
A facile one-pot solvothermal synthesis of CoFe2O4/RGO and its excellent catalytic activity on thermal decomposition of ammonium perchlorate
Teng Chena,
Ping Du*b,
Wei Jiang*a,
Jie Liua,
Gazi Haoa,
Han Gaoa,
Lei Xiaoa,
Xiang Kea,
Fengqi Zhaoc and
Chunlei Xuanc
aNational Special Superfine Powder Engineering Research Center of China, School of Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: superfine_jw@126.com; Fax: +86 25 84315042; Tel: +86 25 84315042
bSchool of Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: dp1314@163.com
cXi'an Modern Chemistry Research Institute, Xi'an 710065, China
First published on 29th August 2016
Abstract
CoFe2O4/RGO hybrids have been successfully fabricated via a facile one-pot solvothermal method, which were characterized by X-ray diffraction (XRD), Raman, Fourier-transform infrared spectroscopy (FT-IR) and X-ray photoelectron spectroscopy (XPS). During this process, graphene oxide was reduced to graphene (RGO) and CoFe2O4 nanoparticles were deposited on the RGO. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) revealed that the average size of CoFe2O4/RGO hybrids was 120 nm, which was smaller than that of bare CoFe2O4, implying that RGO could effectively prevent CoFe2O4 nanoparticles from aggregating. To investigate the catalytic activity of the as-synthesized CoFe2O4 particles and CoFe2O4/RGO hybrids, the thermal decomposition of ammonium perchlorate (AP) was characterized by differential thermal analyser (DTA). Both of the two exothermic processes were merged into a sole exothermic process with the addition of bare CoFe2O4 and CoFe2O4/RGO hybrids, though there was no change in the position of the phase transition temperature of AP. Moreover, the catalytic activity of CoFe2O4/RGO hybrids is higher than that of bare CoFe2O4, due to the large surface area and enhanced properties of RGO in the hybrids. The temperature programmed reduction (TPR) measurements showed that the reduction temperature of CoFe2O4/RGO decreased by 55 °C compared with bare CoFe2O4, which further confirmed the higher catalytic activity of CoFe2O4/RGO of than that of CoFe2O4 nanocomposites. Hence, CoFe2O4/RGO hybrids could be a promising additive in modifying the burning behaviour of AP-based composite propellant.
1. Introduction
In a typical formula, a composite solid propellant consists of a polymer (binder), energetic fuel and oxidizer salts.1–4 Ammonium perchlorate (AP) is one of the main components in composite solid propellants, which has been extensively employed as an oxidizer for rocket propulsion. The high-temperature decomposition (HTD) properties of AP, such as the activation energy, rate and temperature can directly affect the combustion behaviour of a composite solid propellant, including the burning rate and pressure exponent. With the development of modern rockets, an even higher energy release rate is required. Thus it is important to decrease the pyrolysis temperature of AP, because the lower the HTD temperature, the higher the burning rate and the more exothermic heat there will be.5–8In order to decrease the pyrolysis temperature and improve the energy release rate of AP, transition metal particles and metal oxides have been extensively used to catalyse the reaction of AP.9–13 It is known that the particle size of burning rate catalyst could affect the catalytic activity towards AP. Therefore, a nano-sized catalyst is more effective than a micron-sized catalyst, due to its smaller size, larger surface area and high-surface activity. However, nano-sized particles are inclined to agglomerate into large particles which lead to the decrease of their specific surface area and active sites.14 Moreover, the formation of heterogeneous dispersion phase, caused by the agglomeration of nano-sized particles, can also inhibit the catalytic activity in the thermal decomposition process of AP. In order to prevent the nano-sized particles from agglomerating, the most commonly used method is to coat some chemical agents on the surface of nano-sized particles, such as polymers and ligands. The existence of these inert components, which are coated on the surface of nano-sized particles, may decrease their reaction activity and the total energy of composite solid propellant, thus severely limiting their practical application. Hence, it is necessary to explore an alternative method to prevent the aggregation and keep their high reaction activity. With this regards, various supporting materials have been widely studied, of which, graphene could be considered as an ideal substrate for nano-particles to spread and distribute on, due to its large specific surface area and specific properties.15,16
Since graphene was discovered by Andre Geim's team,17 it has attracted tremendous attention from scientific researchers.18–21 It is well known that researchers have paid great attention to carbon nanotubes over the past few decades,22,23 and nowadays, graphene takes a similar level in new applications as carbon nanotubes. Due to the two dimensional structure and extraordinary physical, chemical and mechanical properties of graphene,24 it was considered as a promising nanoscale building blocks for new materials, motivating the development of new materials in many fields,25–29 such as batteries, sensors, catalyst, supercapacitors, and energy storage.
Up till now, numerous facile methods have been developed to prepare graphene sheets, such as mechanical cleavage, thermal expansion of graphite,30–32 chemical vapour deposition17,33 and chemical reduction of exfoliated graphite oxide to reduced graphene oxide (RGO).34,35 Among these methods, chemical reduction of exfoliated graphite oxides is the most convenient way. Several oxygen-containing functional groups, including hydroxyl, carboxyl and epoxy groups, are introduced into graphite oxides, which make it easily to reduce graphite oxides into RGO. The porous structure of RGO can not only improve the stability and dispersion of nano-sized particles, but can also enhance their properties.
At present, the catalytic behaviour of graphene-based metal oxides in the thermal decomposition of AP has been reported by previous studies.28–32 Nevertheless, to the best of our knowledge, there was no other report of CoFe2O4/RGO hybrids.
In this work, CoFe2O4/RGO hybrids were prepared by a facile one-pot solvothermal method and their catalytic behaviour on the decomposition of AP was investigated by varying its content over the range of 1–5% by differential thermal analysis.
2. Experimental
2.1 Materials
Graphene oxide was purchased from Nanjing Jicang Nanotechnology Co., Ltd, ferric chloride hexahydrate (FeCl3·6H2O), cobalt chloride hexahydrate (CoCl2·6H2O), sodium citrate (Na3C6H5O7·2H2O), sodium acetate (CH3COONa), ethylene glycol (HOC2H4OH) were purchased from Sino pharm Chemical Reagent Co., Ltd. All of the chemicals were analytical grade, commercially available and used without further purification.2.2 Synthesis of CoFe2O4/RGO hybrids
The CoFe2O4/RGO hybrids were prepared via a facile one-pot solvothermal in ethylene glycol. In a typical procedure, 90 mg of GO was dispersed in 40 mL ethylene glycol and ultra-sonicated for 2 h. Then 0.34 g of CoCl2·6H2O and 1.06 g of FeCl3·6H2O (the molar ratio of Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co is 2:1) were added to the suspension of GO. The mixture was vigorously stirred with a magnetic stirrer for 30 min to form a stable and homogeneous solution. Subsequently, 0.2 g of sodium citrate and 1.2 g of sodium acetate were slowly added to form a stable suspension. After mechanical stirring for 30 min, the resulting suspension was transferred to a 100 mL Teflon-lined stainless steel autoclave, tightly sealed and maintained at 180 °C for 12 h. After cooling down to room temperature, the obtained precipitate was centrifuged, washed thoroughly with deionized water and dried at 60 °C for 12 h. For comparison purpose, bare CoFe2O4 nanoparticles and RGO were also synthesized under the same experimental condition in the absence of GO or CoCl2·6H2O and FeCl3·6H2O, respectively. The schematic diagram for solvothermal thermal synthesis of nanospheres is illustrated in Fig. 1.
:Co is 2:1) were added to the suspension of GO. The mixture was vigorously stirred with a magnetic stirrer for 30 min to form a stable and homogeneous solution. Subsequently, 0.2 g of sodium citrate and 1.2 g of sodium acetate were slowly added to form a stable suspension. After mechanical stirring for 30 min, the resulting suspension was transferred to a 100 mL Teflon-lined stainless steel autoclave, tightly sealed and maintained at 180 °C for 12 h. After cooling down to room temperature, the obtained precipitate was centrifuged, washed thoroughly with deionized water and dried at 60 °C for 12 h. For comparison purpose, bare CoFe2O4 nanoparticles and RGO were also synthesized under the same experimental condition in the absence of GO or CoCl2·6H2O and FeCl3·6H2O, respectively. The schematic diagram for solvothermal thermal synthesis of nanospheres is illustrated in Fig. 1.
