
3-Aminopropyltrimethoxysilane and graphene oxide/reduced graphene oxide-induced generation of gold nanoparticles and their nanocomposites: electrocatalytic and kinetic activity
P. C. Pandey*,
Shubhangi Shukla and
Yashashwa Pandey
Department of Chemistry, Indian Institute of Technology (BHU), Varanasi-221005, India. E-mail: pcpandey.apc@iitbhu.ac.in
First published on 9th August 2016
Abstract
The role of graphene oxide (GO)/reduced graphene oxide (rGO) in the 3-aminopropyltrimethoxysilane (3-APTMS)-mediated synthesis of gold nanoparticles (AuNPs) in the presence of formaldehyde is reported. It was found that 3-APTMS-functionalized GO/rGO allowed the rapid conversion of gold cations into AuNPs in the presence of formaldehyde. Moreover, the major findings include (i) the process of nanoparticle synthesis is 3-fold faster (2–3 min) in the presence of GO as compared to that of the same process without GO (15–20 min) under similar conditions, (ii) the AuNPs prepared in the presence of GO show a relatively better stability, which is attributed to the synergistic response of oxygen functionalities on the surface of GO and the micellar behaviour of 3-APTMS, (iii) the average size of the Au–GO nanohybrids is significantly reduced to the order of 3–4 nm as compared to that of the same without GO (5–12 nm), (iv) the Au–GO nanohybrid exhibits distinguished homogeneous catalytic behaviour over AuNPs made without GO and (v) the Au–rGO nanohybrid displays heterogeneous electrocatalytic activity due to the conductive nature of rGO and allows nanocomposite formation with Prussian blue nanoparticles. The high catalytic performance of Au–GO and Au–rGO in the homogenous and heterogenous operations is a function of the structural framework of the substrates and the morphological characteristics of the nanoparticles, as evidenced by FTIR, TEM, EDS and UV-Vis analysis.
Introduction
Graphene has become an encouraging substance for many new scientific transformations over the past decade, credited mainly to its conjugated carbon framework and extraordinary properties1–9 for multiple applications. The oxygenated form of graphene, i.e. graphene oxide (GO), together with various oxygen-containing functional groups, contributes to a large range of solution processible applications,10–14 which furthermore can enable the formation of nanohybrids with targetted nanomaterials. In addition, electron delocalisation within the sheets is distorted due to the disturbance in sp2 conjugation, rendering the graphene oxide with non-conductive behaviour,15 although on deoxygenation, the conductive nature is restored,16–19 thus justifying its use in electronic applications.20–22 Recently, the association of graphene with noble metal nanoparticles to generate nanohybrids with enhanced behaviour for use in a range of potential applications has attracted much attention.23–25 Gold nanoparticles have been examined and are recognised as an essential and competent catalyst, for a variety of applications in different fields, such as biotechnology,26 biomedicine, biodiagnostic27 and biodetection,28,29 and are also compatible for use in electroanalytical applications as they have a high surface area to volume ratio and appreciable conductivity.30,31 Despite the multiple potential applications of gold nanoparticles, the stability, persistence and precise control over the size are still a matter of concern, such as for using them in solution catalysis. To address these issues, we studied in detail the importance of 3-aminopropyltrimethoxysilane (3-APTMS) in the synthesis of gold nanoparticles (AuNPs).32–35 Our findings demonstrate that 3-APTMS-capped noble metal ions can be precisely converted into the respective nanoparticles in the presence of another functional alkoxysilane, 3-glycidoxypropyltrimethoxysilane (3-GPTMS), or other suitable organic reducing agents, such as tetrahydrofuranhydroperoxide/cyclohexanone/formaldehyde, etc.32–35 The nature of the organic reducing agents and the micellar behaviour of 3-APTMS allow the practical usability of the as-generated AuNPs in a variety of aqueous and non-aqueous solvents.32–35 Gold nanocrystals thus obtained are highly dispersible in most polar and non-polar solvents, have sound stability at different pH conditions and display good catalytic activity.34The investigations reported earlier demonstrated that 3-APTMS-capped gold cations are converted into AuNPs in the presence of formaldehyde in an alcoholic medium.34 The size of the nanoparticles prepared was nearly 5–12 nm, depending on the concentrations of 3-APTMS, where with time, aggregation of the nanocrystals was observed. In order to eradicate the size and stability related issues, graphene oxide (GO)–Au composites were synthesised using NaBH4 as a reductant.36 Since NaBH4 is a strong reducing agent, it can convert GO to rGO, which turns out to be the primary cause for the nanoparticles to clump together at certain specific sites over the graphene surface. Therefore, it remains a challenge to define a synthetic programme to obtain complete dispersibility along with profound stability. Various other fabrication procedures have been adopted,37 where despite an homogenous distribution of discrete gold nanoparticles over the graphene surface, the size of the nanoparticles38 was around 20 nm in diameter, which rendered the corresponding Au–GO nanocomposites with low catalytic potential. However, the oxygenated form of graphene functionalized with 3-APTMS may act as a building block39–41 and may further control the synthesis of AuNPs since graphene is known to provide the nucleation sites for AuNPs, owing to the availability of oxygen functionalities over the graphene surface,37,42–44 which may allow the size-controlled growth of nanoparticles. Indeed interesting findings on the role of GO/rGO in the 3-APTMS- and formaldehyde-mediated synthesis of AuNPs has been recorded.
The present work is primarily focused on the 3-APTMS and graphene oxide-induced development of gold nanoparticles under ultrasonication conditions, in the presence of formaldehyde. Here, the synthesis of gold nanoparticles took 2–3 min in relation to the time taken in the preparation without the intervention of graphene; also, the size of the nanoparticles decreased, as confirmed by transmission electron microscopy (TEM). Graphene oxide was silanised with 3-APTMS molecules, resulting in covalent bonding between the large amounts of intersheet junctions45 of these moieties. Nanoparticle-embedded graphene oxide sheets displayed great catalytic potential in homogenous catalytic reactions, as demonstrated by the catalytic reduction of p-nitroaniline, and as peroxidase mimetics. Similarly, the electrocatalytic activity of the AuNPs was improved with the fabrication of the Au–rGO nanohybrid. The electrocatalytic activity of the Au–rGO nanohybrids could be further tailored by doping with Prussian blue nanoparticles, which causes the analyte to reduce or oxidise at lower electrode potentials. The results based on cyclic voltammetry and amperometry justifying the significance of Au–rGO–PBNPs are reported herein.
