
Three dimensional hierarchically porous crystalline MnO2 structure design for a high rate performance lithium-ion battery anode†
Shikun Liua,
Xusong Liua,
Jiupeng Zhao*a,
Zhongqiu Tongb,
Jing Wanga,
Xiaoxuan Maa,
Caixia Chia,
Dapeng Sua,
Xiaoxu Liu*ac and
Yao Li*b
aMIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin 150001, PR China. E-mail: jpzhao@hit.edu.cn; liu88062321@163.com
bCenter for Composite Materials, Harbin Institute of Technology, Harbin 150001, PR China. E-mail: yaoli@hit.edu.cn
cHeilongjiang University of Science and Technology, Harbin 150022, PR China
First published on 31st August 2016
Abstract
A reasonably designed anode of hierarchically porous crystalline manganese dioxide on nickel foam has been successfully synthesized by facile anodic electrochemical deposition in combination with heat treatment. The three dimensional structure avoids the application of binder and conductive additives. The Ni foam provides a highly electronically conductive network in conjunction with a large surface area to support well contacted MnO2 nanoparticles and effectively increases the mechanical strength of the MnO2 anode as well as suppresses the aggregation of MnO2 nanoparticles during discharge/charge processes. The hierarchical pores composed of a large amount of macropores and mesopores can not only accommodate the volume change of MnO2 nanoparticles during Li ion insertion/extraction, but also accelerate the penetration of electrolyte and promise fast transport and intercalation kinetics of Li ions. The crystalline MnO2 anode exhibits a higher electrochemical performance than the amorphous one. As a result, the hierarchically porous crystalline MnO2 anode shows a long cycling life of 778.0 mA h g−1 after 200 cycles at a current density of 0.4 A g−1 and high-rate capability of up to 82% capacity retention even after the current density increases 20 times from 0.1 to 2.0 A g−1.
Introduction
Lithium-ion batteries (LIBs), as one of the electrical energy storage devices, have been extensively studied and widely applied in portable electronics, large-scale energy storage and electric vehicles.1–4 However, new electrode materials with higher specific capacity and longer cycling life than graphitic materials (theoretical capacity: 372 mA h g−1),5 need to be developed for next-generation LIBs. Recently, in consideration of their high theoretical capacities, electrochemically active transition metal oxides, such as Fe2O3,6,7 Fe3O4,8,9 SnO2,10,11 Co3O4,12,13 TiO2,14 and NiO,15 have been intensively researched as anode electrodes for LIBs. Among them, MnO2 has attracted significant attention due to its high theoretical specific capacity (1230 mA h g−1), environmental benignity, natural abundance and low cost.16–20 However, the intrinsic low electrical conductivity (10−8 to 10−7 S cm−1) and volume change lead to rapid capacity fading and poor rate performance during repeated discharge/charge processes.21To overcome these challenges, various nanostructured MnO2 materials combined with various conductive matrix have been fabricated for using as anode electrodes in LIBs by many researchers, such as conducting polymer,22 carbon nanohorns,23 carbon nanotubes20,24 and graphene,25–27 which can improve the electronic conductivity and suppress the pulverization of MnO2 anodes. However, the excellent cyclic stability and rate performance for MnO2 related anode, especially stable cycling life at higher current densities, have not been achieved. The excellent electrochemical performance of electrode material depends on the compact attachment between active material and current collector, achieving good electronic conductivity, and sufficient contact between active material and electrolyte, realizing high lithium ion diffusion efficiency.
More recently, three-dimensional (3D) nanostructured composite electrodes, directly growing electrode materials on current collector substrates, have attracted greater attention for LIBs.28,29 The 3D nanostructure avoids the use of binder and conductive additives appearing commonly in electrode materials. In addition, the current collector substrate can tightly bond the active particles to a conductive and robust matrix and can effectively maintain the mechanical strength of the electrode during volume changes. In recent years, the hierarchically porous structure has been an attractive architecture for various applications, including supercapacitors,30 solar cells,31,32 fuel cells,33 and LIBs.34,35 In a hierarchically porous material, the hierarchical pores can not only buffer the large volume variation during Li ions insertion/extraction, but also provide continuous pore spaces for good electrolyte penetration, ensuring the efficient transport of Li ions between the electrolyte and active materials. Some researches have reported that crystalline electrodes show better electrochemical performances than amorphous counterparts.36,37 Transition metal oxides would be easily changed from crystal network structure to nano-crystallite structure after the first discharge.37,38 The nano-crystallite would contribute to the Li ion insertion and lower the electrochemical polarization in the following cycles.37,38 Hence the transition metal oxide electrodes that are directly grown on current collector substrates and possess hierarchically porous crystalline structures would show good electrochemical performances.
In this study, a hierarchically porous crystalline MnO2 (HPC-Mn) anode on Ni foam was successfully fabricated by facile anodic electrochemical deposition into colloidal crystal templates following heat treatment. The HPC-Mn anode shows an improved electrochemical performance than the hierarchically porous amorphous MnO2 (HPA-Mn) anode without annealing. The combined excellent properties of tightly bonding between active materials and conductive substrate, hierarchically porous structure and crystalline structure enable the HPC-Mn anode to have a long cycling life of 778.0 mA h g−1 after 200 cycles at a current density of 0.4 A g−1 and excellent rate capability of up to 82% capacity retention even current density increasing 20 times from 0.1 to 2.0 A g−1.