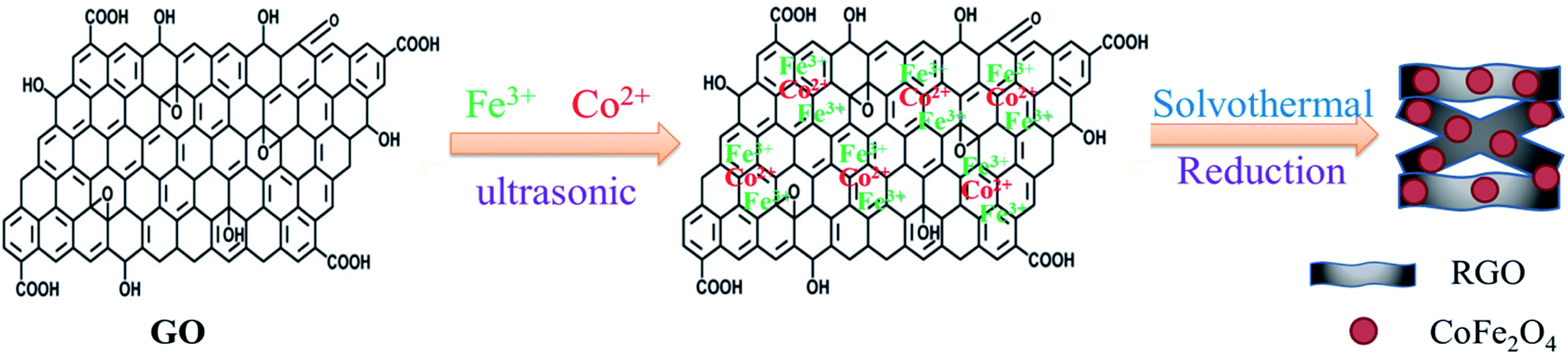 | ||
| Fig. 1 Illustration of synthesis process of CoFe2O4/RGO. | ||
2.3 Characterization
X-ray powder diffraction measurements of the samples were carried out using a Bruker D8-Advanced diffractometer with accelerating voltage 40 kV and current 40 mA of Cu Kα radiation (λ = 0.15406 nm), and the scanning angle of 2θ was in the range from 5° to 80°. Raman scattering spectra was performed to characterize the difference of the as-synthesized products with detecting section ranged from 500 to 4000 cm−1. Fourier-transform infrared (FT-IR) spectra was recorded to detect the chemical bonds on a Bruker Vector 22 spectrometer at room temperature. Scanning electron microscope images (Model-S4800) were taken to investigate the morphology of the samples. The microscopic structure and size were further confirmed by transmission electron microscopy (TEM, Tecnai 12).Information about the element composition was gained from X-ray photoelectron spectroscopy (XPS). The porous characteristics of the as-synthesized samples were measured by the nitrogen physical adsorption of at 77 K. Prior to the experiment, the sample was degassed under vacuum at 200 °C for 5 h. The specific surface areas of the resultant were estimated using the Brunauer–Emmett–Teller (BET) equation. The pore size distribution was obtained from the adsorption branch according to Barrett–Joyner–Halenda (BJH) method.
2.4 Catalytic measurement
The catalytic activity of RGO, CoFe2O4, and CoFe2O4/RGO hybrids on thermal decomposition of AP were investigated using differential thermal analyser (DTA) with a heating rate of 20 °C min−1 in a static nitrogen atmosphere over the temperature range of 50–500 °C. The mass ratio of RGO, CoFe2O4, and CoFe2O4/RGO hybrids in AP were 1%, 3%, 5%, respectively. The activation energies of AP decomposition with and without CoFe2O4/RGO hybrids were measured under different heating rates. The temperature programmed reduction (TPR) analysis was performed in the presence of 5% H2/N2 flow (50 mL min−1) and the temperature was increased from ambient to 1000 °C at 10 °C min−1.3. Results and discussion
The structure of the resulting samples was confirmed by XRD. Fig. 2 shows the XRD patterns of GO, CoFe2O4 and CoFe2O4/RGO. It can be seen that the original GO sample exhibits a sharp diffraction peak at 2θ = 11.05°, corresponding to the (001) reflection. The observed diffraction peak at 30.1°, 35.4°, 43.1°, 53.5°, 56.9°, 62.6° can be indexed to the (220), (311), (400), (422), (511), (440) reflections, respectively. These peaks match well with the standard pattern of cubic spinel structure (JCPDS card 22-1086), indicating the formation of CoFe2O4.36 It's noticeable that the characteristic diffraction peak at 11.05° disappears in the XRD pattern of CoFe2O4/RGO, which suggests that GO has been effectively reduced into graphene sheets. All characteristic diffraction peaks of CoFe2O4/RGO hybrids are accordance with bare CoFe2O4 particles. Meanwhile, no other diffraction peaks related with impurities are detected, which indicate CoFe2O4/RGO hybrids have been synthesized via a facile one-pot solvothermal method.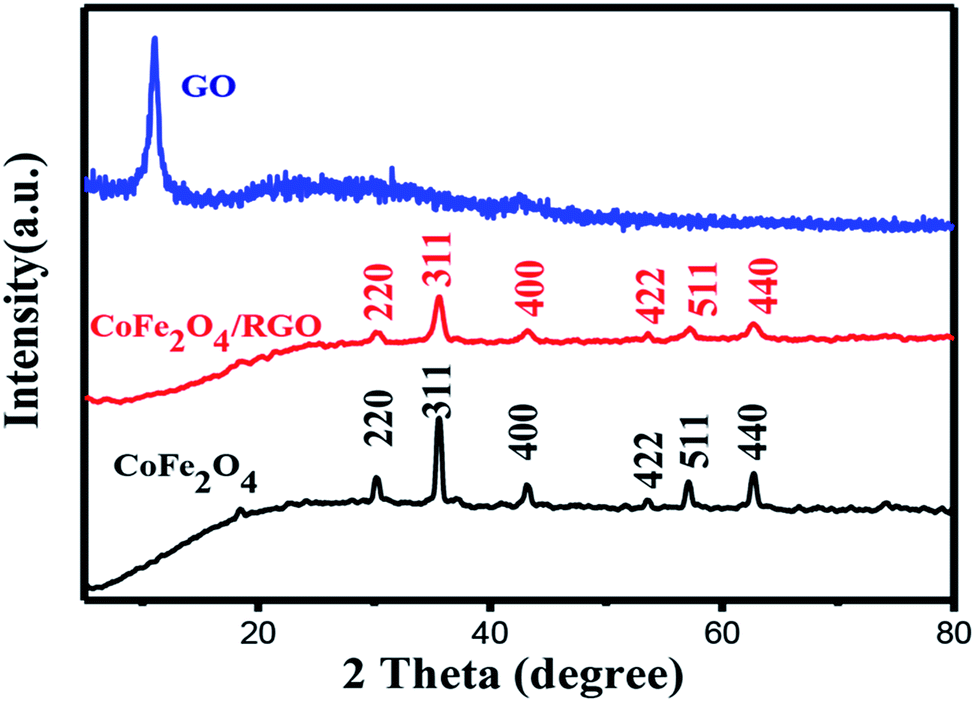 | ||
| Fig. 2 XRD patterns of GO, CoFe2O4 and CoFe2O4/RGO hybrids. | ||
The functionalized groups on graphite oxide and CoFe2O4/RGO were then characterized by FT-IR spectrum. Fig. 3(a) shows the FT-IR spectra of GO and the CoFe2O4/RGO nanocomposites which were recorded between 4000 cm−1 and 500 cm−1. The peak observed at 3620 cm−1 is attributed to the stretching vibration of –OH in the presence of adsorbed water. Several characteristic peaks related with the oxygen-containing functional groups can be found in Fig. 3(a). The peaks at 1721, 1613, 1213 and 1041 cm−1 should be ascribed to the carboxy C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, aromatic CC, epoxy C–O and C–O groups on the surface of graphene sheets respectively. Compared with GO, most characteristic peaks of the oxygen-containing functional groups on GO vanished in the spectrum of CoFe2O4/RGO, which implies that the oxygen-containing functional groups have already been removed from GO in the process of solvothermal.