Experimental
Materials and methods
3-Aminopropyltrimethoxysilane (3-APTMS), tetrachloroauric acid, potassium ferricyanide, ethanol, formaldehyde, cyclohexanone and hydrogen peroxide (H2O2) were obtained from Merck, India. All the experiments were performed using double distilled deionized water.Material characterisation
TEM studies were performed using a Tehnai G2 with an acceleration voltage of 200 kV. UV-Vis data was obtained using an HITACHI U-2900 spectrophotometer. Measurements were done in a 1 cm quartz cuvette in the wavelength range of 200–800 nm. FTIR analysis was carried out using a Bruker ALPHA FTIR spectrometer. Zeta potential measurements were done using a Brookhaven instrument. Electrochemical experiments were performed on an Electrochemical Workstation Model CHI830B, CH Instruments Inc., TX, USA.Synthesis of graphene oxide/reduced graphene oxide
Graphene oxide (GO) was synthesised via a modified Hummer's method.46,47 The graphite was oxygenated by using the strong oxidising agents: sulphuric acid, nitric acid, potassium chlorate and potassium permanganate.1 mg of the prepared GO slurry was suspended in 1 mL of water. To the solution of graphene oxide, 5 μL of hydrazine monohydrate was added, and the mixture was heated at 80 °C for 1 h. The resulting suspension was washed with deionised water to remove any impurities and the residue thus obtained was dried at 50 °C. On reduction, the colour of the graphene oxide changed from brown to dark black, which is the characteristic colour of reduced graphene oxide (rGO).
Synthesis of 3-aminopropyltrimethoxysilane and the GO/rGO-induced conversion of gold cations into Au–GO, Au–rGO nanohybrids
The Au–GO/rGO nanohybrids synthesis proceeds with the addition of 7.5 μL of 3-APTMS to 94 μL of a 10 mg mL−1 suspension of GO/rGO and then sonicated for 10 min to homogenise the contents. 40 μL of this mixture was added to 40 μL of an 0.025 M ethanolic solution of chloroauric acid and the mixture was sonicated for a few minutes. The resulting mixture turned yellowish brown. 100 μL of formaldehyde was then added to this solution and it was sonicated for about 10 min. On sonication, the mixture turned red indicating the formation of gold nanoparticles (AuNPs) as Au–GO or Au–rGO nanohybrids.Synthesis of nanocomposites with Prussian blue nanoparticles
PBNPs were synthesised as described in our previous work.48,49 Originally, 10 μL of 3-APTMS (0.5 M) was added to 50 μL aqueous solutions of potassium ferricyanide (0.05 M) under stirred conditions, followed by the addition of 2 μL of cyclohexanone (9.62 M). The mixture turned deep blue in colour. Further, 5 μL HCl (6.5 M) was added to prevent the conversion of PBNPs to Prussian white. Equal volumes of PBNPs and Au–rGO nanohybrid suspensions were mixed and stirred over a vertex cyclomixer for 5 min, resulting in the formation of an homogeneous Au–rGO–PBNPs sol.Electrochemical measurements
The Au–rGO–PBNPs suspension was adsorbed over graphite (1–2 μm) and dried at 90 °C overnight. The adsorbed material was incorporated in graphite paste electrodes in the following compositions: PB, AuNPs–PBNPs, AuNP–rGO–PBNPs 5% (w/w), graphite powder 65% (w/w), Nujol oil 30% (w/w). The well of the electrode body was filled with active graphite paste. The paste surface was manually smoothed on a clean paper. Electrochemical measurements were performed in a three-electrode configuration with a working volume of 3 mL, equipped with the graphite paste electrode as the working electrode, a Ag/AgCl reference and a platinum plate counter electrode. All the electrochemical experiments were performed in 0.1 M phosphate buffer solution (pH 7.0) containing 0.5 M KCl.Result and discussions
3-APTMS, GO- and rGO-induced synthesis and characterisation of gold nanohybrids
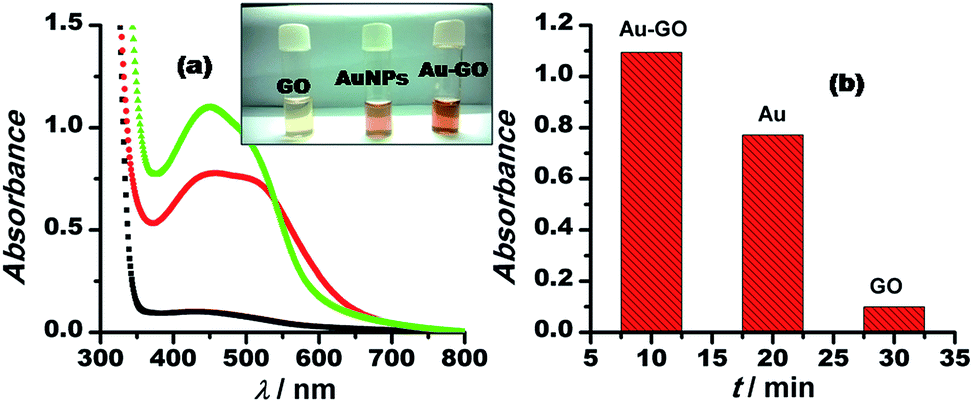