Experimental
Preparation of the HPC-Mn anode
Fig. 1 shows a schematic illustration for the fabrication of HPC-Mn anode. Monodispersed polystyrene (PS) latex spheres of ∼400 nm in diameter were produced using emulsifier-free emulsion polymerization technology.39 PS colloidal crystal templates were grown on pure nickel foams using a controlled vertical drying method.40 The nickel foams were cleaned ultrasonically in ethanol and were soaked in PS sphere suspension (0.5 wt%) in glass vessels. The glass vessels were then put in an incubator at a constant temperature of 60 °C to evaporate the solvent, as shown in Fig. 1a. | ||
| Fig. 1 Schematic illustration for the fabrication of HPC-Mn anode. | ||
Anodic electrochemical deposition of MnO2 into polystyrene colloidal crystal templates was performed at a constant voltage of 0.9 V versus Ag/AgCl for 300 s (Fig. 1b and c). Typically, 0.5 M MnSO4·H2O and 0.5 M Na2SO4 in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture (volume ratio) of distilled water and ethanol was used as electrolyte. Ethanol was used to reduce the surface tension between the electrolyte and the PS spheres surface.40 The electrochemical deposition was carried out in a three electrode configuration with Ag/AgCl as a reference electrode, Pt foil as a counter electrode, and nickel foam coated with PS spheres as a working electrode. The anode and cathode reactions are shown in ESI.† The as-grown samples were washed in ultrapure water and dried at 60 °C in oven. Then the samples were immersed in tetrahydrofuran to remove the PS spheres templates. Finally, the samples were further annealed at 400 °C for 3 h in argon atmosphere to fabricate the HPC-Mn anode (Fig. 1e). ESI Videos 1 and 2† have shown that the annealing has no effects on the mechanical stability and electrical conductivity of Ni foams. The mass loading of MnO2 active material was calculated to be about 16.67 mg cm−3 (the thickness of Ni foam is 0.30 mm) based on the difference value of Ni foam before growing PS template and after annealing. The color change of the samples at different preparation stages was distinct, as shown in Fig. S1.†
:1 mixture (volume ratio) of distilled water and ethanol was used as electrolyte. Ethanol was used to reduce the surface tension between the electrolyte and the PS spheres surface.40 The electrochemical deposition was carried out in a three electrode configuration with Ag/AgCl as a reference electrode, Pt foil as a counter electrode, and nickel foam coated with PS spheres as a working electrode. The anode and cathode reactions are shown in ESI.† The as-grown samples were washed in ultrapure water and dried at 60 °C in oven. Then the samples were immersed in tetrahydrofuran to remove the PS spheres templates. Finally, the samples were further annealed at 400 °C for 3 h in argon atmosphere to fabricate the HPC-Mn anode (Fig. 1e). ESI Videos 1 and 2† have shown that the annealing has no effects on the mechanical stability and electrical conductivity of Ni foams. The mass loading of MnO2 active material was calculated to be about 16.67 mg cm−3 (the thickness of Ni foam is 0.30 mm) based on the difference value of Ni foam before growing PS template and after annealing. The color change of the samples at different preparation stages was distinct, as shown in Fig. S1.†
Characterization
The morphology and nanostructures of the samples were characterized by scanning electron microscopy (SEM), transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM). SEM images of the samples were collected by a SU8000 scanning electron microscope with an accelerating voltage of 15 kV. TEM-HRTEM was carried out on TEM (JEOL JEM-2010). X-Ray Diffraction (XRD) measurements were performed at a Rigaku D/max-rB X-ray diffractometer with Cu Kα (λ = 0.15418 nm) incident radiation. The diffraction patterns were collected at room temperature in the 2θ ranges of 10 to 90°. An X-ray photoelectron spectroscopy (XPS) study was conducted with a PHI 5700 ESCA system using Al Kα radiation (1486.6 eV). Raman spectroscopy was performed with a laser micro-Raman spectrometer (Renishaw inVia, Renishaw, 532 nm excitation wavelength). Brunauer–Emmett–Teller (BET) analysis was carried out using a N2 adsorption–desorption apparatus (3H-2000PS1, Bass). The small angle X-ray scattering (SAXS) tests were carried out at Shanghai Synchrotron Radiation Facility, by using a wavelength of 0.124 nm, a sample to detector distance of 5 m, and an exposure time of 10 s.Electrochemical measurements
The electrochemical properties of the samples were evaluated using CR 2032 coin cells. The coin cells were assembled in a glove box filled with high-purity argon, where the as-fabricated HPC-Mn and HPA-Mn anodes were used as the working electrodes, metallic lithium foil as the counter/reference electrode, a polypropylene (PP) film (Celgard 2400) as the separator, and 1 M LiPF6 in ethylene carbonate (EC)–dimethyl carbonate (DMC) (1:1 in volume) as the electrolyte, without any additives. The discharge–charge performance of the batteries was tested using a channels battery analyzer (Neware CT-3008) with the voltage cut-off between 0.01 and 3.0 V vs. Li/Li+. The electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) measurements were performed using a CHI660E electrochemical workstation. EIS was recorded with a frequency ranging from 100 kHz to 10 mHz and a AC signal of 5 mV in amplitude as the perturbation. The voltage range of the CV measurements was 0.01–3.0 V and the scanning rate was 0.5 mV s−1. All the tests were performed at room temperature.