O, aromatic CC, epoxy C–O and C–O groups on the surface of graphene sheets respectively. Compared with GO, most characteristic peaks of the oxygen-containing functional groups on GO vanished in the spectrum of CoFe2O4/RGO, which implies that the oxygen-containing functional groups have already been removed from GO in the process of solvothermal.
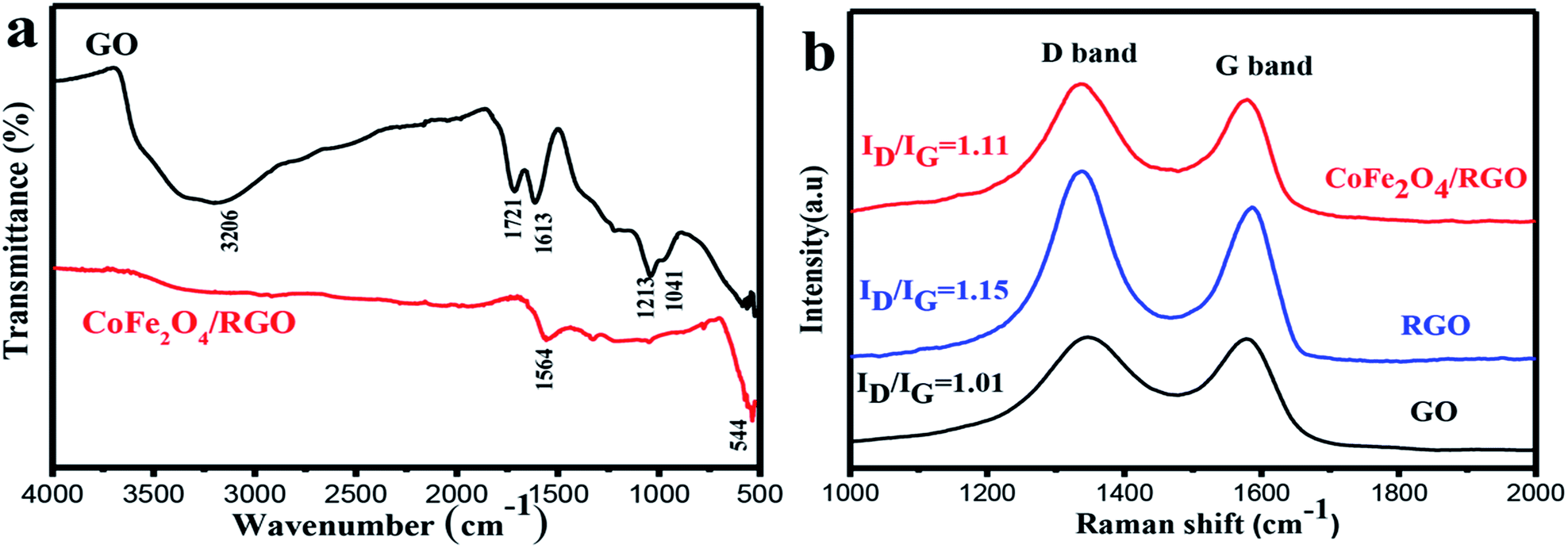 | ||
| Fig. 3 FT-IR spectra of GO and CoFe2O4/RGO (a) and Raman spectra of GO, RGO and CoFe2O4/RGO (b). | ||
In contrast to the peak at 1612 cm−1 in GO, a red shift peak around 1564 cm−1 can be observed in CoFe2O4/RGO, indicating the restoration of π–π conjugation of graphene sheets. The new absorption peak appeared at lower frequency around 554 cm−1 in CoFe2O4/RGO suggests that the Fe (Co)–O bonds have been formed in the presence of cobalt ferrite.37 From the FT-IR spectrum, we can assume that graphite oxide has been reduced to RGO and the target product of CoFe2O4/RGO has been synthetized.
Raman spectroscopy is an efficient tool for detecting the structure of carbonaceous materials. The Raman spectra of GO, RGO and CoFe2O4/RGO hybrids are shown in Fig. 3(b). It is obvious to observe that all of the spectra for GO, RGO and CoFe2O4/RGO display two peaks, which represent the G- and D-bands respectively. The G-band corresponds to the first-order scattering of the E2g mode for sp2-hybridized carbon and the D-band originates from intervalley scattering of disordered structure. Compared with those of GO, the peaks of D-bands shifted to lower frequencies. The D-band shifted from 1346 to 1336 cm−1, indicating that GO has been reduced to RGO.38 The intensity ratio (ID/IG) of D-band to G-band is correlative with the disorder and defects in graphene. It can be observed that the ID/IG ratio of RGO (1.15) is higher than that of GO (1.01), suggesting the reduction of GO into RGO and more defects generated in RGO.39–42 Metal or metal oxides nanoparticles can be easily trapped by the surface defects of RGO, which will result in the decrease of the defect density. It can be observed that ID/IG ratio of RGO in CoFe2O4/RGO is 1.11, which is lower than that of 1.15 in pristine RGO, implying that the defect density of RGO decreases after the decoration of CoFe2O4 nanoparticles on the surface of RGO.43–45
The microstructure and morphology of the as-synthesized CoFe2O4/RGO hybrids were characterized by SEM and TEM. From the SEM images in Fig. 4, it can be clearly seen that graphene sheets are distributed between the CoFe2O4 nanoparticles, which can inhibit CoFe2O4 nanoparticles from aggregation. The TEM images of CoFe2O4/RGO are displayed in Fig. 5. It can be observed that CoFe2O4 nanoparticles are densely and uniformly decorated on the surface of crumpled graphene sheets. It should be pointed out that, no CoFe2O4 particles fell off graphene sheets after sonication for a long time in the process of preparing for TEM specimen. It indicates that the defect structures of RGO could trap CoFe2O4 nanoparticles,43,44 which is accordance with result of the Raman analyses.
 | ||
| Fig. 4 SEM images of the as-synthesized CoFe2O4 (a); CoFe2O4/RGO hybrids (b). | ||
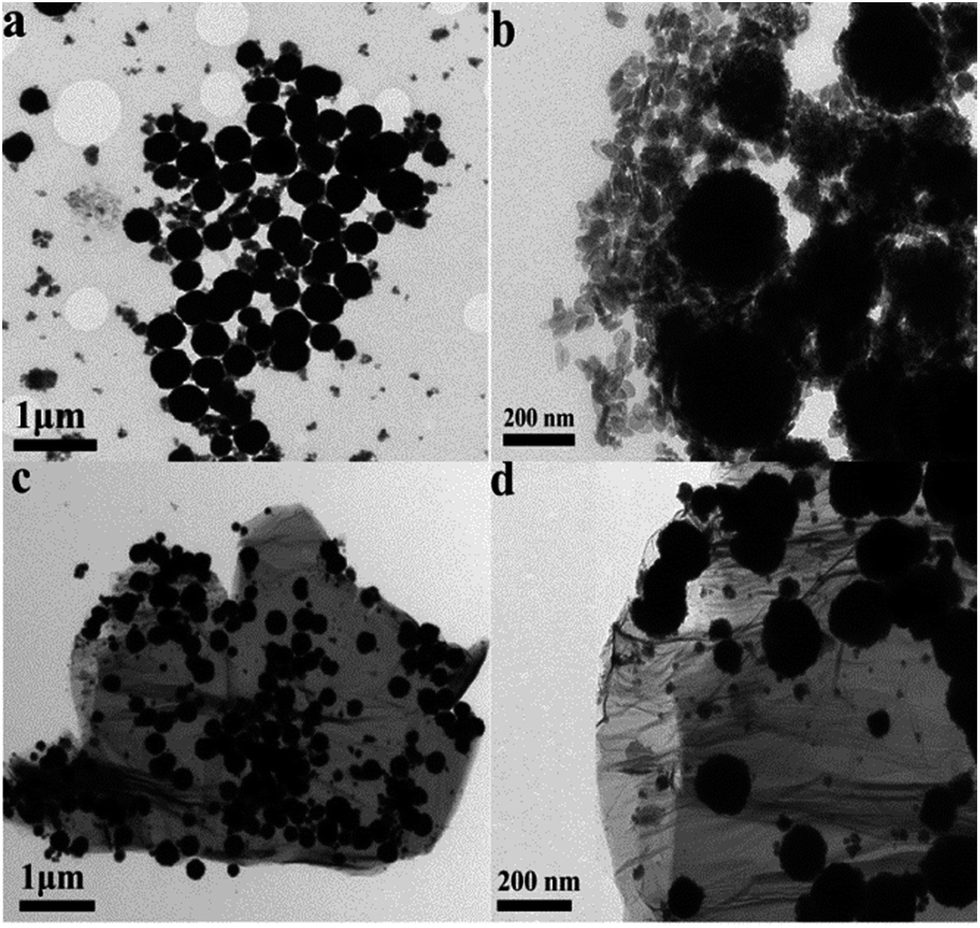 | ||
| Fig. 5 TEM images of the as-synthesized CoFe2O4 (a and b); CoFe2O4/RGO hybrids (c and d). | ||
Further evidence for chemical composition and valence state of elements in CoFe2O4/RGO hybrids were obtained by X-ray photoelectron spectroscopy (XPS). The wide scan XPS spectrum reveals the existence of Co (Co 2p1/2 and Co 2p3/2), Fe (Fe 2p1/2 and Fe 2p3/2), O (O 1s) and C (C 1s) element in CoFe2O4/RGO hybrids. The deconvolution of the Co 2p peak in CoFe2O4 and CoFe2O4/RGO are shown in Fig. 6(a). The peaks located at 780.5 eV and 796.3 eV in are assigned to Co 2p3/2 and Co 2p1/2, respectively. The accompanied shake up satellite peaks at 786.6 eV and 803.5 eV confirm the oxidation state of Co2+ in the samples.46 As illustrated in Fig. 6(a), the Fe 2p3/2 peak is centered at 710.9 eV and the Fe 2p1/2 peak is located at 724.8 eV. Two peaks appeared at 718.8 eV and 733.4 eV are ascribed to the shake up satellites of Fe 2p3/2 and Fe 2p1/2 respectively, indicating the existence of Fe3+.47 Compared with the Co 2p and Fe 2p spectra in CoFe2O4 and CoFe2O4/RGO, it can be inferred there is no shift in the binding energy of the peaks after decoration of CoFe2O4 on graphene sheets. The O 1s spectra in CoFe2O4 and CoFe2O4/RGO (Fig. 6(d)) exhibit a large peak at 529.3 eV and 529.9 eV, respectively, corresponding to M–O–M, which is ascribed to the lattice oxygen in CoFe2O4 phase.48 The other O 1s peak at 531.5 eV indicates the presence of oxygen containing groups which bonded with C atoms in RGO.49 Fig. 6(e) shows the C 1s spectra of GO, RGO and CoFe2O4/RGO. From the spectrum of GO, four de-convoluted peaks can be observed, which are related to CC and C–C bonds (284.8 eV) in the aromatic rings, C–O bond (285.5 eV) of epoxy and alkoxy, CO bond (287.7 eV) and O–CO groups (288.8 eV), respectively.50 In comparison with GO, the peak intensity of carbon–oxygen species in RGO decreases remarkably, indicating a effective reduction in the solvothermal process. Similar phenomenon can be observed in the spectrum of CoFe2O4/RGO. To further investigate the reduction degree of GO in CoFe2O4/RGO, the peak area ratios of oxygen-containing groups to total area are calculated according to the C 1s spectrum and the results are depicted in Table 1. It can be concluded that the percentage of oxygen-containing groups has a remarkably decrease, which further confirms the reduction of GO in CoFe2O4/RGO hybrids.
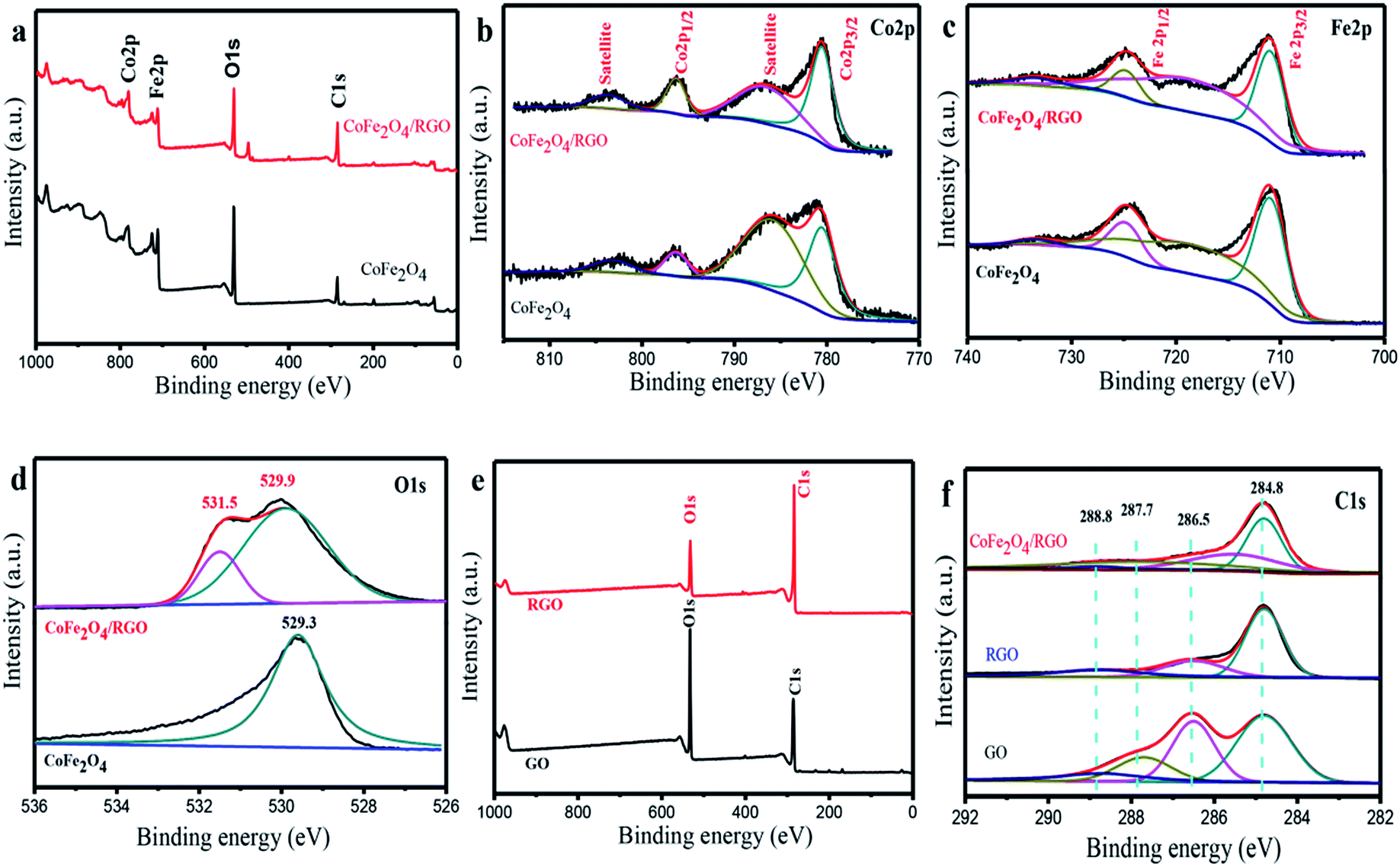 | ||
| Fig. 6 XPS survey spectra of the CoFe2O4/RGO and CoFe2O4 (a); XPS spectra for Co 2p of the CoFe2O4/RGO and CoFe2O4 (b); XPS spectra for Fe 2p of the CoFe2O4/RGO and CoFe2O4 (c); XPS spectra for O 1s of the CoFe2O4/RGO and CoFe2O4 (d); XPS survey spectra of the RGO and GO (e); XPS spectra for C 1s of the CoFe2O4/RGO, RGO and GO (f). | ||
| Sample | C–C | C–O | CO |
COOH |
|---|---|---|---|---|
| GO | 44.3 | 28.9 | 19.9 | 6.9 |
| RGO | 59.8 | 22.4 | 12.0 | 5.8 |
| CoFe2O4/RGO | 68.0 | 19.2 | 7.6 | 5.4 |
The specific surface area and the porosity distribution characteristic of the as-synthesized sample were investigated by N2 adsorption–desorption at 77 K using N2 as adsorbent. Fig. 7 shows the N2 adsorption–desorption isotherms and porosity distribution curves. The isotherm demonstrates a typical “type IV” isotherm with H3 hysteresis loops, revealing the existence of mesoporous in CoFe2O4/RGO hybrids. The porosity distribution curve of CoFe2O4/RGO hybrids inserted in Fig. 7(b) shows a narrow pore-size distribution at 3.04 nm and a broad pore-size distribution around 15.28 nm, indicating that the hybrids virtually comprise mesoporous structure and a minor fraction of microporous structure. The Brunauer–Emmett–Teller (BET) surface area is calculated to be 242.5 m2 g−1 and the average pore size is 7.25 nm. Nevertheless, the specific surface area and the average pore size of bare CoFe2O4 nanoparticles are 60.2 m2 g−1 and 18.79 nm, respectively. These results indicate that the surface area of CoFe2O4/RGO hybrids is relatively larger and the distribution of pore size is more uniform than those of bare CoFe2O4 nanoparticles.
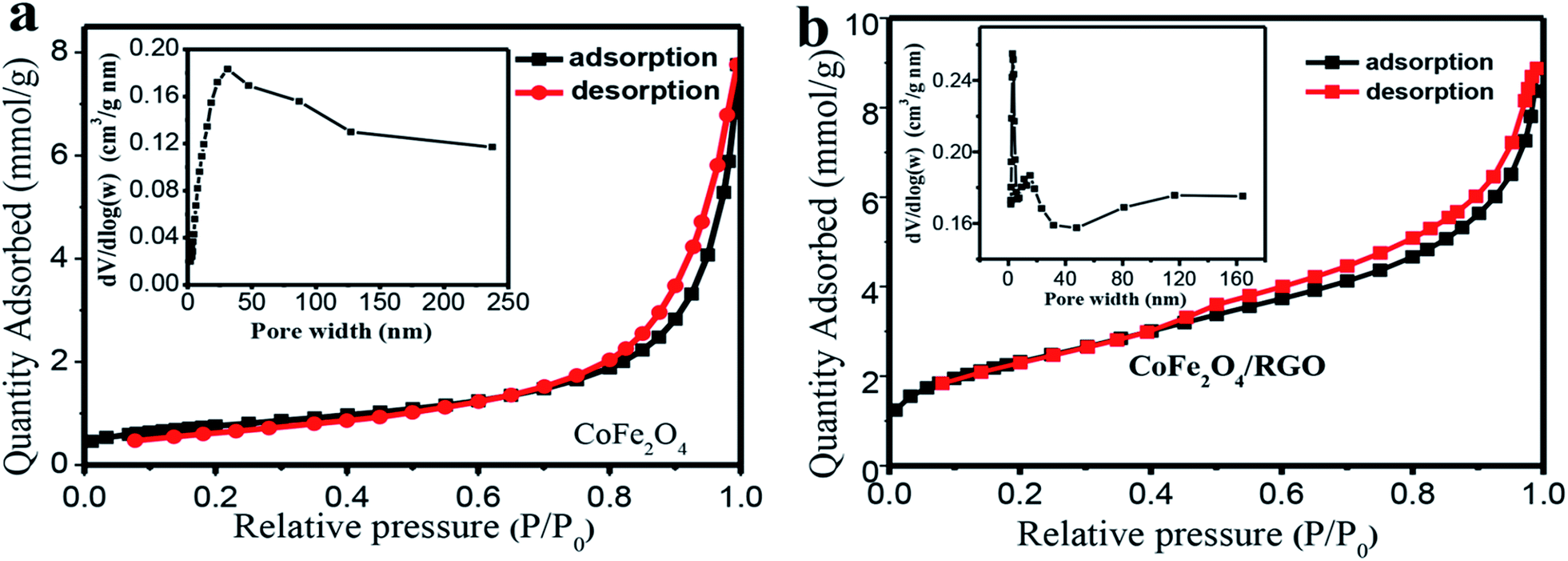 | ||
| Fig. 7 Nitrogen adsorption–desorption isotherms of CoFe2O4 (a) and CoFe2O4/RGO hybrids (b). | ||
The catalytic effect of the as-prepared CoFe2O4/RGO hybrids with different ratio in the thermal decomposition of AP was further investigated by DTA measurements at a heating rate of 20 °C min−1. The DTA and TGA (thermogravimetric analysis) curves of pure AP and the as-prepared CoFe2O4/RGO hybrids are shown in Fig. 8. An endothermic peak centered at about 240 °C in all DTA curves corresponds to the crystallographic transition of AP from the orthorhombic to cubic form. Two obvious exothermic peaks appeared at about 320 °C and 440 °C for pure AP, represent the low-temperature decomposition (LTD) and high-temperature decomposition (HTD), respectively. Pure RGO has no catalytic behaviour on the decomposition of AP, as shown in Fig. 8(a). When adding bare CoFe2O4 and CoFe2O4/RGO hybrids with different ratio to AP, two exothermic processes merge into a sole exothermic process, though there was no change in the position of the phase transition temperature of AP. For thermogravimetric analysis (TGA) of pure AP, two weight loss steps are observed as shown in Fig. 8(d), which are attributed to the partial decomposition and complete decomposition of AP, respectively. While only one weight loss step is presented for AP mixed with CoFe2O4/RGO hybrids, which is consistent with DTA curves. Moreover, the catalytic activity is highly related with the amount of catalyst added, the decomposition temperature of AP gradually decreases with increasing the amount of catalyst. In the presence of bare CoFe2O4 (1%, 3%, 5%), the HTD temperature of AP is reduced by 77.6 °C, 89.5 °C, 95.2 °C, respectively. Whereas, with the 1%, 3%, 5% addition of CoFe2O4/RGO hybrids, the HTD temperature of AP decreases by 92.3 °C, 104.7 °C, 113.6 °C, respectively. All the results manifest that both CoFe2O4 and CoFe2O4/RGO hybrids can promote the decomposition of AP. However, the catalytic activity of CoFe2O4/RGO hybrids is higher than that of bare CoFe2O4, due to the large surface area and enhanced properties of RGO in the hybrids discussed above, which is consistent with the theoretical expectation. Moreover, compared with the result reported in literature,51 the catalytic behaviour of CoFe2O4/RGO is higher than that of Fe2O3/graphene. The HTD temperature of AP mixed with 2% Fe2O3/graphene, is 367.0 °C, whereas, the HTD temperature of AP mixed with 1% CoFe2O4/RGO is 347.6 °C. This may be ascribed to the synergetic effect of Co2+ and Fe3+.
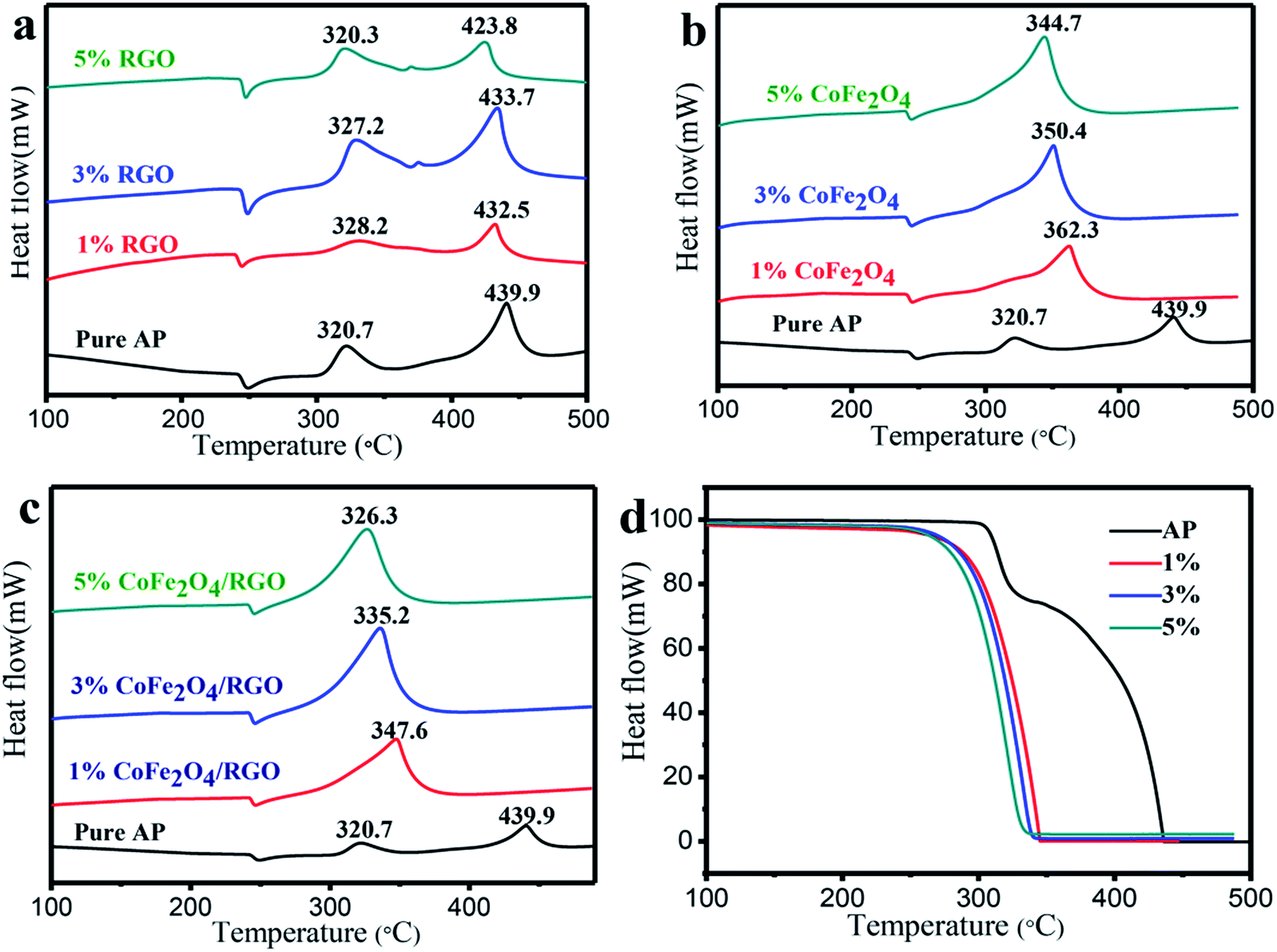 | ||
| Fig. 8 DTA curves of pure AP and AP mixed with RGO (1%, 3%, and 5%) (a), pure AP and AP mixed with CoFe2O4 (1%, 3%, and 5%) (b), pure AP and AP mixed with CoFe2O4/RGO hybrids (1%, 3%, and 5%) (c), TGA curves of pure AP and mixture of AP with CoFe2O4/RGO hybrids (1%, 3%, and 5%) (d). | ||
To further investigate the catalytic behaviour of the as-sythesized CoFe2O4/RGO hybrids, DTA test with different heating rate were performed. Fig. 9 shows the DTA curves of pure AP and the mixtures of AP with CoFe2O4 and CoFe2O4/RGO at different heating rates.
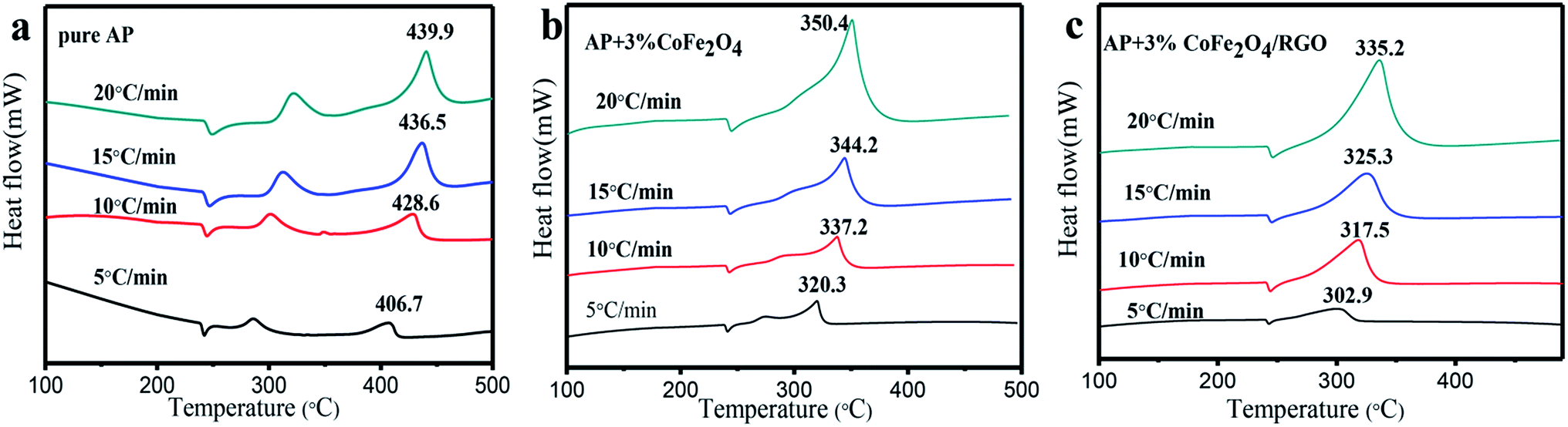 | ||
| Fig. 9 DTA curves of pure AP (a), AP mixed with 3% CoFe2O4 (b) and AP mixed with 3% CoFe2O4/RGO hybrids (c) at different heating rates. | ||
The HTD kinetic parameters of AP with and without catalyst are calculated from the plot of exothermic peak temperature dependence as a function of heating rate.
The relationship between the decomposition temperature and heating rate can be dipicted by Kissinger correlation52 and Arrhenius equation.
 | (1) |
 | (2) |
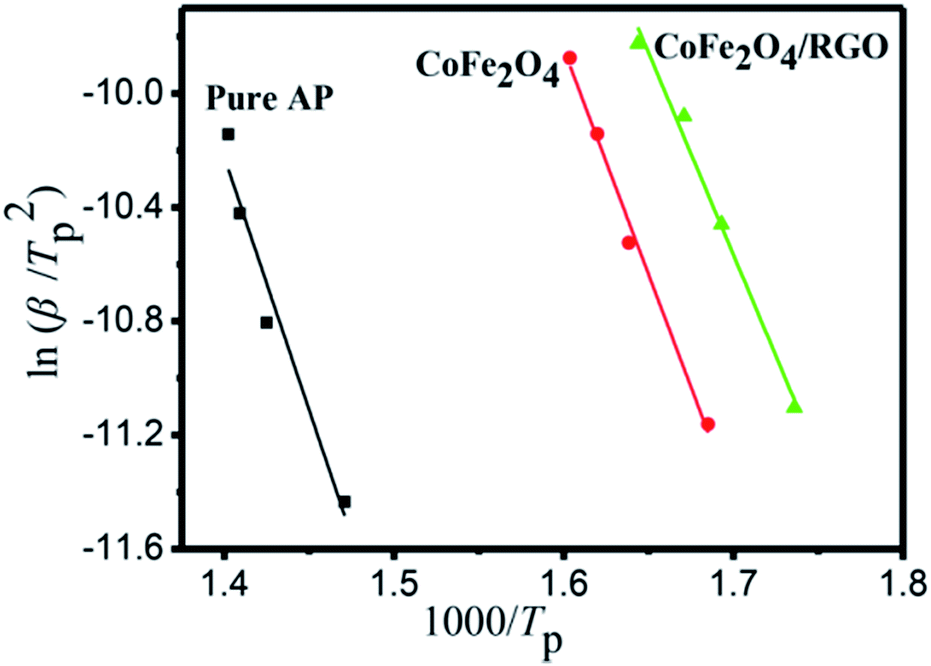 | ||
| Fig. 10 Dependence of ln(β/Tp2) on 1/Tp for AP and mixtures of AP with 3% additives. Scatter points are experimental data and lines denotes the linear fitting results. | ||
| Sample | Ea/kJ mol−1 | A/min−1 | k/s−1 |
|---|---|---|---|
| Pure AP | 147.62 | 4.02 × 1010 | 2.35 × 10−3 |
| AP + CoFe2O4 | 131.47 | 8.15 × 1010 | 8.53 × 10−2 |
| AP + CoFe2O4/RGO | 117.91 | 1.08 × 1010 | 1.27 × 10−1 |
The particle size of catalyst and interaction between active centre and catalyst support can affect its reaction activity.54 To probe the principle of high activity of CoFe2O4/RGO hybrids, H2-TPR measurements were performed to investigate the reducibility of the catalysts. Fig. 11 showed the H2-TPR profiles of CoFe2O4 nanoparticles and CoFe2O4/RGO hybrids. As shown in Fig. 11, two reduction peaks can be observed for CoFe2O4 and CoFe2O4/RGO: the first one can be ascribed to the reduction of CoO to metallic cobalt (Co0) and Fe2O3 to Fe3O4, the second one might be interpreted as a reduction of Fe3O4 to FeO and Fe0.55 Compared with that of CoFe2O4 nanoparticles, the reduction temperature of CoFe2O4/RGO decreases by 26.2 °C. The downward shift in temperature implies that RGO can promote the reduction of CoFe2O4, hence, the CoFe2O4/RGO hybrids show the higher catalytic activity than CoFe2O4 nanocomposites.
 | ||
| Fig. 11 H2-TPR profiles of CoFe2O4 nanoparticles and CoFe2O4/RGO hybrids. | ||
Numerous researches were performed to reveal the mechanism of thermal decomposition of AP.56–59 In this article, the mechanism of thermal decomposition of AP catalysed by CoFe2O4/RGO hybrids, has been explicated by adopting electron transfer mechanism.
The decomposition of AP undergoes three primary steps, the detail can be described as follows:
(1) The first step refers to the endothermic process, in which crystals of AP transform from the low-temperature orthorhombic phase to the high-temperature cubic phase.
(2) The second step represents the exothermic low-temperature decomposition (LTD) process of AP (300–330 °C), in which decomposition and sublimation take place. The scheme is shown as follows:
| NH4ClO4 ↔ NH4+ + ClO4− ↔ NH3 (g) + HClO4 (g) ↔ NH3 (s) + HClO4 (s) |
(3) In the third step, the exothermic high-temperature decomposition (HTD) process of AP (450–480 °C) would take place. In the HTD step, the heterogeneous decomposition of deprotonized HClO4 would generate from the surface of reactant with the main products of N2O, O2, Cl2, H2O and a little NO.
In view of the above experimental findings, it can be inferred that CoFe2O4/RGO hybrids enhance both the LTD and HTD, due to the presence of RGO. A mechanism is proposed, as depicted in Fig. 12.
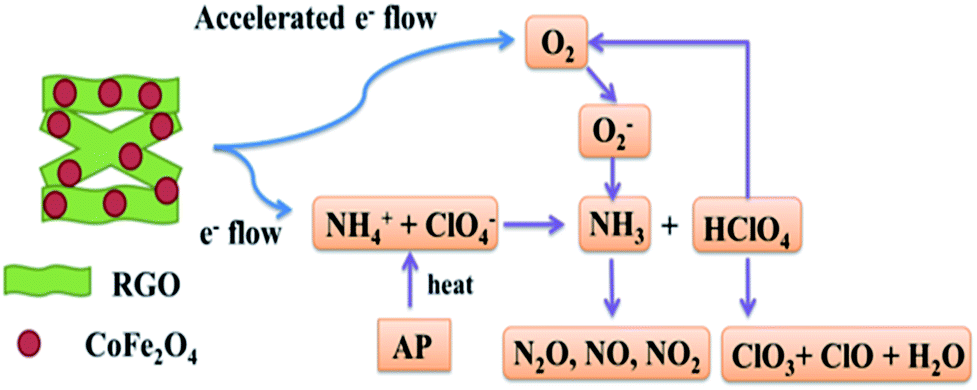 | ||
| Fig. 12 Illustration of catalytic thermal decomposition process of AP by CoFe2O4/RGO hybrids. | ||
Several hypotheses have been proposed to explain the decomposition mechanism of the AP catalysed by metal oxide additives, including the formation of easily melting eutectics between metal oxide additives and AP or intermediate amine compounds, and the release of O2− ion.59 In fact, the decomposition of AP involves two crucial steps: ammonia oxidation and dissociation of ClO4− species into ClO3− and O2. In first step, metal oxides show high and stable catalytic activity and selectivity toward ammonia oxidation, thus promoting the AP decomposition. In second step, the ClO4− species in the presence of the oxide additives would decompose.60
For the low-temperature decomposition (LTD) process, in which the controlling step should be the electron transformation from ClO4− to NH4+, both gas and solid phase occur, involving dissociation and sublimation. While for HTD, it is associated with the transformation of O2 to superoxide (O2−). According to the traditional electron-transfer theory, the presence of partially filled 3d orbit in Fe or Co atom provides help in an electro-transfer process. Positive hole in Fe or Co atom can accept electrons from AP ion and its intermediate products, by which the thermal decomposition of AP is accelerated. Due to the extraordinary conductivity of graphene, electrons can travel a lot faster and longer distance than they do among metal atoms without being scattered.17,27 Thus, it can be concluded that the as-synthesized CoFe2O4/RGO hybrids with excellent electrical and conductive properties could provide more effective electrons and accelerate the two above mentioned thermal decomposition processes of AP. Namely, with the help of more effective and accelerated electrons, NH4+ and ClO4− can be easily transformed to NH3 and ClO4. Superoxide (O2−), which is transformed in a more efficient way from O2 generated by HClO4, could help NH3 decompose into N2O, O2, Cl2, H2O and a little NO, completely.
In the absence of graphene, aggregated bare CoFe2O4 nanoparticles with low specific surface area and fewer active sites for adsorption gas phase molecules (NH3, HClO4) inhibit the decomposition of AP. However, when deposited on graphene sheet, which is a perfect catalyst carrier with a large surface area, the CoFe2O4 nanoparticles can fully unfold on the graphene sheet to form more effective active sites to react with NH3 and HClO4, and thus the catalysis effect is enhanced.
4. Conclusions
In conclusion, CoFe2O4/RGO hybrids have been successfully prepared by a facile one-pot solvothermal method. The average diameter of CoFe2O4/RGO hybrids is estimated to be 120 nm, which was confirmed by SEM and TEM, indicating that RGO could effectively prevent the CoFe2O4 nanoparticles from aggregating. The as-synthesized CoFe2O4/RGO hybrids exhibit highly catalytic activity on the thermal decomposition of AP, and the HTD temperature gradually decreases with the increase of catalyst. With 3% of CoFe2O4/RGO hybrids, the HTD temperature of AP is reduced by 104.7 °C, which is lower than that of bare CoFe2O4 under the same proportion. The results give the clear evidence that RGO can enhance the catalytic activity of CoFe2O4 and promote the thermal decomposition process, owing to its large surface area and enhanced properties of RGO. The facile preparation method depicted in this paper can be a promising and scalable technology for fabrication of other metal oxide/RGO hybrids with high catalytic activity for AP and AP based composite propellants. The improved catalytic activity, which is ascribed to the synergistic effect of nanoparticles and RGO may broaden a new application in modifying the burning behaviour of AP base composite propellant.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Project No. 41101287), the Scientific and Technical Supporting Programs of Jiangsu province (BE2012758), Priority Academic Program Development of Jiangsu Higher Education Institutions and the Qin Lan Project.Notes and references
- P. W. M. Jacobs and H. M. Whitehead, Chem. Rev., 1969, 69, 551–590 CrossRef CAS.
- A. G. Ajaz, J. Hazard. Mater., 1995, 42, 303–306 CrossRef CAS.
- I. P. S. Kapoor, P. Srivastava and G. Singh, Propellants, Explos., Pyrotech., 2009, 34, 351–356 CrossRef CAS.
- G. Singh, I. P. Singh, S. M. Mannan and J. Kaur, J. Hazard. Mater., 2000, 79, 1–18 CrossRef CAS PubMed.
- V. Boldyrev, Thermochim. Acta, 2006, 443, 1–36 CrossRef CAS.
- S. Vyazovkin and C. A. Wight, Chem. Mater., 1999, 11, 3386–3393 CrossRef CAS.
- M. Shusser, F. E. C. Culick and N. S. Cohen, J. Propul. Power, 2002, 18, 1093–1100 CrossRef CAS.
- R. P. Fitzgerald and M. Q. Brewster, Combust. Flame, 2004, 136, 313–326 CrossRef CAS.
- A. Dey, V. Nangare, P. V. More, M. A. S. Khan, P. K. Khanna, A. K. Sikder and S. Chattopadhyay, RSC Adv., 2015, 5, 63777–63785 RSC.
- J. Zhao, Z. S. Liu, Y. L. Qin and W. B. Hu, CrystEngComm, 2014, 16, 2001–2008 RSC.
- D. Yan, H. Y. Zhao, Y. Liu, X. Wu and J. Y. Pei, CrystEngComm, 2015, 17, 9062–9069 RSC.
- Q. Li, Y. He and R. F. Peng, New J. Chem., 2015, 39, 8703–8707 RSC.
- J. Cheng, R. X. Zhang, Z. L. Liu, L. X. Li, F. Q. Zhao and S. Y. Xu, RSC Adv., 2015, 5, 50278–50288 RSC.
- X. M. Chen, G. H. Wu, J. M. Chen, X. Chen, X. Chen, Z. X. Chen and X. R. Wang, J. Am. Chem. Soc., 2011, 133, 3693–3695 CrossRef CAS PubMed.
- Z. S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS PubMed.
- J. L. Yang, J. J. Wang, Y. J. Tang, D. N. Wang, X. F. Li, Y. H. Hu, R. Y. Li, G. X. Liang, T. K. Sham and X. L. Sun, Energy Environ. Sci., 2013, 6, 1521–1528 CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2009, 110, 132–145 CrossRef PubMed.
- S. Niyogi, E. Bekyarova, M. Itkis, J. McWilliams, M. Hamon and R. Haddon, J. Am. Chem. Soc., 2006, 128, 7720–7721 CrossRef CAS PubMed.
- C. N. R. Rao, A. K. Sood, R. Voggu and K. S. Subrahmanyam, J. phys. chem. Lett., 2010, 1, 572–580 CrossRef CAS.
- J. Wu, M. Agrawal, H. A. Becerril, Z. Bao, Z. liu, Y. Chen and P. Peumans, ACS Nano, 2010, 4, 43–48 CrossRef CAS PubMed.
- B. Yue, Y. W. Ma, H. S. Tao, L. S. Yu, G. Q. Jian, X. Z. Wang, X. S. Wang, Y. N. Lu and Z. Hu, J. Mater. Chem., 2008, 18, 1747–1750 RSC.
- N. Mackiewicz, G. Surendran, H. Remita, B. Keita, G. Zhang, L. Nadjo, A. Hagege, E. Doris and C. Mioskowsk, J. Am. Chem. Soc., 2008, 130, 8110–8111 CrossRef CAS PubMed.
- J. Chen, M. Ishigami, C. Jiang, D. R. Hines, M. S. Fuhrer and E. D. Williams, Adv. Mater., 2007, 19, 3623–3627 CrossRef CAS.
- P. Blake, P. D. Brimicombe, R. R. Nair, T. J. Booth, D. Jiang, F. Schedin, L. A. Ponomarenko, S. V. Morozov, H. F. Gleeson and E. W. Hill, Nano Lett., 2008, 8, 1704–1708 CrossRef PubMed.
- A. Kezzima, N. Nasrallaha, A. Abdi and M. Trari, Energy Convers. Manage., 2011, 52, 2800–2806 CrossRef.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- L. P. Zhou, J. Xu, H. Miao, F. Wang and X. Q. Li, Appl. Catal., 2005, 292, 223–236 CrossRef CAS.
- L. Su, W. J. Qin, H. G. Zhang, Z. U. Rahman, C. L. Ren, S. D. Ma and X. G. Chen, Biosens. Bioelectron., 2015, 63, 384–391 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- H. C. Schniepp, J. L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS PubMed.
- A. Reina, X. T. Jia, J. Ho, D. Nezich, H. B. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, Nano Lett., 2009, 9, 30–35 CrossRef CAS PubMed.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- X. B. Fan, W. C. Peng, Y. Li, X. Y. Li, S. L. Wang, G. L. Zhang and F. B. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef CAS.
- I. C. Nlebedim, N. Ranvah, P. I. Williams, Y. Melikhov, J. E. Snyder, A. J. Moses and D. C. Jiles, J. Magn. Magn. Mater., 2010, 322, 1929–1933 CrossRef CAS.
- X. Fan, J. G. Guan, X. F. Cao, W. Wang and F. Z. Mou, Eur. J. Inorg. Chem., 2010, 3, 419–426 CrossRef.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- W. D. Yang, Y. R. Li and Y. C. Lee, Appl. Surf. Sci., 2016, 380, 249–256 CrossRef CAS.
- D. L. Zhao, X. Gao, C. N. Wu, R. Xie and S. J. Feng, Appl. Surf. Sci., 2016, 384, 1–9 CrossRef CAS.
- S. Q. Song, B. Cheng, N. S. Wu, A. Y. Meng, S. W. Cao and J. G. Yu, Appl. Catal., B, 2016, 181, 71–78 CrossRef CAS.
- G. Q. Luo, X. J. Jiang, M. J. Li, Q. Shen, L. M. Zhang and H. G. Yu, ACS Appl. Mater. Interfaces, 2013, 5, 2161–2168 CAS.
- J. A. Rodriguez-Manzo, O. Cretu and H. Banhart, ACS Nano, 2010, 4, 3422–3428 CrossRef CAS PubMed.
- S. J. Jiang and S. Q. Song, Appl. Catal., B, 2013, 140–141, 1–8 CrossRef CAS.
- C. C. Yeh and D. H. Chen, Appl. Catal., B, 2014, 150–151, 298–304 CrossRef CAS.
- S. A. Chambers, R. F. C. Farrow, S. Maat, M. F. Toney, L. Folks, J. G. Catalano, T. P. Trainor and G. E. Brown Jr, J. Magn. Magn. Mater., 2002, 246, 124–139 CrossRef CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- W. Y. Bian, Z. R. Yang, P. Strasser and R. Z. Yang, J. Power Sources, 2014, 250, 196–203 CrossRef CAS.
- M. Zong, Y. Huang and N. Zhang, Appl. Surf. Sci., 2015, 345, 272–278 CrossRef CAS.
- H. Estrade-Szwarckopf, Carbon, 2004, 42, 1713–1721 CrossRef CAS.
- Y. Yuan, W. Jiang, Y. J. Wang, P. Shen, F. S. Li, P. Y. Li, F. Q. Zhao and H. X. Gao, Appl. Surf. Sci., 2014, 303, 354–359 CrossRef CAS.
- H. E. Kissinger, Anal. Chem., 1957, 29, 1702–1706 CrossRef CAS.
- G. Tang, S. Q. Tian, Z. X. Zhou, Y. W. Wen, A. M. Pang, Y. G. Zhang, D. W. Zeng, H. T. Li, B. Shan and C. S. Xie, J. Phys. Chem. C, 2014, 118, 11833–11841 CAS.
- G. Fierro, M. L. Jacono and M. Inversi, Appl. Catal., A, 1996, 137, 327–348 CrossRef CAS.
- C. A. Chagas, E. F. de Souza, M. C. N. A. de Carvalho, R. L. Martins and M. Schmal, Appl. Catal., A, 2016, 519, 139–145 CrossRef CAS.
- P. W. M. Jacobs and A. Russell-Jones, J. Phys. Chem., 1968, 72, 202–207 CrossRef CAS.
- S. S. Joshi, P. R. Patil and V. N. Krishnamurthy, Def. Sci. J., 2008, 586, 721–726 CrossRef.
- L. Song, S. Zhang, B. Chen, J. Ge and X. Jia, Colloids Surf., A, 2010, 360, 1–5 CrossRef CAS.
- V. V. Boldyrev, Thermochim. Acta, 2006, 443, 1–36 CrossRef CAS.
- Y. F. Zhang and C. G. Meng, J. Alloys Compd., 2016, 674, 259–265 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |