by 31%. However, when it is added to sugar-containing food, the glycerol concentration is typically lower than sugar concentration.
Table 2 Lower concentration of interfering species that influences the analytical signal for 1 mM of glucose and the variation of the analytical signal produced
| Interfering species |
Concentration (%variation) |
| Citric acid |
— |
| Lactic acid |
— |
| Ascorbic acid |
1 mM (+22%) |
| Ethanol |
5 mM (+13%) |
| Glycerol |
0.25 mM (+31%) |
3.6. Determination of sugars in real samples
The performance of the non-enzymatic electrode for the determination of reducing sugars in food samples such as orange juice and honey was evaluated. Standard additions method was employed in order to minimize the matrix effects and a known amount of glucose (50, 100 and 200 μM) was added to the different samples diluted in 0.1 M NaOH. For the orange juice samples, a concentration of 0.49 ± 0.04 g L−1 of sugar was estimated (value statistically similar to the 0.49 g L−1 specified in the nutritional information). For the honey sample, a concentration of 6.8 ± 0.9 g kg−1 of sugar was found (compared to the 7.8 g kg−1 specified in the nutritional information and 6.3 ± 0.1 g kg−1 obtained with glucose and fructose sensors previously published by our group40,41). These results show that the NiNFSPE electrochemical device is able to determine with good accuracy the concentration of reducing sugars in complex food samples.
4. Conclusions
In this work, we were able to generate in situ nickel nanoflowers on screen-printed carbon electrodes by a galvanostatic electrodeposition methodology. Nickel nanoflowers have a quasi-spherical geometry but with different edges that increase its surface area compared to typical nanoparticles. Nickel nanoflowers electrodeposited on screen-printed electrodes showed a great electrocatalytic effect towards the oxidation of reducing sugars. The non-enzymatic device is very promising for the determination of reducing sugars in food samples, even at low μM concentrations, in a short analysis time and with low sample consumption. The excellent stability presented by this nanostructured device, even being able to reuse the same device on different days without loss of the electrochemical response, could mean notable cost savings if the device were to be implanted in the food industry.
Acknowledgements
This work has been supported by the FC-15-GRUPIN-021 project from the Asturias Regional Government and the CTQ2014-58826-R project from the Spanish Ministry of Economy and Competitiveness (MEC). Daniel Martín-Yerga thanks the MEC for the award of a FPI grant (BES-2012-054408). Authors thank the Spectroscopy Unit of the Scientific and Technical Services of the University of Oviedo for the help with the XPS measurements.
References
- D. R. Whiting, L. Guariguata, C. Weil and J. Shaw, Diabetes Res. Clin. Pract., 2011, 94, 311–321 CrossRef PubMed.
- H. Greenfield and D. A. T. Southgate, Food composition data, Elsevier Science Publishers, Rome, Italy, 2003 Search PubMed.
- J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS PubMed.
- D. W. Kimmel, G. LeBlanc, M. E. Meschievitz and D. E. Cliffel, Anal. Chem., 2012, 84, 685–707 CrossRef CAS PubMed.
- S. Park, H. Boo and T. D. Chung, Anal. Chim. Acta, 2006, 556, 46–57 CrossRef CAS PubMed.
- G. Wang, X. He, L. Wang, A. Gu, Y. Huang, B. Fang, B. Geng and X. Zhang, Microchim. Acta, 2012, 180, 161–186 CrossRef.
- B. Pérez-López and A. Merkoçi, Trends Food Sci. Technol., 2011, 22, 625–639 CrossRef.
- Y. Mu, D. Jia, Y. He, Y. Miao and H. L. Wu, Biosens. Bioelectron., 2011, 26, 2948–2952 CrossRef CAS PubMed.
- A. Ciszewski and I. Stepniak, Electrochim. Acta, 2013, 111, 185–191 CrossRef CAS.
- L. A. Hutton, M. Vidotti, A. N. Patel, M. E. Newton, P. R. Unwin and J. V. Macpherson, J. Phys. Chem. C, 2011, 115, 1649–1658 CAS.
- J. Wang, W. Bao and L. Zhang, Anal. Methods, 2012, 4, 4009 RSC.
- M. García and A. Escarpa, Biosens. Bioelectron., 2011, 26, 2527–2533 CrossRef PubMed.
- L.-M. Lu, L. Zhang, F.-L. Qu, H.-X. Lu, X.-B. Zhang, Z.-S. Wu, S.-Y. Huan, Q.-A. Wang, G.-L. Shen and R.-Q. Yu, Biosens. Bioelectron., 2009, 25, 218–223 CrossRef CAS PubMed.
- G. H. Yue, Y. C. Zhao, C. G. Wang, X. X. Zhang, X. Q. Zhang and Q. S. Xie, Electrochim. Acta, 2015, 152, 315–322 CrossRef CAS.