Results and discussion
The morphology of the PS template on the Ni foam substrate is shown in Fig. 2a. The PS spheres present well-ordered and the average size of PS spheres is about 400 nm. Fig. 2c and d show the morphology of HPC-Mn. It is clearly seen that, after anodic electrochemical deposition, removing the PS template and annealing, ordered macroporous structure could be formed. The inner surface of macropores is rough and the pore walls are composed of MnO2 nanoparticles, with average particle size of about 20 nm. The nano-sized MnO2 particles will enable the HPC-Mn anode to possess high surface electrochemical reactivity.38 The smallest thickness of the pore walls is about 10 nm, which gives effective transport pathways for electron and Li ions conduction.40 This nonuniformity of the pore walls' thicknesses is because that the PS spheres template is grown on cambered surfaces of Ni foam (Fig. S2a†) that is difficult to form uniform voids between PS spheres compared to flat surfaces.40,41 The macroporous structures are conducive to complete infiltration of the electrolyte, hence improving the diffusion rate of Li ions from the electrolyte to MnO2 nanoparticles. Moreover, the macropores can act as buffers for continuous volume expansion/contraction of MnO2 during lithiation/delithiation. Additionally, the directly grown of MnO2 nanoparticles onto Ni foam ensures a high electron conductivity. | ||
| Fig. 2 SEM images of (a) PS template, (b) HPA-Mn sample and (c and d) HPC-Mn sample. | ||
Fast ion/electron transfer would lead to fast reaction kinetics. This is especially important for the application of high-rate LIBs.42 For comparison, high resolution SEM image of the sample without annealing treatment (HPA-Mn) is shown in Fig. 2b. It can be clearly seen that the inner surface of the macropores is smooth and compact, which may reduce the diffusion rate and intercalation kinetics of Li ions. Fig. S2† shows that the whole surfaces of Ni foams have been covered by the porous MnO2 active materials.
The microstructure and composition of HPC-Mn sample were characterized by TEM-HRTEM micrographs, elemental mapping and energy dispersive X-ray spectroscopy (EDX). The SAED pattern shown in the top right inset of Fig. 3a indicates the polycrystalline characteristic of HPC-Mn sample. The blue labeled in the zone is selected for HRTEM (Fig. 3b), and the lattice fringes show the interplanar spacings are about 0.22, 0.25 and 0.31 nm, which are corresponding to the (330), (211) and (310) planes of tetragonal α-MnO2 respectively. The (310) plane are observed consistently at an angle of ∼59.5° to the (211) plane. Furthermore, the lattice fringe of Ni under fringes of α-MnO2 can also be obviously seen from Fig. 3b. The interplanar spacing is about 0.17 nm, which is corresponding to the (200) plane of Ni. This corroborates that the electrodeposited MnO2 nanoparticles are firmly connected to the Ni foam after annealing, which ensures high electronic conductivity and mechanical strength for the HPC-Mn anode.43 The elemental mapping (Fig. 3c and c1–c3) in the orange square area can also demonstrate the tightly bonding between MnO2 nanoparticles and Ni foam. The manganese and nickel elements occur at the same area (Fig. 3c1 and c2), indicating that the MnO2 nanoparticles are still bonded to Ni foam even after the strongly ultrasonic process when prepared the TEM sample. This is well agreement with Fig. 3b. It is notable that the TEM sample was prepared by scraping MnO2 nanoparticles from Ni foam using a blade. Hence, Ni particles would also be scraped along with MnO2 nanoparticles due to the strong adhesion. The strong adhesion and mechanical strength can be further intuitively seen from the ESI Video 3.† We tested the mechanical adhesion under a tape test, and no MnO2 active materials were visualized on the tape, indicating good adhesion of MnO2 nanoparticles on the Ni foam. In addition, EDX spectrum was conducted to determine the composition of the HPC-Mn sample (Fig. 3d), which confirms that the sample is composed of manganese, oxygen and nickel. The carbon and copper peaks derive from the carbon-coated copper grid.