We demonstrated the one-pot synthesis of Au–GO/rGO nanocomposites in two simple steps, whereby at first, graphene undergoes silanisation or chemical modification using an organo-functional silane 3-APTMS; second, the reduction of gold cations is achieved by applying a mild reducing agent, formaldehyde. The chemically exfoliated sheets of graphene oxide consist of oxygen functionalities in bulk, as depicted from the FTIR spectra shown in Fig. 1. The characteristic absorption bands in the FTIR spectra observed are: C–OH stretching at 3360.7 cm−1, corresponding to hydroxyl groups and also due to the N–H stretching of 3-APTMS, which was used for silanization,3 either belonging to alcoholic or carboxylic functional groups; a band at 1646 cm−1, relating to the sp2 framework of graphene, and bands at 1093 cm−1, 1017.73 cm−1 and 905 cm−1, which refer to the C–O epoxy stretching.50,51 The FTIR study confirmed the oxygen-functionalised nature of graphene oxide as described in previous works. UV-Vis spectroscopy was employed to investigate the structural differences between the GO/rGO nanocomposites. The Au–GO nanocomposites showed a surface plasmon band at 515 nm, while Au–rGO displayed a band at nearly 535 nm (Fig. 1). This red shift of about 20 nm in the case of Au–rGO is attributed to the deoxygenation of GO52 along with an increase in the sizes of gold nanoparticles. The differences in absorbance predict the diversity in the number of nanoparticles in both cases.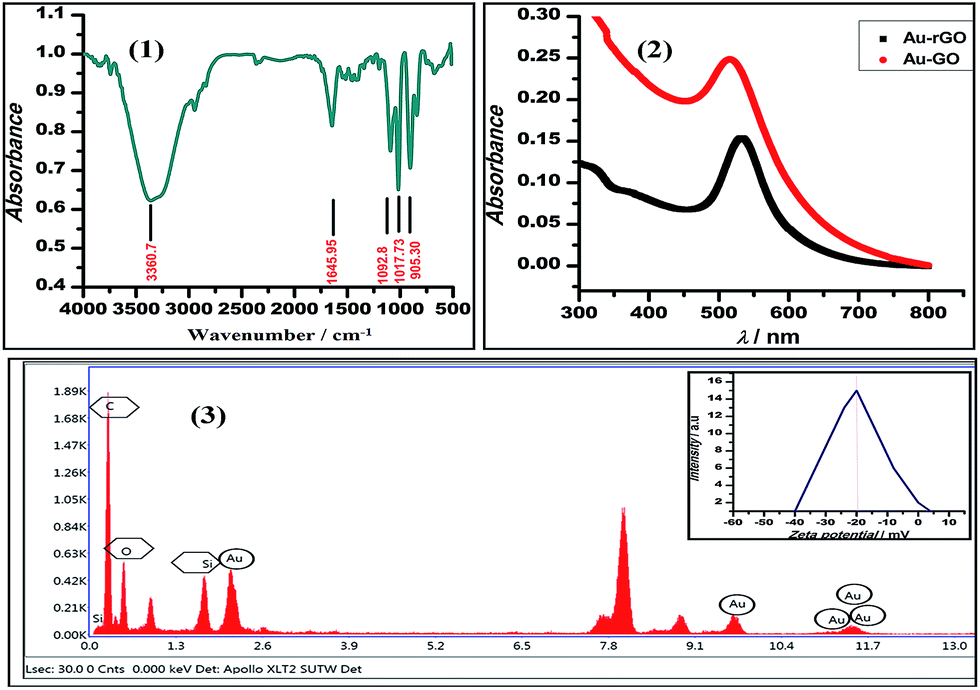 | ||
| Fig. 1 FTIR (1) and EDS (3) spectra of Au–GO nanocomposite; (2) UV-Vis spectra of Au–GO and Au–rGO nanocomposites. Inset to this figure (3) shows the results of the zeta potential of the Au–GO nanocomposites. | ||
3-APTMS was functionally immobilised on the graphene oxide surface, as is clear from the FTIR study. 3-APTMS and graphene oxide both being hydrophilic in character are blended to give an homogenous dispersion. It is known that the residual groups of the organo-functional silane (3-APTMS) and the oxygen functionalities act as the active sites for the nucleation Au–GO nanocomposites. 3-APTMS has been proven to be a versatile capping agent for the Au3+ ions,33 holding a precise control over the nucleation of gold nanoparticles and Au3+ ions to ensure they get individually hooked up at the oxygen moieties on the graphene surface,39 as reported in the literature. Thus, both the groups facilitate the regulation of the sizes of the in situ budding nanoparticles over the graphene surface. As evident from the UV-Vis and the transmission electron microscopy data, the size of the fabricated gold nanoparticles is relatively small, and the zeta potential measurements, as discussed later, ensure the stability of the Au–GO dispersion.
The synthetic procedure for Au–GO nanohybrids is compared with that of the AuNPs synthesised without GO. The gold nanoparticles wrapped with GO are formed within a shorter span of time, comparatively. The kinetics of the sonochemical synthesis of AuNPs and Au–GO nanohybrids were recorded and are shown in Fig. 2. The concentrations of the reactants were kept constant, and the time-dependent formation of AuNPs was studied in the absence (Fig. 2a and b) and the presence of 1 mg mL−1 GO (Fig. 2c and d). The variation in absorbance as a function of time is shown in the form of bar diagrams, and confirmed that GO promotes the rate of AuNPs formation under similar conditions. The reasons for such variation may be further explained as follows.
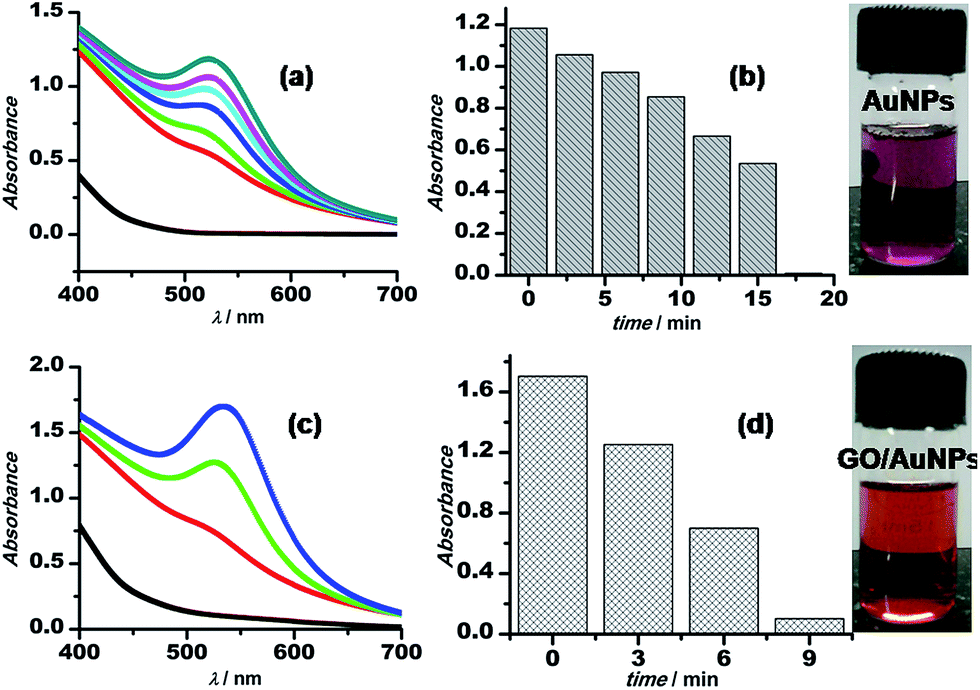 | ||
| Fig. 2 Time-dependent absorption spectra of 3-APTMS and formaldehyde-mediated synthesis of AuNPs in the absence (a) and the presence of (c) GO. (b) and (d) show the bar diagram representing the increase in absorbances as a function of time. | ||
The micellar behaviour of 3-APTMS is responsible for the controlled morphology and sizes of the nanoparticles.33 Graphene (GO/rGO) sheets act as a supporting material for the dispersion of metal nanoparticles, and due to the presence of a number of functional groups containing “O” heteroatom with lone pairs, which configures the non-bonding interactions with the charged metal ions, are also accountable for the controlled nucleation of the nanospheres and for preventing the nanoparticles becoming in close proximity to coalesce.