- Y. Zhang, Y. Liu, Y. Guo, Y. X. Yeow, H. Duan, H. Li and H. Liu, Mater. Chem. Phys., 2015, 151, 160–166 CrossRef CAS.
- Z. H. Ibupoto, K. Khun, V. Beni and M. Willander, Soft Nanosci. Lett., 2013, 03, 46–50 CrossRef CAS.
- H. Yang, G. Gao, F. Teng, W. Liu, S. Chen and Z. Ge, J. Electrochem. Soc., 2014, 161, B216–B219 CrossRef CAS.
- M. García and A. Escarpa, Anal. Bioanal. Chem., 2012, 402, 945–953 CrossRef PubMed.
- M. Hjiri, R. Dhahri, N. Ben Mansour, L. El Mir, M. Bonyani, A. Mirzaei, S. G. Leonardi and G. Neri, Mater. Lett., 2015, 160, 452–455 CrossRef CAS.
- J. Yang, J.-H. Yu, J. Rudi Strickler, W.-J. Chang and S. Gunasekaran, Biosens. Bioelectron., 2013, 47, 530–538 CrossRef CAS PubMed.
- W.-Y. Jeon, Y.-B. Choi and H.-H. Kim, Sensors, 2015, 15, 31083–31091 CrossRef CAS PubMed.
- C.-H. Lien, J.-C. Chen, C.-C. Hu and D. S.-H. Wong, J. Taiwan Inst. Chem. Eng., 2014, 45, 846–851 CrossRef CAS.
- X. Niu, M. Lan, H. Zhao and C. Chen, Anal. Chem., 2013, 85, 3561–3569 CrossRef CAS PubMed.
- G. Martínez-Paredes, M. B. González-García and A. Costa-García, Electrochim. Acta, 2009, 54, 4801–4808 CrossRef.
- A. Sánchez Calvo, C. Botas, D. Martín-Yerga, P. Álvarez, R. Menéndez and A. Costa-García, J. Electrochem. Soc., 2015, 162, B282–B290 CrossRef.
- P. F. Luo, T. Kuwana, D. K. Paul and P. M. Sherwood, Anal. Chem., 1996, 68, 3330–3337 CrossRef CAS PubMed.
- E. S. Lambers, C. N. Dykstal, J. M. Seo, J. E. Rowe and P. H. Holloway, Oxid. Met., 1996, 45, 301–321 CrossRef CAS.
- N. Amiri and H. Behnejad, J. Chem. Phys., 2016, 144, 144705 CrossRef PubMed.
- E. Sharifi, A. Salimi, E. Shams, A. Noorbakhsh and M. K. Amini, Biosens. Bioelectron., 2014, 56, 313–319 CrossRef CAS PubMed.
- J. Yang, M. Cho and Y. Lee, Sens. Actuators, B, 2016, 222, 674–681 CrossRef CAS.
- F. Muench, M. Oezaslan, M. Rauber, S. Kaserer, A. Fuchs, E. Mankel, J. Brötz, P. Strasser, C. Roth and W. Ensinger, J. Power Sources, 2013, 222, 243–252 CrossRef CAS.
- H. Li, W. Hao, J. Hu and H. Wu, Biosens. Bioelectron., 2013, 47, 225–230 CrossRef CAS PubMed.
- R. Gusmão, V. López-Puente, I. Pastoriza-Santos, J. Pérez-Juste, M. F. Proença, F. Bento, D. Geraldo, M. C. Paiva and E. González-Romero, RSC Adv., 2015, 5, 5024–5031 RSC.
- C. Zhao, C. Shao, M. Li and K. Jiao, Talanta, 2007, 71, 1769–1773 CrossRef CAS PubMed.
- N. Qiao and J. Zheng, Microchim. Acta, 2012, 177, 103–109 CrossRef CAS.
- T. You, O. Niwa, Z. Chen, K. Hayashi, M. Tomita and S. Hirono, Anal. Chem., 2003, 75, 5191–5196 CrossRef CAS PubMed.
- K. Toghill and R. Compton, Int. J. Electrochem. Sci., 2010, 5, 1246–1301 CAS.
- V. Ganesh, S. Farzana and S. Berchmans, J. Power Sources, 2011, 196, 9890–9899 CrossRef CAS.
- J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry, Pearson Education Limited, Essex, England, 2010 Search PubMed.
- J. Biscay, E. Costa Rama, M. B. González García, J. M. Pingarrón Carrazón and A. Costa-García, Electroanalysis, 2011, 23, 209–214 CrossRef CAS.
- J. Biscay, E. Costa Rama, M. B. González García, A. Julio Reviejo, J. M. Pingarrón Carrazón and A. C. García, Talanta, 2012, 88, 432–438 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra15578b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Agustín Costa-García
* and
Agustín Costa-García