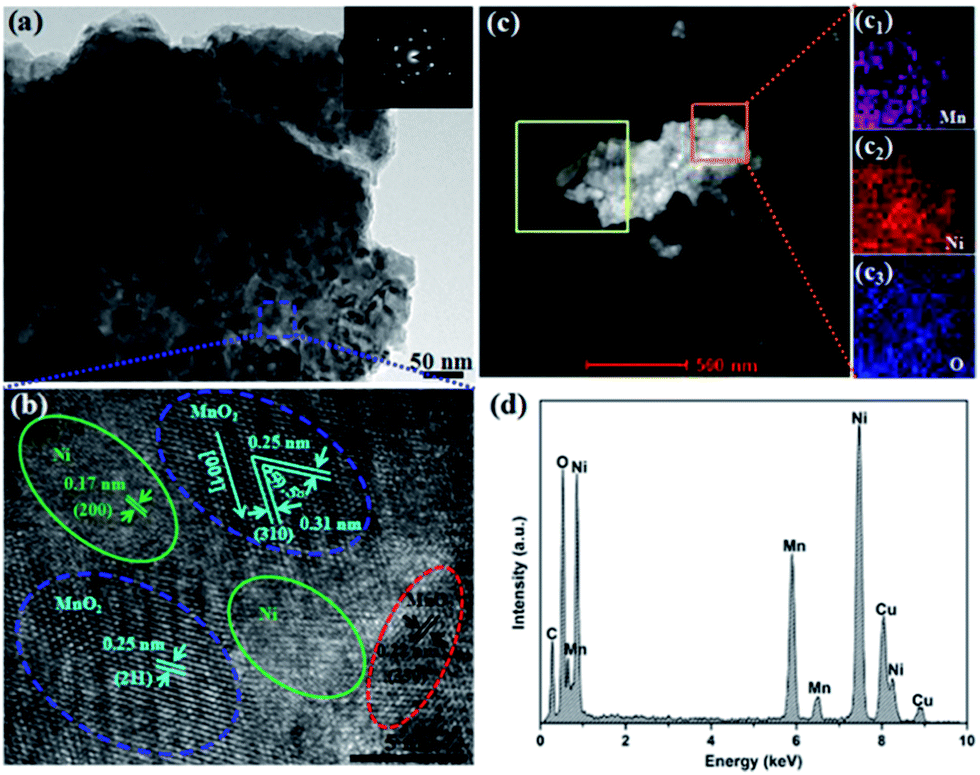 | ||
| Fig. 3 TEM-HRTEM micrographs of HPC-Mn sample. (a) TEM image, the inset is the SAED pattern and (b) HRTEM images of the labeled zone in (a). (c) Elemental mapping of Mn (c1), Ni (c2) and O (c3) corresponding to the outlined area by the orange square. (d) EDX spectrum of the HPC-Mn sample. | ||
The phases and crystal structures of various samples were monitored and characterized by the XRD pattern, Raman and XPS spectra. Fig. 4a shows the XRD patterns of HPC-Mn and HPA-Mn, Ni foam for comparison. All samples show three strong diffraction peaks at 44.6°, 52.0° and 76.4°, which are corresponding to (111), (200) and (220) crystal planes of Ni respectively, consistent with the HRTEM results. The diffraction peaks of HPC-Mn at 18.1°, 29.0°, 32.6°, 36.2° and 60.2° can be indexed to the diffractions from (200), (310), (101), (211) and (521) crystal planes of body-centered tetragonal structure α-MnO2 (JCPDS 44-0141), respectively. While the XRD pattern of HPA-Mn without annealing expresses no obvious peaks, indicating an amorphous structure. The HPC-Mn sample has been further characterized by Raman spectroscopy, as shown in Fig. 4b. The two main peaks at 556 and 655 cm−1 are attributed to the Mn–O stretching mode of MnO6 octahedra.44,45 While the weak peak at 345 cm−1 is assigned to Mn2O3 or Mn3O4 formed during the spectrum acquisition due to the local heating of the sample.46 The XPS spectrum was conducted to confirm the oxidation state of Mn in the HPC-Mn sample. The wide survey XPS spectrum of the sample is shown in Fig. S3,† which shows predominant signals of Mn, O and C. Note that the elemental carbon comes from surrounding atmosphere. Mn 2p spectra of the as-prepared sample is shown in Fig. 4c. The spin energy separation of Mn 2p3/2 and Mn 2p1/2 centered at 641.8 and 653.5 eV, respectively, is 11.7 eV, which is good consistent with reported data for MnO2, indicating the Mn4+ in the HPC-Mn sample.47,48 Fig. 4d exhibits the O 1s spectra of the as-prepared sample. The peaks located at 530.0 eV and 532.0 eV are related to binding energy of O 1s in anhydrous (Mn–O–Mn) and hydrated (Mn–O–H) manganese oxides, respectively.29
 | ||
| Fig. 4 (a) XRD patterns of pure Ni foam, HPA-Mn and HPC-Mn samples. (b) Raman spectrum of HPC-Mn sample. (c and d) XPS of HPC-Mn sample, in which (c) corresponds to the Mn 2p states and (d) corresponds to the O 1s states. (e) 2D SAXS pattern of HPC-Mn anode. (f) Mesoporous size distributions of HPC-Mn and HPA-Mn anodes. | ||
Mesoporous structures of the HPC-Mn and HPA-Mn anodes were measured through SAXS experiment. Fig. 4e and S4† show the two-dimensional (2D) SAXS patterns of HPC-Mn anode and HPA-Mn anode respectively. The 2D SAXS patterns confirm that the both anodes possess mesopores,49–51 forming the hierarchically porous structures with macropores. Fig. S5† shows the schematic representation of the mesoporous formation in HPA-Mn anode. During electrochemical synthesis of MnO2, water electrolysis would occur and give rise to the formation of oxygen on working electrode that explains the formation of mesopores.52,53 ESI Video 4† shows the occurrence of gas bubbles on working electrode, further confirming the formation of oxygen. The mesoporous size distributions of the two anodes are shown in Fig. 4f. The mesoporous diameters of HPC-Mn and HPA-Mn anodes are both around 5 and 14 nm, while the proportions of the two kinds of mesopores are obviously different for the both anodes. It can be clearly seen that the amount of mesopore of 14 nm in diameter decrease while the amount of mesopore of 5 nm in diameter increase after annealing at 400 °C. During annealing process, the compact HPA-Mn sample would fracture, resulting in the decrease of larger mesopores. Then the newly formed crystalline MnO2 nanoparticles would accumulate in situ to form large amounts of smaller mesopores, as shown in the illustration (Fig. 1d and e). The obvious morphology changes before and after annealing can also be observed from Fig. 2b and d. The BET tests were further carried out to study the porous structure of the HPC-Mn and HPA-Mn anodes. The N2 adsorption–desorption isotherms are shown in Fig. S6† and exhibit the isotherm of type IV with distinct hysteresis loops, indicating the presence of mesopores in the two anodes.51,54