This inference is supported by the stability studies carried out by zeta potential measurements.52 The zeta potential of Au–GO nanocomposite was found to be near about −25 mV (Fig. 1), which shows that the prepared nanohybrid is significantly stable, and has a relatively better shelf-life than the homogeneous suspension.
Transmission electron microscopic analysis of the nanocomposites reveals the presence of opaque and extremely fine sheets of graphene (Fig. 3a and 4a), and the successful loading of spherical gold nanocrystals in both cases (Au–GO and Au–rGO). Interpretation of the HRTEM micrographs (Fig. 3d and 4) show a consistent, non-overlapping and dense distribution of individual gold nanocrystals over the surfaces of graphene oxide/reduced graphene oxide. This ensures the non-aggregation of the particles at any site and thereby justifies the promotional effect of graphene oxide on the morphology and structural arrangement of AuNPs. The diameters of the gold nanoparticles obtained were 2–5 nm in Au–GO and 7–9 nm in the case of Au–rGO nanocomposites. Furthermore, the nanoparticles do not coalesce into one another to form bigger particles, which proves the significant roles of 3-APTMS and the oxygen functionalities.
 | ||
| Fig. 3 TEM images (a)–(c) and HRTEM image (d) of Au–GO nanohybrids. Inset shows the magnified image of a nanoparticle with visible fringes. | ||
 | ||
| Fig. 4 TEM image of graphene sheet (a) and Au–rGO nanohybrids (b and c); SAED pattern (d) of Au–rGO nanohybrids; inset to (a) shows the diffraction pattern of graphene. | ||
The mean spacing between the distinguished lattice fringes (Fig. 5) detected in the HRTEM images was nearly 0.330 nm. The average diameters of the Au–GO and Au–rGO nanohybrids were 4.5 nm and 7.2 nm, respectively. The prepared nanohybrids were considerably smaller, which is in agreement with respect to the statistical data available from previous studies,38,47 where the reported nanoparticles are above 20 nm in diameter. This demonstrates that the synthetic route followed here generates a material with superior quality over the conventional pathways reported earlier.38,47 The polydisperse character of both the dispersions (Au–GO/Au–rGO) are well indicated by the variation in sizes of the nanocrystals and corresponding lattice spacings. The selected area electron diffraction (SAED) patterns, as shown in Fig. 4d and 6, provides a deeper insight into the crystallographic structure of the materials. The concentric rings obtained in both GO/rGO-based gold nanohybrids confirm the polycrystalline nature of the nanocomposites. The SAED patterns are well indexed with highly resolved lattice planes at corresponding d-values, namely, 0.230 nm (111), 0.201 nm (200), 0.14 nm (220), 0.095 nm (400), and 0.0798 nm (422) within Au–GO, and 0.223 nm (111), 0.13 nm (220) and 0.121 nm (311) in the Au–rGO nanohybrids. The lattice planes obtained here are in fair agreement with those reported in the literature.
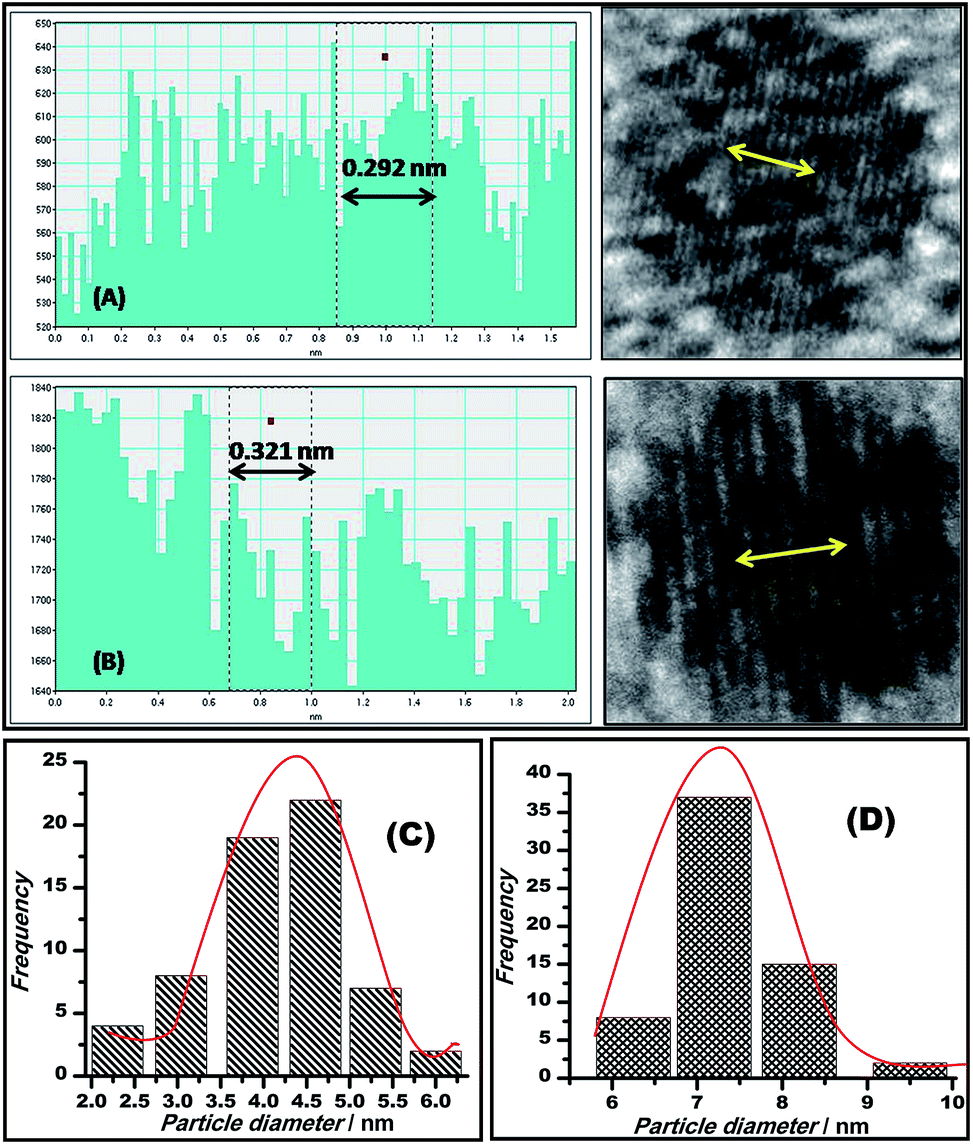 | ||
| Fig. 5 Plots showing the lattice spacings of Au–GO (A) and Au–rGO (B); bar diagrams displaying the average diameter of particles, 4.5 nm of Au–GO (C) and 7.2 nm of Au–rGO (D). | ||
 | ||
| Fig. 6 SAED pattern of Au–GO nanocomposites indexed with the lattice planes. | ||
Further surface analysis of the nanocomposites was performed using energy dispersive X-ray spectroscopy (EDX). The spectra obtained (Fig. 1) give information about a number of constituent materials, such as carbon (skeleton of graphene), oxygen (functional groups), Si (organosilane) and Au (nanoparticle), present in the prepared nanohybrids. Table 1 presents the Au content in the Au–GO nanohybrids.