The electrochemical performance of the HPC-Mn anode in LIBs was investigated, together with the comparison with HPA-Mn anode. Fig. 5a shows the representative CV curves of HPC-Mn anode measured between 0.01 and 3.0 V (versus Li/Li+) at a constant scan rate of 0.5 mV s−1. It is noted that the shape of the first cathodic curve varies essentially from the subsequent ones. An irreversible reduction peak at 0.15 V has a relatively broad bottom, corresponding to the formation of a solid electrolyte interface (SEI) layer and the reduced reaction of MnO2 with Li ions.29 From the second cycle, two reduction peaks located at 1.05 and 0.45 V occur, which are related to the reduction of Mn4+ to Mn2+ and the complete reduction of Mn2+ to metallic Mn, respectively.29,55 In the anodic cycle, there is a mild broad peak centered at around 1.3 V and a sharper peak centered at 2.0 V, which corresponds to the oxidation of metallic Mn to Mn2+ and the further oxidation of Mn2+ to Mn4+, respectively. It can be clearly seen that the voltages of reduction peaks become higher after the first CV cycle, indicating the reduced electrochemical polarization. This may be the reason that the crystal structure of HPC-Mn anode have changed to nano-crystallite structure after the first lithiation.37,38
 | ||
| Fig. 5 (a and c) CV curves of the HPC-Mn and HPA-Mn anodes between 0.01 and 3.0 V (versus Li/Li+) at a scan rate of 0.5 mV s−1. (b and d) The 1st, 2nd and 3rd galvanostatic discharge/charge curves at 0.1 A g−1 between 0.01 and 3.0 V of the HPC-Mn and HPA-Mn anodes. | ||
The voltage plateaus for the first three discharge/charge curves of HPC-Mn shown in Fig. 5b are in good agreement with the redox peaks of CV curves. There is one flat plateau in the first discharge process, confirming the reduced reaction of MnO2 with Li ions in one step, while two steep plateaus occurring in the following two discharge processes demonstrate the reduced reaction of MnO2 with Li ions in two steps. The higher voltage plateaus after the first discharge also demonstrate the reduced electrochemical polarization due to the structural transformation. Two charge plateaus can also be observed, corresponding to different oxidation reactions during the Li ion extraction. The first discharge and charge specific capacities are 1357.7 and 916.6 mA h g−1 at the current density of 0.1 A g−1, respectively, and the initial coulombic efficiency is 67.5%. It is noted that the first discharge specific capacity has higher value than the theoretical capacity, which might be attributed to the irreversible reaction of the electrode to form the SEI layer on the surface of the electrode.23,47
Different from the HPC-Mn anode, there is only one reduction peak or one discharge plateau after the first cycle for HPA-Mn anode, as shown in Fig. 5c and d, indicating one step reduced reaction of MnO2 with Li ions (from Mn4+ to metallic Mn). While the CV curves (Fig. 5c) show two oxidation peaks located at 1.1 V and 2.2 V, corresponding to the charge plateaus (Fig. 5d), indicating two step oxidation reactions (from metallic Mn to Mn2+ and from Mn2+ to Mn4+ respectively). The first discharge and charge specific capacities of HPA-Mn anode are 1218.4 and 769.8 mA h g−1 at the current density of 0.1 A g−1, respectively, and the initial coulombic efficiency is 63.2%, lower than that of HPC-Mn anode. The unchanged voltage plateau indicates that the electrochemical polarization of the HPA-Mn anode remains the same values after the first discharge due to the larger overpotential of amorphous structure than crystal structure when change to nano-crystallite structure in the first lithiation.37 The larger polarization value of HPA-Mn anode may also ascribe to the lower conductivity than that of HPC-Mn anode (Fig. S7†). The heat treatment of electrodeposited MnO2 sample enhances the adhesion between hierarchically porous MnO2 nanostructures and Ni foam, which increases the electrical conductivity and results in relatively lower electrochemical impedance.43 Compared to HPA-Mn anode, there is a better overlap for HPC-Mn anode, not only in the CV curves but also in the discharge/charge profiles. It indicates that the HPC-Mn anode has better electrochemical reversibility and structural stability.
Fig. 6a, b and d compare the rate performance of these two different anodes, in which the current density is up to 10 A g−1 from 0.1 A g−1. It is clear that the HPC-Mn anode has better rate capability than the HPA-Mn anode. The HPC-Mn anode can deliver a reversible specific capacity of 973.8, 956.2, 925.1, 865.0, 864.2, 798.8, 677.9, 508.8, 391.8 mA h g−1 at a current density of 0.1, 0.2, 0.4, 0.8, 1.0, 2.0, 4.0, 8.0, and 10.0 A g−1, respectively, indicating a good rate capacity. While the HPA-Mn anode delivers a lower reversible specific capacity of 614.9, 395.4, 265.1, 192.8, 178.5, 136.7, 86.1, 35.7, 33.5 mA h g−1 at the same current densities. The better rate performance of HPC-Mn anode should be resulted from the nano-sized MnO2 particles with high surface electrochemical reactivity and the structural change, from crystal structure to nano-crystallite structure, which may promote the intercalation kinetics of Li ions.37,38 In addition, the increased adhesion between MnO2 nanoparticles and Ni foam, due to heat treatment process, ensures the high electronic conductivity and mechanical strength and the hierarchically porous structure accelerates the electrolyte penetration that would improve the diffusion rate of Li ions. When the current density again returns to 0.1 A g−1, the HPC-Mn anode can still deliver a reversible capacity of 955.8 mA h g−1. When the current density increases 20 times from 0.1 to 2.0 A g−1, the HPC-Mn anode has high capacity retention ratio of 82% (Fig. 6d). Compared to other reported MnO2 based anodes, the HPC-Mn anode reveals excellent rate performance, as shown in Table S1.†
 | ||
| Fig. 6 The galvanostatic discharge/charge curves (a and b) at various current densities and (c) at different cycles, (d) rate capabilities, and (e) cycling performances at 0.4 A g−1 of HPC-Mn and HPA-Mn anodes. | ||
The cycling performances of HPC-Mn and HPA-Mn anodes are given in Fig. 6e. The two anodes were discharged and charged for 200 cycles at the current density of 0.4 A g−1. For the HPA-Mn anode, the reversible specific capacity decreases obviously at first 40 cycles and maintains a low reversible specific capacity in the following cycles. The discharge/charge profiles of HPC-Mn anode at 0.4 A g−1 for different cycles are shown in Fig. 6c. The first discharge and charge specific capacities are 1161.8 and 797.1 mA h g−1, respectively, with an initial coulombic efficiency of 68.6%. In the first several cycles, the capacity drop may ascribe to the irreversible conversion reaction and the formation of SEI layers.56 The increasing trend during cycling may be resulted from the activation of the electrode materials and/or the reversible growth of a polymeric gel-like film from kinetically activated electrolyte degradation, which has been presented in other reports.29,56–58 After 200 cycles, the reversible specific capacity of HPC-Mn anode is 778.0 mA h g−1, retaining 97.6% of its initial value. The better cycling performance of HPC-Mn anode should be ascribed to the strong adhesion between active materials and Ni foam after annealing, suppressing the degradation of MnO2 active materials and the exfoliation from the current collector of Ni foam, and the hierarchically porous structure, accommodating the volume expansion of MnO2 nanoparticles during cycling processes. Table S2† compares the cycling performance with other MnO2 systems reported in literatures and shows the good cycling capability of HPC-Mn anode.
In order to further understand the enhanced electrochemical performance of HPC-Mn anode relative to HPA-Mn anode, electrochemical impedance spectroscopy (EIS) measurements were carried out for these two samples, as shown in Fig. S7.† Both the EIS spectra exhibit the typical Nyquist plots, which is composed of a semicircle at the high-to-medium frequency region and a slope line following at the low frequency region. The former corresponds to the ohmic resistance and the charge transfer resistance between the electrolyte and the electrodes, while the latter represents the Warburg impedance of Li ions diffusion within electrodes. It can be obviously discovered that the reaction resistance of HPC-Mn anode is much less than that of HPA-Mn anode, demonstrating the excellent rate capability and long cycling life for HPC-Mn anode.
The good electrochemical performance of the HPC-Mn anode results from its unique crystal architecture of hierarchical pores. As mentioned above, it could be summarized as follows (1) the tightly bonding of MnO2 nanoparticles onto conductive Ni foam ensures high electron conductivity and mechanical strength; (2) the hierarchically porous structure contributes to the electrolyte penetration, hence ensuring well contact of electrolyte and active materials and improving the diffusion rate of Li ions from electrolyte to MnO2 nanoparticles, besides it can also accommodate the volume change of MnO2 nanoparticles during cycles; (3) the bicontinuous nanoscaled walls give effective transport pathways for electron and Li ion conduction; (4) the formed nano-sized MnO2 particles upon annealing may enhance the surface electrochemical reactivity; (5) the structural transformation from crystal structure to nano-crystallite structure in the first lithiation should facilitate the Li ion insertion and lower the electrochemical polarization. The combination of the novel crystalline structure with the abundant hierarchical pores improves the electrochemical performance of the HPC-Mn anode.
Conclusions
In summary, we construct a MnO2 anode with 3D hierarchically porous crystalline structure on Ni foam via a facile anodic electrochemical deposition and anneal process. The designed nanostructure of the electrode increases the contact area between electrode and electrolyte, shortens the transport pathways of Li ions, improves the electronic conductivity, increases the mechanical strength and surface electrochemical reactivity of MnO2 nanoparticles, lower the electrochemical polarization, and is able to accommodate the volume variation during lithiation/delithiation, and markedly increases the rate capability and cycle life of the electrode. The electrode exhibits improved electrochemical performance that the reversible specific capacity reaches 778.0 mA h g−1 after 200 cycles at a current density of 0.4 A g−1 and excellent rate capability of up to 82% capacity retention when the current density increases 20 times from 0.1 to 2.0 A g−1. Therefore, this reasonable design of 3D hierarchically porous crystalline structure will deeply promote future researches and applications for other electrodes with low conductivity and large volume expansion during cycling.Acknowledgements
We thank National Natural Science Foundation of China (No. 51572058, 91216123, 51174063, 51502057), the Natural Science Foundation of Heilongjiang Province (No. E201436, E2016062), the International Science & Technology Cooperation Program of China (2013DFR10630, 2015DFE52770) and Specialized Research Fund for the Doctoral Program of Higher Education (SRFDP 20132302110031), the China Postdoctoral Science Foundation (General Financial Grant No. 2014M561345), the Heilongjiang Postdoctoral Science Foundation (LBH-Z14105), the Scientific Research Foundation for the Returned Overseas Chinese Scholars of the State Education Ministry (No. 20151098), the University Nursing Program for Young Scholars with Creative Talents in Heilongjiang province (No. 2015082), and the Open Project Program of the Key Laboratory for Photonic and Electric Band Gap Materials of the Ministry of Education of Harbin Normal University.References
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- W. J. Lee, T. H. Hwang, J. O. Hwang, H. W. Kim, J. Lim, H. Y. Jeong, J. Shim, T. H. Han, J. Y. Kim, J. W. Choi and S. O. Kim, Energy Environ. Sci., 2014, 7, 621 CAS.
- X. Liu, D. Chao, Y. Li, J. Hao, X. Liu, J. Zhao, J. Lin, H. J. Fan and Z. X. Shen, Nano Energy, 2015, 17, 43 CrossRef CAS.
- R. B. Cervera, N. Suzuki, T. Ohnishi, M. Osada, K. Mitsuishi, T. Kambara and K. Takada, Energy Environ. Sci., 2014, 7, 662 CAS.
- S. Liu, W. He, X. Zhang, H. Li, S. Zhang and Y. Wang, Energy Environ. Focus, 2015, 4, 178 CrossRef.
- D. Yang, S. Xu, S. Dong, J. Liu, A. Guo, X. Yan and F. Hou, RSC Adv., 2015, 5, 106298 RSC.
- G. W. Zhou, J. Wang, P. Gao, X. Yang, Y. S. He, X. Z. Liao, J. Yang and Z. F. Ma, Ind. Eng. Chem. Res., 2012, 52, 1197 CrossRef.
- H. Geng, Q. Zhou, J. Zheng and H. Gu, RSC Adv., 2014, 4, 6430 RSC.
- S. Zhang, W. He, X. Zhang, G. Yang, J. Ma, X. Yang and X. Song, Electrochim. Acta, 2015, 174, 1175 CrossRef CAS.
- Y. S. Lin, J. G. Duh and M. H. Hung, J. Phys. Chem. C, 2010, 114, 13136 CAS.
- H. Zhang, L. Gao and S. Yang, RSC Adv., 2015, 5, 43798 RSC.
- Z. S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187 CrossRef CAS PubMed.
- J. He, Y. Liu, Y. Meng, X. Sun, S. Biswas, M. Shen, Z. Luo, R. Miao, L. Zhang, W. E. Mustain and S. L. Suib, RSC Adv., 2016, 6, 24320 RSC.
- Z. Weng, H. Guo, X. Liu, S. Wu, K. W. K. Yeung and P. K. Chu, RSC Adv., 2013, 3, 24758 RSC.
- W. Wen, J. M. Wu and M. H. Cao, Nano Energy, 2013, 2, 1383 CrossRef CAS.
- J. B. Chen, Y. W. Wang, X. M. He, S. M. Xu, M. Fang, X. Zhao and Y. M. Shang, Electrochim. Acta, 2014, 142, 152 CrossRef CAS.
- J. Yue, X. Gu, L. Chen, N. N. Wang, X. L. Jiang, H. Y. Xu, J. Yang and Y. T. Qian, J. Mater. Chem. A, 2014, 2, 17421 CAS.
- L. H. Li, C. Y. Nan, J. Lu, Q. Peng and Y. D. Li, Chem. Commun., 2012, 48, 6945 RSC.
- K. F. Chen, Y. D. Noh, K. Y. Li, S. Komarneni and D. F. Xue, J. Phys. Chem. C, 2013, 117, 10770 CAS.
- A. L. M. Reddy, M. M. Shaijumon, S. R. Gowda and P. M. Ajayan, Nano Lett., 2009, 9, 1002 CrossRef CAS PubMed.
- M. Kundu, C. C. A. Ng, D. Y. Petrovykh and L. F. Liu, Chem. Commun., 2013, 49, 8459 RSC.
- W. Xiao, J. S. Chen, Q. Lu and X. W. Lou, J. Phys. Chem. C, 2010, 114, 12048 CAS.
- H. Lai, J. X. Li, Z. G. Chen and Z. G. Huang, ACS Appl. Mater. Interfaces, 2012, 4, 2325 CAS.
- H. Xia, M. O. Lai and L. Lu, J. Mater. Chem., 2010, 20, 6896 RSC.
- C. X. Guo, M. Wang, T. Chen, X. W. Lou and C. M. Li, Adv. Energy Mater., 2011, 1, 736 CrossRef CAS.
- X. L. Li, H. F. Song, H. Wang, Y. L. Zhang, K. Du, H. Y. Li and J. M. Huang, J. Appl. Electrochem., 2012, 42, 1065 CrossRef CAS.
- A. P. Yu, H. W. Park, A. Davies, D. C. Higgins, Z. W. Chen and X. C. Xiao, J. Phys. Chem. Lett., 2011, 2, 1855 CrossRef CAS.
- Y. Zhang, Z. P. Luo, Q. Z. Xiao, T. L. Sun, G. T. Lei, Z. H. Li and X. J. Li, J. Power Sources, 2015, 297, 442 CrossRef CAS.
- J. W. Deng, L. F. Chen, Y. Y. Sun, M. H. Ma and L. Fu, Carbon, 2015, 92, 177 CrossRef CAS.
- X. H. Xia, J. P. Tu, X. L. Wang, C. D. Gu and X. B. Zhao, J. Mater. Chem., 2011, 21, 671 RSC.
- J. T. Park, D. K. Roh, R. Patel, E. Kim, D. Y. Ryu and J. H. Kim, J. Mater. Chem., 2010, 20, 8521 RSC.
- J. T. Park, J. H. Koh, J. A. Seo and J. H. Kim, J. Mater. Chem., 2011, 21, 17872 RSC.
- Z. M. He, J. Liu, Y. Qiao, C. M. Li and T. T. Y. Tan, Nano Lett., 2012, 12, 4738 CrossRef CAS PubMed.
- Y. Tao, D. B. Kong, C. Zhang, W. Lv, M. X. Wang, B. H. Li, Z. H. Huang, F. Y. Kang and Q. H. Yang, Carbon, 2014, 69, 169 CrossRef CAS.
- K. Xi, S. Cao, X. Y. Peng, C. Ducati, R. V. Kumar and A. K. Cheetham, Chem. Commun., 2013, 49, 2192 RSC.
- S. Li, Q. Xu, E. Uchaker, X. Cao and G. Cao, CrystEngComm, 2016, 18, 2532 RSC.
- Y. Xu, G. Jian, Y. Liu, Y. Zhu, M. R. Zachariah and C. Wang, Nano Energy, 2014, 3, 26 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS PubMed.
- M. A. McLachlan, N. P. Johnson, R. M. De La Rue and D. W. McComb, J. Mater. Chem., 2004, 14, 144 RSC.
- Z. Q. Tong, J. Hao, K. Zhang, J. P. Zhao, B. L. Su and Y. Li, J. Mater. Chem. C, 2014, 2, 3651 RSC.
- Z. Tong, H. Yang, L. Na, H. Qu, X. Zhang, J. Zhao and Y. Li, J. Mater. Chem. C, 2015, 3, 3159 RSC.
- M. H. Chen, X. H. Xia, J. F. Yuan, J. H. Yin and Q. G. Chen, J. Power Sources, 2015, 288, 145 CrossRef CAS.
- X. Hu, X. Han, Y. Hu, F. Cheng and J. Chen, Nanoscale, 2014, 6, 3522 RSC.
- S. H. Liang, F. T. G. Bulgan, R. L. Zong and Y. F. Zhu, J. Phys. Chem. C, 2008, 112, 5307 CAS.
- M. Sun, B. Lan, T. Lin, G. Cheng, F. Ye, L. Yu, X. L. Cheng and X. Y. Zheng, CrystEngComm, 2013, 15, 7010 RSC.
- J. Luo, H. T. Zhu, H. M. Fan, J. K. Liang, H. L. Shi, G. H. Rao, J. B. Li, Z. M. Du and Z. X. Shen, J. Phys. Chem. C, 2008, 112, 12594 CAS.
- Y. Y. Li, Q. W. Zhang, J. L. Zhu, X. L. Wei and P. K. Shen, J. Mater. Chem. A, 2014, 2, 3163 CAS.
- L. Li, A. R. O. Raji and J. M. Tour, Adv. Mater., 2013, 25, 6298 CrossRef CAS PubMed.
- X. Liu, J. Yin, M. Chen, W. Bu, W. Cheng and Z. Wu, Nanosci. Nanotechnol. Lett., 2011, 3, 1 CrossRef.
- X. Liu, J. Yin, Y. Kong, M. Chen, Y. Feng, K. Yan, X. Li, B. Su and Q. Lei, Thin Solid Films, 2013, 544, 352 CrossRef CAS.
- Y. Si, T. Ren, B. Ding, J. Yu and G. Sun, J. Mater. Chem., 2012, 22, 4619 RSC.
- M. Ghaemi and L. Binder, J. Power Sources, 2002, 111, 248 CrossRef CAS.
- G. R. Li, Z. P. Feng, Y. N. Ou, D. Wu, R. Fu and Y. X. Tong, Langmuir, 2010, 26, 2209 CrossRef CAS PubMed.
- P. P. Wang, B. Bai, S. Hu, J. Zhuang and X. Wang, J. Am. Chem. Soc., 2009, 131, 16953 CrossRef CAS PubMed.
- J. X. Li, M. Z. Zou, Y. Zhao, Y. B. Lin, H. Lai, L. H. Guan and Z. G. Huang, Electrochim. Acta, 2013, 111, 165 CrossRef CAS.
- J. Yue, X. Gu, X. L. Jiang, L. Chen, N. N. Wang, J. Yang and X. J. Ma, Electrochim. Acta, 2015, 182, 676 CrossRef CAS.
- S. Grugeon, S. Laruelle, L. Dupont and J. M. Tarascon, Solid State Sci., 2003, 5, 895 CrossRef CAS.
- X. Xu, Z. Y. Fan, X. Y. Yu, S. J. Ding, D. M. Yu and X. W. Lou, Adv. Energy Mater., 2014, 4, 1400902 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra16430g |
| This journal is © The Royal Society of Chemistry 2016 |