| Element | Weight% | Atomic% |
|---|---|---|
| C K | 65.86 | 72.47 |
| O K | 32.32 | 26.70 |
| Si L | 1.67 | 0.78 |
| Au L | 0.15 | 0.05 |
Monophasic catalysis
The graphene oxide sheets are chemically functionalized with oxygen groups, such as hydroxyls and epoxides, that stabilise the dispersion of the nanosheets in polar solvents. This characteristic feature is exploited in preparing an inorganically modified colloidal solution of Au–GO nanohybrids, with such an architectural framework to facilitate the perfect homogenisation of the contents. Since the resultant model presented no signs of aggregation in the solution phase, it was significantly used here to catalyse the degradation of p-nitroaniline to p-diaminobenzene and to act as a peroxidase mimetic in the reduction of H2O2. Furthermore, 3-APTMS used here acts as a silanising agent, and owing to the presence of an amino group (electron-donating group) it also acts as a potential capping agent for nanoparticles and has a strong tendency to form an imine linkage with the aldehydes, which are catalytic in nature.53 Thus, the dominant and efficient catalytic behaviour of the Au–GO nanohybrids is attributed to the advanced structural arrangements between the precursors.AuNPs and Au–GO nanohybrids were investigated for their catalytic ability in the degradation of p-nitroaniline with the addition of equal volumes of NaBH4 at regular intervals.54,55 The progress of the reaction was monitored using UV-Vis spectroscopy, whereby a characteristic peak at 380 nm was dissipated and a plasmon band at about 300 nm (p-diaminobenzene) appeared. Time-dependent studies revealed that the Au–GO nanohybrid suspension is more sensitive towards the catalytic degradation of nitro compounds, compared to their analogues (AuNPs). The UV-Vis spectra in Fig. 7 clearly demonstrate the faster reduction of p-nitroaniline to p-diaminobenzene by Au–GO nanohybrid relative to AuNPs.
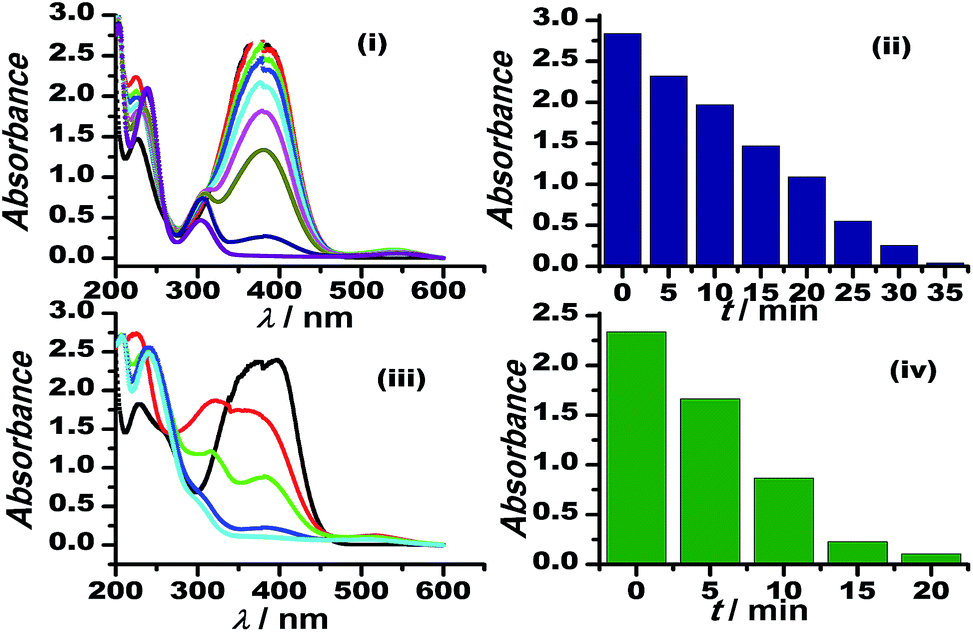 | ||
| Fig. 7 Time-dependent UV-Vis spectral analysis of p-nitroaniline reduction by AuNPs (i), (ii) and Au–GO (iii) and (iv). | ||
Moreover, the catalytic potential of Au–GO nanohybrids was further demonstrated by evaluating its peroxidase mimetic ability, since the nanoparticles have previously shown their potential in peroxidase replacement as artificial biocatalysts due to some deliberate disadvantages of the enzymes.56,57 In the model reaction, H2O2 was reduced in the presence of nanocatalysts marked with the appearance of a characteristic colour of the o-dianisidine dye added, at a wavelength of 430 nm. GO stacked with gold nanoparticles (Au–GO) displayed enhanced catalytic performance (Fig. 8). Au–GO nanohybrids subsequently proved to be more highly catalytic than the individual components (GO and AuNPs), as shown in the corresponding bar diagram (Fig. 8b).
 | ||
| Fig. 8 (a) UV-Vis spectra displaying the peroxidise mimetic activity of GO, AuNPs and Au–GO NPs; bar diagram representing the relative efficiency of the peroxidase mimetic activity under similar conditions for GO, AuNPs and Au–GONPs (b); inset to this figure displays the characteristic colour change shown by GO, AuNPs and Au–GONPs, respectively. | ||
The catalytic performances were indirectly related to the surface structures of the nanoparticles and the diverse orientations of lattice planes along the axes. HRTEM clearly demonstrates the influence of the surface properties on the catalytic aptitude of the nanomaterials.
The inferences drawn from the experimental evidence suggest that the Au–GO nanohybrids have the high catalytic aptitude. Since Au–GO nanohybrids are used in solution catalysis, they cannot be recycled, but only a small amount is required for catalysing the reactions, therefore, their non-recyclable nature does not override their importance.
Electrocatalytic reduction of H2O2
With a chemical reduction (using hydrazine hydrate), GO could be reduced to restore the graphene structure (rGO) and its conductive properties. Thus, the Au–rGO nanohybrids are considered suitable for electrocatalytic and semiconductor applications. Au–rGO nanohybrids were doped with Prussian blue nanoparticles to further improve the properties of the nanohybrids. We subsequently examined the electrochemical behaviour of the Au–rGO–PB nanocomposite-modified electrode. Fig. 9a–c show the cyclic voltammograms of the PBNPs (a), the Au–PB nanocomposite (b) and the Au–rGO–PB nanocomposite (c) modified electrode in 0.1 M phosphate buffer (pH 7.0) in the absence and presence of 5 mM H2O2. There was an increase in cathodic current on the addition of H2O2, as is clear from Fig. 9. However, the Au–rGO–PB nanocomposite-modified system showed the highest reduction current (Fig. 9c) as compared to PB (Fig. 9a), and the PB–AuNPs system (Fig. 9b), indicating the enhanced electrocatalytic activity of the nanocomposite towards H2O2.58,59 In order to study the quantitative determination of H2O2, the amperometric responses for these systems were recorded upon the addition of varying concentrations of H2O2 (5 nM to 2 mM) in 0.1 M phosphate buffer (pH 7.0) at a working potential of 0.0 V vs. Ag/AgCl, as shown in Fig. 10, which clearly visualises the amplified response in the case of the Au–rGO–PB nanocomposite (Fig. 10c) over the PBNPs (Fig. 10a) and Au–PB nanocomposite60 (Fig. 10c), which again confirmed the enhanced electrocatalytic behaviour of the composite material towards the reduction of H2O2.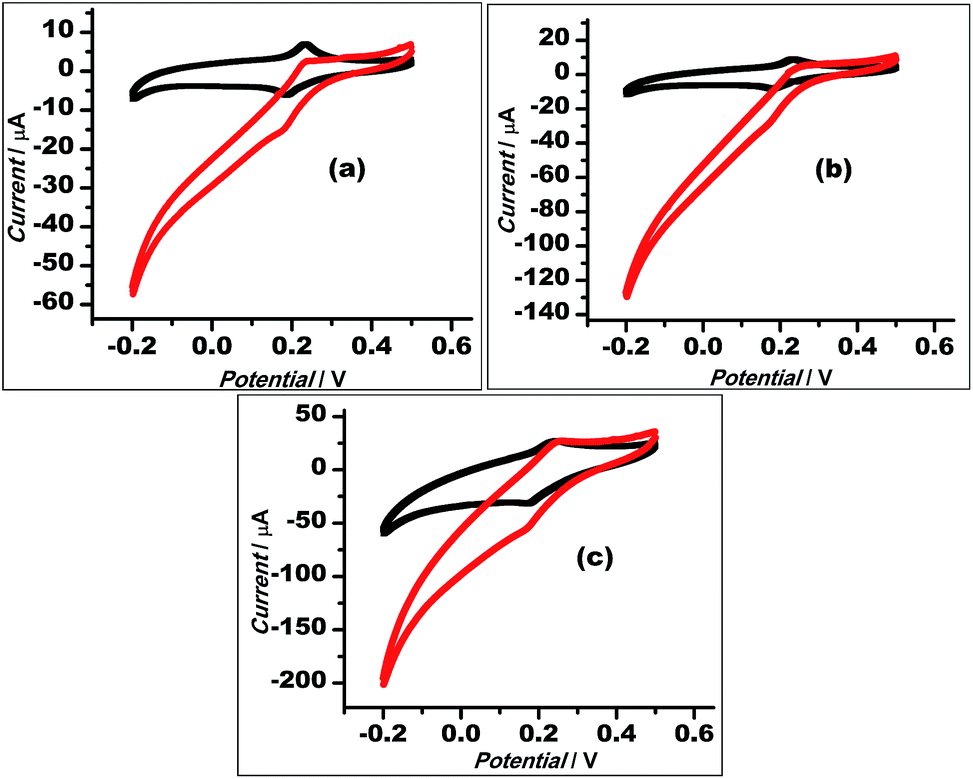 | ||
| Fig. 9 Cyclic voltammograms of PBNPs (a), AuNPs–PBNP nanocomposite (b), rGO–AuNP–PBNPs nanocomposite (c), modified electrodes in the absence (1) and presence (2) of 5 mM H2O2 in 0.1 M phosphate buffer (pH 7.0) containing 0.5 KCl. | ||
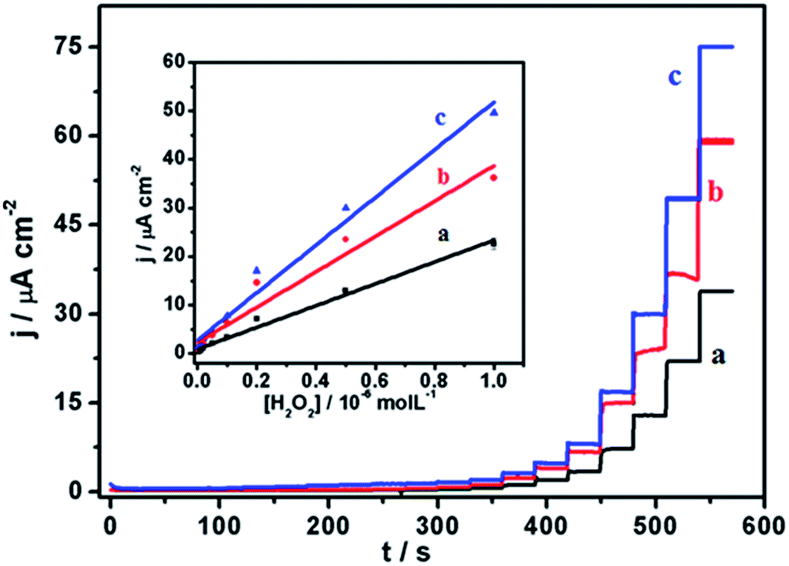 | ||
| Fig. 10 Amperometric response of PBNPs (a), AuNPs–PBNP nanocomposite (b), rGO–AuNP–PBNPs nanocomposite (c), modified electrode upon the addition of H2O2 between 0.005 and 2 mM at 0.0 V vs. Ag/AgCl in 0.1 M phosphate buffer (pH 7.0) containing 0.5 KCl. The inset to this figure shows the respective calibration plots for H2O2 analysis. | ||
The calibration curves for H2O2 analysis at the modified electrodes were constructed using the average currents recorded for each concentration. The inset to Fig. 10 depicts the calibration curves for H2O2 for PBNP (curve a), the Au–PB nanocomposite (curve b) and the Au–rGO–PB nanocomposite (curve c) modified electrodes. The sensitivity of PBNPs, the Au–PB nanocomposite and the Au–rGO–PB nanocomposite-modified system towards H2O2 was also evaluated. The lowest detection limits for H2O2 analysis for the three systems were found to be 2 μm, 1 μM and 50 nM, respectively.
The electrocatalytic stability of the catalyst (Au–rGO–PB nanohybrid) was investigated by recording the electrochemical behaviour of the modified electrode using cyclic voltammetry, as illustrated in the literature.61,62 The current density was found to increase even up to the 120th cycle (Fig. 11), which shows that the catalyst retained its ultimate catalytic aptitude after 120 cycles. Usually the current for the 1st two segments is observed to be higher than that in following segments where it declines gradually, but here, the Au–rGO–PB nanocatalyst showed a progressive current density over successive cycles. Thus, the present nanohybrid can be considered a potential candidate over the other conventional gold nanoparticle-based nanomaterials.
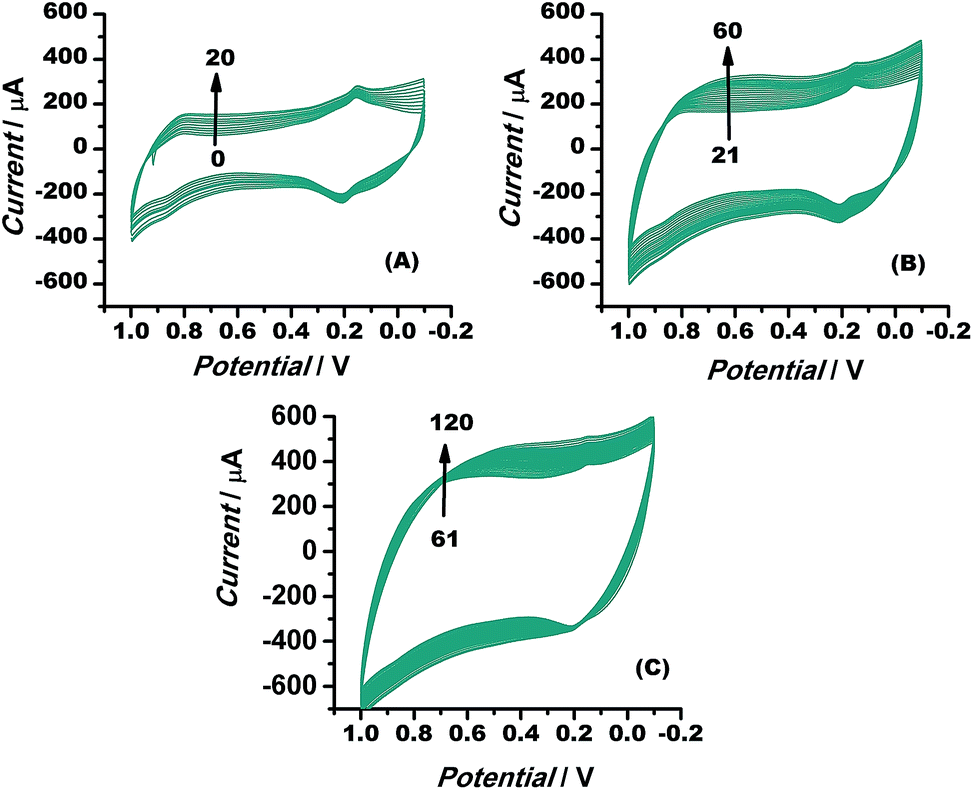 | ||
| Fig. 11 Cyclic voltammograms of Au–rGO–PB nanocomposite from 1 to 120 cycles at a scan rate of 10 mV s−1, illustrating the stability of the nanocomposite. | ||
Conclusion
To summarise, this work mainly comprised the fabrication of graphene-based gold nanohybrids, their detailed characterisation and their implementation in various potential applications. The synthetic procedure involved the active participation of GO/rGO, 3-aminopropyltrimethoxysilane and formaldehyde. 3-APTMS acts as a surface modification and capping agent, while GO/rGO sheets provide nucleation sites for the gold nanoparticles to grow over the surface. Graphene oxide accelerates the synthesis of gold nanoparticles. Graphene oxide and 3-APTMS have a mutual control over the particle size and do not permit agglomeration. Considering the selectivity of reduced graphene oxide towards the electrochemical applications, the corresponding gold analogue, Au–rGO nanohybrid in combination with PBNPs tested well for electrochemical behaviour. Thus, the versatile nature, of the nanocomposites can be effectively explored for most of the potential applications.Acknowledgements
Thankful to Central Instrumental Facility Centre (CIFC), IIT (BHU), for providing characterisation facility. Thanks to UGC for one time grant.References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- S. Saxena and T. A. Tyson, ACS Nano, 2010, 4, 3515–3521 CrossRef CAS PubMed.
- M. Xue, G. Chen, H. Yang, Y. Zhu and D. Wang, J. Am. Chem. Soc., 2012, 134, 6536–6539 CrossRef CAS PubMed.
- C. Tan, X. Huang and H. Zhang, Mater. Today, 2013, 16, 29–36 CrossRef CAS.
- P. C. Pandey, A. Prakash and A. K. D. Pandey, J. Anal. Bioanal. Tech., 2014, S7–S012 Search PubMed.
- W. Hong, H. Bai, Y. Xu, Z. Yao, Z. Gu and G. Shi, J. Phys. Chem. C, 2010, 114, 1822–1826 CAS.
- W. Choi, I. Lahiri, R. Seelaboyina and Y. S. Kang, Crit. Rev. Solid State Mater. Sci., 2010, 35, 52–71 CrossRef CAS.
- A. W. Tsen, L. Brown, M. P. Levendorf, F. Ghahari and P. Y. Huang, Science, 2012, 336, 1143–1146 CrossRef CAS PubMed.
- J. Fang, B. Zhang, Q. Yao, Y. Yang, J. Xie and N. Yan, Coord. Chem. Rev., 2016, 322, 1–29 CrossRef CAS.
- Y. Gao, P. Tang, H. Zhou, W. Zhang, H. Yang, N. Yan, H. Gang, D. Mei, J. Wang and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 3124–3128 CrossRef CAS PubMed.
- J. Li, C. Liu and Y. Liu, J. Mater. Chem., 2012, 22, 8426–8430 RSC.
- H. Li, S. Liu, J. Tian, L. Wang, W. Lu, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, ChemCatChem, 2012, 4, 1079–1083 CrossRef CAS.
- H. D. Jang, S. K. Kim, H. Chang, J. W. Choi and J. Huang, Mater. Lett., 2013, 106, 277–280 CrossRef CAS.
- T. Qian, C. Yu, S. Wu and J. Shen, Colloids Surf., B, 2013, 112, 310–314 CrossRef CAS PubMed.
- K. T. Nguyen and Y. Zhao, Nanoscale, 2014, 6, 6245–6266 RSC.
- B. Z. Jang and A. J. Zhamu, J. Mater. Sci., 2008, 43, 5092–5101 CrossRef CAS.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- M. Hirata, T. Gotou, S. Horiuchi, M. Fujiwara and M. Ohba, Carbon, 2004, 42, 2929–2937 CAS.
- M. J. McAllister, J. L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Caro, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- R. Muszynski, B. Seger and P. V. Kamat, J. Phys. Chem. C, 2008, 112, 5263–5266 CAS.
- Y. Fang, S. Guo, C. Zhu, Y. Zhai and E. Wang, Langmuir, 2010, 26, 11277–11282 CrossRef CAS PubMed.
- W. J. Hong, H. Bai, Y. X. Xu, Z. Y. Yao, Z. Z. Gu and G. Q. Shi, J. Phys. Chem. C, 2010, 114, 1822–1826 CAS.
- Y. C. Si and E. T. Samulski, Chem. Mater., 2008, 20, 6792–6797 CrossRef CAS.
- R. Muszynski, B. Seger and P. V. Kamat, J. Phys. Chem. C, 2008, 112, 5263–5266 CAS.
- C. Xu, X. Wang and J. W. Zhu, J. Phys. Chem. C, 2008, 112, 19841–19845 CAS.
- A. P. Alivisatos, K. P. Johnsson, X. G. Peng, T. E. Wilson, C. J. Loweth, M. P. Bruchez and P. G. Schultz, Nature, 1996, 382, 609–611 CrossRef CAS PubMed.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS PubMed.
- E. Katz and I. Willner, Angew. Chem., 2004, 116, 6166–6235 CrossRef.
- S. G. Penn, L. He and M. J. Natan, Curr. Opin. Chem. Biol., 2003, 7, 609–615 CrossRef CAS PubMed.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- D. L. Feldheim and C. D. Keating, Chem. Soc. Rev., 1998, 27, 1–12 RSC.
- P. C. Pandey and D. S. Chauhan, Analyst, 2012, 137, 376–385 RSC.
- P. C. Pandey and G. Pandey, J. Mater. Chem. B, 2014, 2, 3383–3390 RSC.
- P. C. Pandey, A. K. Pandey and G. Pandey, J. Nanosci. Nanotechnol., 2014, 14, 6606–6613 CrossRef CAS PubMed.
- P. C. Pandey, D. Panday and G. Pandey, RSC Adv., 2014, 4, 60563–60572 RSC.
- H. Zhang, D. Hines and D. L. Akins, Dalton Trans., 2014, 43, 2670–2675 RSC.
- G. Goncalves, A. A. P. Paula Marques, M. Carlos Granadeiro, I. S. Helena Nogueira, M. K. Singh and J. Gracio, Chem. Mater., 2009, 21, 4796–4802 CrossRef CAS.
- Y. Cui, D. Zhou, Z. Sui and B. Han, Chin. J. Chem., 2015, 33, 119–124 CrossRef CAS.
- A. Halder, M. Zhang and Q. Chi, Advanced Catalytic Materials, 2015, DOI:10.5772/61808.
- T. Y. Perry, S. Shah, M. Chhowalla and K. B. Lee, Chem. Rev., 2015, 115, 2483–2531 CrossRef PubMed.
- B. Dong, G. Liu, J. Zhou, A. Wang, J. Wang, R. Jina and H. Lva, RSC Adv., 2015, 5, 97798–97806 RSC.
- L. Fan, K. Wang, J. Wei, M. Zhong, D. Wub and H. Zhu, J. Mater. Chem. A, 2014, 2, 13123–13128 CAS.
- C. Chung, Y. K. Kim, D. Shin, S. R. Ryoo, B. H. Hong and D. H. Min, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed.
- X. M. Wang and W. H. Zhang, New Res. Carbon Mater., 2013, 28, 321–327 CAS.
- Z. Xu, J. Zhang, M. Shan, Y. Li, B. Li, J. Niu, B. Zhou and X. Qian, J. Membr. Sci., 2014, 458, 1–13 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- J. Sun, N. Yang, Z. Sun, M. Zeng, L. Fu, C. Hu and S. Hu, ACS Appl. Mater. Interfaces, 2015, 7, 21356–21363 CAS.
- P. C. Pandey and A. K. Pandey, Electrochim. Acta, 2013, 87, 1–8 CrossRef CAS.
- P. C. Pandey, A. Prakash and A. K. Pandey, Electrochim. Acta, 2014, 127, 132–138 CrossRef CAS.
- D. Konios, M. M. Stylianakis, E. Stratakis and E. Kymakis, J. Colloid Interface Sci., 2014, 430, 108–112 CrossRef CAS PubMed.
- J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- J. R. Lombardi and R. L. Birke, J. Phys. Chem. C, 2008, 112, 5605–5617 CAS.
- J. T. G. Overbeek, J. Colloid Interface Sci., 1977, 58, 408–422 CrossRef CAS.
- P. C. Pandey and G. Pandey, Catal. Sci. Technol., 2016, 6, 3911–3917 CAS.
- S. Gu, S. Wunder, Y. Lu, M. Ballauf, R. Fenger, K. Rademann, B. Jaquet and A. Zaccone, J. Phys. Chem. C, 2014, 118, 18618–18625 CAS.
- Z. D. Pozun, S. E. Rodenbusch, K. Emily, T. Kelly, T. Wenjie, K. J. Stevenson and G. Henkelman, J. Phys. Chem. C, 2013, 117, 7598–7604 CAS.
- H. Wei and E. Wang, Anal. Chem., 2008, 80, 2250–2254 CrossRef CAS PubMed.
- W. Chen, S. Cai, Q. Q. Ren, W. Wen and Y. D. Zhao, Analyst, 2012, 137, 49–58 RSC.
- S. Chen, R. Yuan, Y. Chai and F. Hu, Microchim. Acta, 2013, 180, 15–32 CrossRef CAS.
- H. S. Marinhoa, C. Real, L. Cyrnea, H. Soaresa and F. Antunesa, Redox Biol., 2014, 2, 535–562 CrossRef PubMed.
- M. Luo, Y. Dou, H. Kang, Y. Ma, X. Ding, B. Liang, B. Ma and L. Li, J. Solid State Electrochem., 2015, 19, 1621–1631 CrossRef CAS.
- W. Ye, Y. Chen, Y. Zhou, J. Fu, D. Gao, F. Zhou, C. Wang and D. Xue, Electrochim. Acta, 2014, 142, 18–24 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |