
Deletion of the side chain assembly reveals diverse post-PKS modifications in the biosynthesis of ansatrienins†
Xiaomei Lia,
Jing Zhub,
Guoyin Shib,
Mingwei Suna,
Zhixing Guoa,
Haoxin Wangb,
Chunhua Lu*a and
Yuemao Shen*ab
aKey Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Shandong University, No. 44 West Wenhua Road, Jinan, Shandong 250012, P. R. China. E-mail: ahua0966@sdu.edu.cn; Tel: +86-531-88382108
bState Key Laboratory of Microbial Technology, School of Life Sciences, Shandong University, Jinan, Shandong 250100, P. R. China. E-mail: yshen@sdu.edu.cn; Tel: +86-531-88382108
First published on 31st August 2016
Abstract
Seven new ansatrienin-type ansamycins, namely, ansatrienols B–H (1–7), along with four known ones (8–11), were isolated from the strain Streptomyces sp. XZQH13OEΔastC constructed by deleting the astC gene responsible for the assembly of the N-cyclohexanoyl D-alanyl side chain. Their structures were determined by analysis of the 1D and 2D NMR spectroscopic data and high-resolution ESIMS. This study revealed the diverse post-PKS modifications during the biosynthesis of ansatrienins.
Introduction
Ansamycins are a family of bioactive macrolactams, including antituberculosis rifamycin,1 antitumor maytansine,2,3 and Hsp90 inhibitor geldanamycin.4 These macrolactams are constructed by the multidomain modular type I PKSs using 3-amino-5-hydroxybenzoic acid (AHBA) as the starter unit,5 followed by various post-PKS modifications. By analogy to the biosynthesis of other type I polyketides such as macrolactones, we speculate that there could be more new ansamycins awaiting discovery. The potential structural diversities and promising bioactivity of ansamycins encouraged us to search for more new ansamycins using a genetic approach. In the course of our PCR screening for ansamycins on the basis of the AHBA synthase gene, a library of plant-associated and marine-derived actinomycetes was obtained,6 which was followed by the isolation of new hygrocins,7,8 divergolides,9,10 ansamycins juanlimycins,11 and neoansamycins.12 Moreover, among these AHBA synthase gene-positive strains, Streptomyces sp. XZQH13 was activated to produce the ansatrienin-type ansamycins.13 Among ansamycins, ansatrienins are a unique group having an N-cyclohexanoyl D-alanyl side chain attached to the hydroxyl group of the 21-membered macrocyclic lactam ring.14,15 In our continuing studies on the biosynthesis of ansatrienins, we succeeded in confirming that the astC gene is required for the attachment of the N-cyclohexanoyl D-alanyl side chain onto the ansatrienin ansa-ring and identifying the metabolic intermediate thiazinotrienomycinol E (11) accumulated by the XZQH13OEΔastC strain.14 Furthermore, to exploit the diversity of post-PKS modifications in the biosynthesis of ansatrienins, we carried out systematical isolation of the fermentation products of the mutant strain, XZQH13OEΔastC. Eleven ansatrienins without the N-cyclohexanoyl D-alanyl side chain but with diverse post-PKS modifications were obtained, including 7 new ones, namely, ansatrienols B–H (1–7), along with four known ones, mycotrienol II (8),16 trienomycinol (9),17 demethyltrienomycinol (10),18 and thiazinotrienomycinol E (11).14 Herein, the isolation, structure elucidation and bioactivity of seven new compounds are reported. The structures were determined by analysis of the high-resolution ESIMS, 1D and 2D NMR spectroscopic data.Results and discussion
The strain Streptomyces sp. XZQH13OEΔastC was cultured on YMG agar plates for 4 d at 28 °C. The 30 L fermented agar cakes were diced and extracted three times overnight with EtOAc–MeOH (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20, v/v) at room temperature. After removing the organic solvents, the crude extract was partitioned between water and EtOAc (1:1, v/v) and then the EtOAc extract was partitioned between 95% aqueous MeOH and petroleum ether (PE). The MeOH layer was concentrated under vacuum to afford the defatted MeOH extract (4.5 g), which was subjected to column chromatography over Sephadex LH-20, MPLC over reversed-phase C18 silica gel and finally HPLC to yield compounds 1–11.
:20, v/v) at room temperature. After removing the organic solvents, the crude extract was partitioned between water and EtOAc (1:1, v/v) and then the EtOAc extract was partitioned between 95% aqueous MeOH and petroleum ether (PE). The MeOH layer was concentrated under vacuum to afford the defatted MeOH extract (4.5 g), which was subjected to column chromatography over Sephadex LH-20, MPLC over reversed-phase C18 silica gel and finally HPLC to yield compounds 1–11.
Compound 1 was isolated as a white powder. Its molecular formula was determined to be C26H33NO6 on the basis of high-resolution ESIMS at m/z 456.2383 [M + H]+ (calcd for C26H34NO6+, 456.2381) and 478.2199 [M + Na]+ (calcd for C26H33NO6Na+, 478.2200) (ESI Fig. S48†). Analysis of the 1H–1H COSY correlations demonstrated the presence of a three-carbon (C-15 to C-17) and a thirteen-carbon (C-2 to C-13, and C-24) fragment (Fig. 2 and ESI Table S1†). The 1H and 13C NMR indicated four oxy-substitutions and one nitrogen-substitution (δC 66.2, C-4) in the thirteen-carbon fragment (Table 1). The linkage of the two fragments via C-14 was revealed by the HMBC correlations from H-25 (δH 1.77) to C-13 (δC 70.7), C-14 (δC 138.0) and C-15 (δC 130.6). The HMBC correlations of H-21 (δH 7.22) to C-19 (δC 140.0), C-20 (δC 125.5), C-22 (δC 151.3) and C-23 (δC 115.5), and H-23 (δH 6.45) to C-19, C-21 (δC 107.2) and C-22 indicated the presence of a tetra-substituted benzene ring (Fig. 2 and ESI Table S1†). Oxy-substitution was revealed by the assignment of δC 151.3 at C-22. The C-17 methylene substitution at C-18 was determined on the basis of the HMBC correlations from H-17 (δH 2.87, 2.22) to C-18 (δC 132.6), C-19 and C-23. The ether bond between C-5 and C-19 was determined by the chemical shift δC 140.0 assigned to C-19 and the HMBC correlations from H-5 (δH 4.63) to C-19 (Fig. 2 and ESI Table S1†). The nitrogen substitution at C-20, C-4 and C-1 was indicated by the HMBC correlations from H-4 (δH 3.63) to C-1 (δC 172.6) and C-20. The linkage between C-1 and C-2 was supported by the HMBC correlations from H-2 (δH 2.93, 2.62) and H-3 (δH 4.03) to C-1, further indicating the presence of a γ-lactam (Fig. 2 and ESI Table S1†). Hence, the chromophore moiety was determined to be a derivative of benzoxazole fused with a pyrrolidinone ring (Fig. 1), showing a similar backbone to that of benzoxazomycin,19 except for the absence of the N-cyclohexanoyl D-alanyl side chain. Thus, compound 1 was elucidated to be decyclohexanoyl-D-alanyl benzoxazomycin, namely, ansatrienol B (Fig. 1).
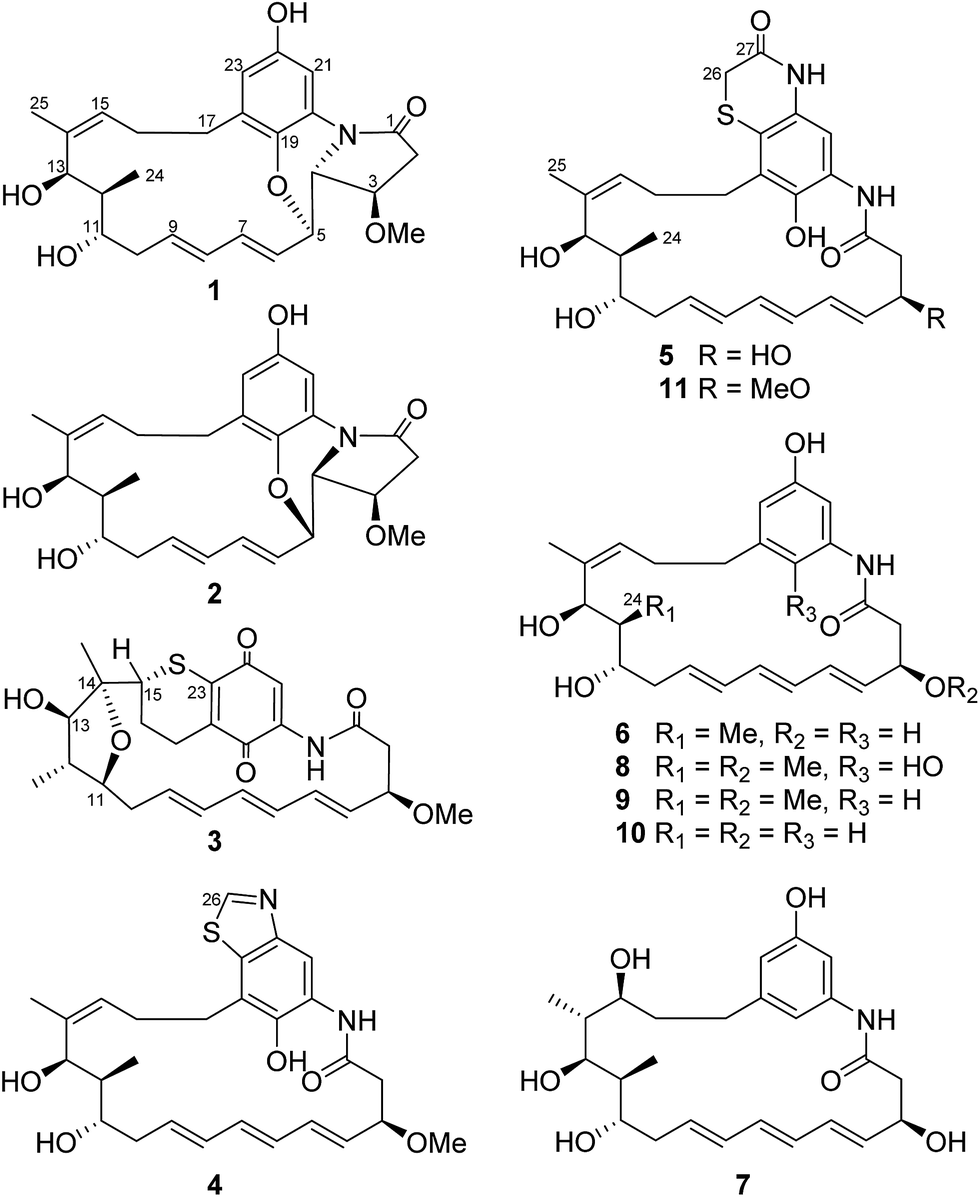 | ||
| Fig. 1 Structures of compounds 1–11. | ||
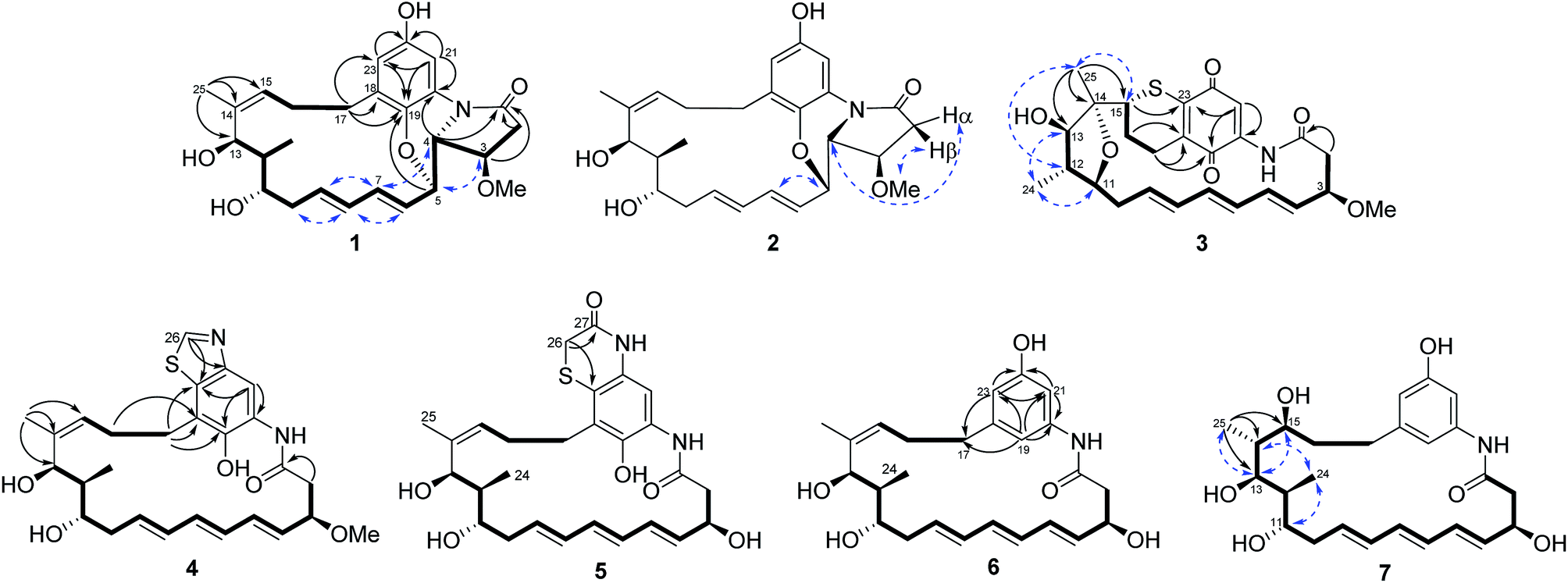 | ||
| Fig. 2 Selected 1H–1H COSY (━), HMBC (→) and NOE (← →) correlations for compounds 1–7. | ||
| Pos. | 1 (CD3OD) | 2 [(CD3)2CO] | ||
|---|---|---|---|---|
| δH (mult., J in Hz) | δC mult. | δH (mult., J in Hz) | δC mult. | |
| 1 | 172.6s | 171.7s | ||
| 2α | 2.93 (dd, 17.5, 7.9) | 38.8t | 2.79 (dd, 17.3, 5.3) | 38.4t |
| 2β | 2.62 (dd, 17.5, 4.9) | 2.59 (d, 17.5) | ||
| 3 | 4.03 (m) | 77.0d | 4.20 (t, 4.9) | 75.0d |
| 4 | 3.63 (dd, 6.1, 4.2) | 66.2d | 3.97 (dd, 7.5, 4.9) | 63.4d |
| 5 | 4.63 (dd, 8.5, 6.3) | 78.5d | 4.94 (t, 7.0) | 75.1d |
| 6 | 6.07 (dd, 15.8, 6.2) | 126.1d | 5.95 (dd, 15.7, 6.3) | 126.9d |
| 7 | 6.69 (dd, 15.8, 10.5) | 140.3d | 6.65 (dd, 15.6, 10.3) | 138.5d |
| 8 | 6.29 (dd, 15.1, 10.7) | 132.1d | 6.23 (dd, 15.1, 10.3) | 131.6d |
| 9 | 5.64 (ddd, 15.4, 10.7, 4.9) | 135.1d | 5.81 (dt, 15.2, 7.6) | 134.8d |
| 10α | 2.52 (m) | 38.2t | 2.45 (m) | 37.8t |
| 10β | 2.39 (m) | 2.38 (m) | ||
| 11 | 3.60 (m) | 72.2d | 3.68 (m) | 72.8d |
| 12 | 2.00 (m) | 44.3d | 1.93 (m) | 42.9d |
| 13 | 4.38 (d, 9.4) | 70.7d | 4.44 (d, 3.1) | 70.2d |
| 14 | 138.0s | 138.9s | ||
| 15 | 5.44 (dd, 10.2, 4.1) | 130.6d | 5.23 (t, 5.6) | 129.1d |
| 16α | 3.02 (m) | 29.6t | 2.06 (m) | 28.5t |
| 16β | 1.81 (m) | 2.47 (m) | ||
| 17α | 2.87 (m) | 34.0t | 2.82 (dt, 12.6, 5.5) | 33.0t |
| 17β | 2.22 (m) | 2.28 (dt, 12.5, 4.6) | ||
| 18 | 132.6s | 131.9s | ||
| 19 | 140.0s | 138.4s | ||
| 20 | 125.5s | 127.5s | ||
| 21 | 7.22 (d, 2.8) | 107.2d | 7.56 (d, 2.4) | 105.3d |
| 22 | 151.3s | 151.6s | ||
| 23 | 6.45 (d, 2.8) | 115.5d | 6.42 (d, 2.7) | 113.5d |
| 24 | 1.10 (d, 6.9) | 10.6q | 1.00 (d, 6.9) | 11.3q |
| 25 | 1.77 (s) | 19.3q | 1.76 (s) | 20.1q |
| MeO-3 | 3.35 (s) | 57.6q | 3.35 (s) | 57.0q |
Compound 2 was isolated as a white powder. Its molecular formula was determined to be C26H33NO6 on the basis of high-resolution ESIMS at m/z 456.2383 [M + H]+ (calcd for C26H34NO6+, 456.2381) (ESI Fig. S49†). The HRESIMS and NMR comparison with that of 1 indicated the presence of the same planar structure as 1 but showing different chemical shifts (Table 1) and NOE correlations (Fig. 2). The H-3/H-5 and H-4/H-7 NOE correlations (Fig. S9†) were observed in 1, whereas different NOE correlations (Fig. S15 and S16†) were observed in 2, i.e. the H-2β/MeO-3, H-2α/H-4, and H-5/H-7 NOE correlations, suggesting that compounds 1 and 2 have the opposite stereochemistry at C-4 and C-5. This difference between 1 and 2 may originate from the formation of an oxazole ring via a putative hetero-Diels–Alder reaction in ansatrienol A (Fig. 3). Therefore, compound 2 was designated to be ansatrienol C (Fig. 1).
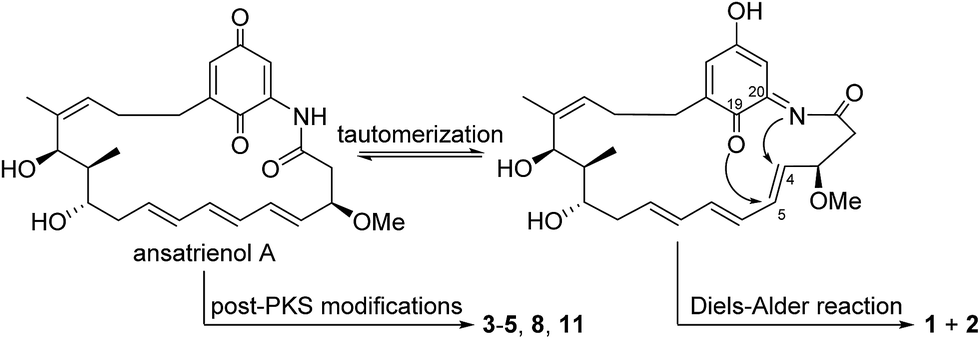 | ||
| Fig. 3 Putative post-PKS modifications in the biosynthesis of compounds 1–5, 8, and 11. | ||
Compound 3 was isolated as a red powder. Its molecular formula was determined to be C26H31NO6S on the basis of high-resolution ESIMS at m/z 486.1949 [M + H]+ (calcd for C26H32NO6S+, 486.1945) (ESI Fig. S50†). The NMR comparison with that of mycotrienol I revealed that the structure of 3 was different from mycotrienol I in the C-14/C-15 double bond and phenolic chromophore moiety (Table 2).20 The HMBC correlations from H-25 (δH 1.14) to C-13 (δC 84.0), C-14 (δC 85.2) and C-15 (δC 48.9) suggested the absence of a C14/C15 double bond (Fig. 2 and ESI Table S3†). Furthermore, the assignment of δC 84.2 for C-11, δC 84.0 for C-13 and δC 85.2 for C-14, along with dehydration of one molecule of H2O required by the molecular formula suggested that C-11 and C-14 were dehydrated and linked via an ether bond (Table 2 and Fig. 2). Moreover, C-15 was connected to C-23 via a sulfur atom according to the HMBC correlations from H-15 (δH 3.38) to C-23 (δC 147.9) (Fig. 2 and ESI Table S3†). The relative configurations of C-14 and C-15 were determined both to be the R*-form with the support of the NOE correlations of H-11/H-25/H-13 and H-12/H-24/H-15 (Fig. 2), whereas the stereochemistry of C-3, C-11, C-12 and C-13 was assumed to be the same as that of other ansatrienins on the basis of biosynthetic logic.21 Thus, compound 3 was elucidated as ansatrienol D (Fig. 1).
| Pos. | 3 | 4 | 5 | |||
|---|---|---|---|---|---|---|
| δH (mult., J in Hz) | δC mult. | δH (mult., J in Hz) | δC mult. | δH (mult., J in Hz) | δC mult. | |
| 1 | 172.5s | 172.0s | 172.0s | |||
| 2α | 2.78 (m) | 45.5t | 2.88 (dd, 12.7, 4.4) | 44.1t | 2.76 (m) | 45.9t |
| 2β | 2.73 (dd, 11.4, 5.6) | 2.71 (dd, 12.5, 10.2) | 2.66 (dd, 12.7, 9.6) | |||
| 3 | 4.00 (dt, 9.7, 5.6) | 82.1d | 4.15 (dt, 9.5, 4.4) | 81.3d | 4.56 (m) | 71.6d |
| 4 | 5.57 (dd, 15.6, 8.8) | 130.6d | 5.58 (dd, 15.3, 8.0) | 131.1d | 5.64 (dd, 15.3, 7.3) | 133.0d |
| 5 | 6.04 (dd, 15.6, 9.8) | 135.5d | 6.27 (m) | 136.1d | 6.21 (dd, 15.2, 10.0) | 133.6d |
| 6 | 5.86 (dd, 14.8, 9.8) | 128.3d | 6.11 (m) | 130.2d | 6.10 (m) | 130.4d |
| 7 | 5.98 (dd, 14.8, 10.3) | 137.0d | 6.10 (m) | 135.8d | 6.13 (dd, 14.7, 9.8) | 135.5d |
| 8 | 5.90 (dd, 14.6, 10.5) | 131.7d | 6.07 (m) | 133.7d | 6.06 (m) | 133.8d |
| 9 | 5.52 (ddd, 14.7, 9.1, 6.7) | 136.0d | 5.74 (ddd, 14.7, 8.9, 6.1) | 132.1d | 5.73 (m) | 131.8d |
| 10α | 2.27 (dd, 13.3, 6.6) | 37.6t | 2.41 (m) | 37.5t | 2.45 (m) | 37.6t |
| 10β | 1.85 (dt, 13.3, 9.5) | 2.31 (m) | 2.32 (m) | |||
| 11 | 3.28 (t, 10.2) | 84.2d | 3.65 (dt, 6.7, 3.2) | 72.8d | 3.65 (m) | 72.9d |
| 12 | 1.67 (m) | 43.5d | 1.81 (m) | 41.8d | 1.81 (m) | 42.0d |
| 13 | 3.82 (d, 10.0) | 84.0d | 4.69 (d, 4.8) | 70.5d | 4.67 (d, 4.9) | 70.6d |
| 14 | 85.2s | 140.2s | 139.6s | |||
| 15 | 3.38 (t, 2.9) | 48.9d | 5.19 (t, 8.0) | 125.9d | 5.20 (d, 8.0) | 126.5d |
| 16α | 2.50 (ddd, 14.2, 6.4, 3.6) | 22.8t | 2.39 (m) | 27.6t | 2.31 (m) | 27.9t |
| 16β | 1.72 (dt, 14.2, 3.9) | 2.18 (m) | 2.03 (m) | |||
| 17α | 2.77 (m) | 19.6t | 2.97 (m) | 33.0t | 2.81 (m) | 29.9t |
| 17β | 2.44 (ddd, 18.4, 13.8, 4.7) | 2.84 (m) | 2.76 (m) | |||
| 18 | 134.5s | 123.7s | 130.1s | |||
| 19 | 179.0s | 146.3s | 144.3s | |||
| 20 | 140.5s | 128.5s | 126.4s | |||
| 21 | 7.15 (s) | 114.0d | 8.14 (s) | 114.4d | 7.10 (s) | 110.4d |
| 22 | 186.3s | 147.0s | 131.1s | |||
| 23 | 147.9s | 133.5s | 119.1s | |||
| 24 | 1.03 (d, 6.4) | 13.0q | 0.87 (d, 6.9) | 10.5q | 0.89 (d, 6.8) | 10.5q |
| 25 | 1.14 (s) | 20.5q | 1.74 (s) | 20.6q | 1.75 (s) | 20.5q |
| 26 | 9.02 (s) | 154.5d | 3.35 (m), 3.33 (m) | 30.7t | ||
| 27 | 168.3s | |||||
| MeO-3 | 3.32 (s) | 56.7q | 3.36 (s) | 56.8q | ||
Compound 4 was isolated as a light yellow powder. Its molecular formula was determined to be C27H34N2O5S on the basis of high-resolution ESIMS at m/z 499.2267 [M + H]+ (calcd for C27H35N2O5S+, 499.2261) and 521.2079 [M + Na]+ (calcd for C27H34N2O5SNa+, 521.2081) (ESI Fig. S51†). The 1H–1H COSY spectra of 4 were the same as that of 1 at the fragment from C-2 to C-13 and C-15 to C-17 (Fig. 2 and ESI Table S4†). The HMBC correlations from H-17 (δH 2.97, 2.84) to C-18 (δC 123.7), C-19 (δC 146.3) and C-23 (δC 133.5), and H-21 (δH 8.14) to C-19, C-20 (δC 128.5) and C-23 suggested a penta-substituted benzene ring (Fig. 2 and ESI Table S4†). The HMBC correlations from H-26 (δH 9.02) to C-22 (δC 147.0) and C-23 indicated that C-26 was linked to C-22 via a nitrogen atom and to C-23 through a sulfide bond, which was supported by the high-resolution ESIMS data (Fig. 2 and ESI Table S4†). Hence, the chromophore moiety of 4 was determined to be a derivative of benzothiazole (Fig. 1). Furthermore, the NMR comparison with that of thiazinotrienomycin G (11) demonstrated that compound 4 was decyclohexanoyl-D-alanyl thiazinotrienomycin G (Table 2),19 namely, ansatrienol E (Fig. 1).
Compound 5 was isolated as a light yellow powder. Its molecular formula was determined to be C27H34N2O6S on the basis of high-resolution ESIMS at m/z 537.2029 [M + Na]+ (calcd for C27H34N2O6SNa+, 537.2030) and 497.2107 [M + H − H2O]+ (calcd for C27H33N2O5S+, 497.2105) (ESI Fig. S52†). Its structure was similar to thiazinotrienomycinol E with the ansa moiety from C-1 to C-17 and the phenolic chromophore moiety benzothiazine, except for the absence of a methoxyl group, which was confirmed by the 1H–1H COSY correlations and the HMBC correlations from H-26 (δH 3.33, 3.35) to C-27 (δC 168.3) and C-23 (δC 119.1), and the disappearance of the signal at δH 3.35 (s, MeO–) (Fig. 2 and ESI Table S5†). Hence, compound 5 was determined to be 3-O-demethyl thiazinotrienomycinol E (Table 2),14 namely, ansatrienol F (Fig. 1).
Compound 6 was isolated as a light yellow powder. Its molecular formula was determined to be C25H33NO5 on the basis of high-resolution ESIMS at m/z 445.2698 [M + NH4]+ (calcd for C25H33NO5NH4+, 445.2697) and 450.2250 [M + Na]+ (calcd for C25H33NO5Na+, 450.2251) (ESI Fig. S53†). The NMR studies on the 1H–1H COSY and the HMBC correlations (ESI Table S6†) demonstrated that 6 was partially identical to 5 (from C-1 to C-17). The HMBC correlations from H-19 (δH 6.42) to C-17 (δC 37.5), C-20 (δC 140.1), C-21 (δC 107.1) and C-23 (δC 112.8), H-21 (δH 7.02) to C-19 (δC 113.1), C-20 and C-22 (δC 158.6), and H-23 (δH 6.38) to C-17, C-19, C-21 and C-22 revealed a meta-trisubstituted benzene ring (Fig. 2). Moreover, the NMR comparison with that of trienomycinol revealed that compound 6 was 3-O-demethyltrienomycinol (Table 3),17 namely, ansatrienol G (Fig. 1).
| Pos. | 6 | 7 | ||
|---|---|---|---|---|
| δH (mult., J in Hz) | δC mult. | δH (mult., J in Hz) | δC mult. | |
| 1 | 171.2s | 171.1s | ||
| 2α | 2.68 (dd, 12.0, 4.3) | 46.9t | 2.72 (dd, 12.1, 4.4) | 47.0t |
| 2β | 2.51 (m) | 2.46 (m) | ||
| 3 | 4.51 (m) | 72.0d | 4.52 (m) | 72.1d |
| 4 | 5.69 (dd, 14.4, 7.7) | 134.3d | 5.65 (dd, 14.9, 7.8) | 134.7d |
| 5 | 6.11 (m) | 132.7d | 6.16 (m) | 132.8d |
| 6 | 6.16 (m) | 130.9d | 6.18 (m) | 131.3d |
| 7 | 6.06 (m) | 135.0d | 6.17 (m) | 134.6d |
| 8 | 6.09 (m) | 133.8d | 6.10 (m) | 133.3d |
| 9 | 5.70 (m) | 131.7d | 6.02 (m) | 132.2d |
| 10α | 2.41 (m) | 37.7t | 2.68 (m) | 41.8t |
| 10β | 2.31 (m) | 2.33 (dd, 14.6, 10.1) | ||
| 11 | 3.66 (m) | 73.0d | 3.88 (d, 5.9) | 77.0d |
| 12 | 1.85 (m) | 42.5d | 1.72 (m) | 39.2d |
| 13 | 4.63 (d, 6.1) | 70.7d | 3.97 (d, 10.1) | 74.7d |
| 14 | 139.1s | 1.76 (m) | 43.9d | |
| 15 | 5.25 (dd, 8.2, 4.9) | 127.1d | 3.74 (m) | 72.4d |
| 16α | 2.29 (m) | 30.6t | 1.59 (m) | 33.3t |
| 16β | 2.01 (m) | 1.46 (m) | ||
| 17α | 2.48 (m) | 37.5t | 2.82 (m) | 33.4t |
| 17β | 2.44 (m) | 2.45 (m) | ||
| 18 | 145.1s | 145.7s | ||
| 19 | 6.42 (t, 1.7) | 113.1d | 6.58 (s) | 112.7d |
| 20 | 140.1s | 140.0s | ||
| 21 | 7.02 (t, 2.0) | 107.1d | 6.86 (s) | 107.0d |
| 22 | 158.6s | 158.6s | ||
| 23 | 6.38 (t, 1.7) | 112.8d | 6.42 (s) | 113.4d |
| 24 | 0.95 (d, 6.9) | 10.5q | 1.05 (d, 7.0) | 12.1q |
| 25 | 1.76 (s) | 20.3q | 0.69 (d, 6.8) | 10.6q |
Compound 7 was obtained as a white powder. Its molecular formula was determined to be C25H35NO6 on the basis of high-resolution ESIMS at m/z 446.2540 [M + H]+ (calcd for C25H36NO6+, 446.2537) and 468.2355 [M + Na]+ (calcd for C25H35NO6Na+, 468.2357) (ESI Fig. S54†). The NMR comparison with that of 6 indicated that both the compounds had the same scaffold except the absence of two olifenic carbons and the presence of one aliphatic carbon at δC 43.9 and one oxy-carbon at δC 72.4 in 7 (Table 3), suggesting that the C-14/C-15 double bond in 7 was saturated by the addition of one molecule of H2O, which was further supported by the inspection of the 1H–1H COSY and HMBC correlations (Fig. 2 and ESI Table S7†). The relative configurations of C-14 and C-15 were suggested on the basis of the NOE correlations of H-25/H-13/H-15, H-24/H-11, and H-24/H-14 (Fig. 2). Thus, compound 7 was determined to be ansatrienol H (Fig. 1).
Ansamycins are well known for their antibacterial activities, such as rifamycins,1 and their antitumor activities such as maytansine.2,3 Therefore, compounds 1–11 were assayed for their antimicrobial activities against Staphylococcus aureus ATCC 25923, Bacillus subtilis ACCC 11060 and Pseudomonas aeruginosa PA01. With the exception of 3 and 10, which presented modest activities against Staphylococcus aureus ATCC 25923 and Bacillus subtilis ACCC 11060, none of the other compounds showed activities on the strains tested. Compounds 1–7 were also tested for their cytotoxicity against human cancer HeLa and H1299 cell lines. None of them showed significant cytotoxic activity (ESI Fig. S55†).
In view of lack of cytotoxicity against the test bacteria and human cancer cell lines, compounds 1–7 were further assayed for their activities of inhibiting the type three secretion system (T3SS) of Salmonella enterica. The inhibitors of T3SS have no effects on bacterial growth, thus it has become an interesting drug target for the development of novel anti-virulence agents.22,23 T3SS is a significant virulence factor, which is an injector-like secretion apparatus to transport effector proteins from the bacterial cytoplasm into eukaryotic host cells and facilitate the invasion and dissemination of pathogens.24 Its structure and function are highly conserved among different Gram-negative bacteria.25,26 Salmonella enterica is one of the bacteria, which can cause intestinal diseases for humans and animals. The T3SSs of S. enterica are encoded by the Salmonella pathogenicity island 1 (SPI-1) and SPI-2. To screen novel anti-bacterial agents targeting SPI-1, compounds 1–7 were assayed for their activities of inhibiting the T3SSs of S. enterica using Csn-B (cytosporone B) as the positive control, as described.27 Compound 3 showed evident inhibitory activity against the secretion of SPI-1 effectors SipA/B/C/D and no effects on FliC, whereas the other compounds displayed weak or no inhibitory effects at the concentration of 100 μM (Fig. 4). Therefore, it is worth to further study the mechanism of action of 3, inhibiting T3SS for developing novel anti-virulence agents.22,23
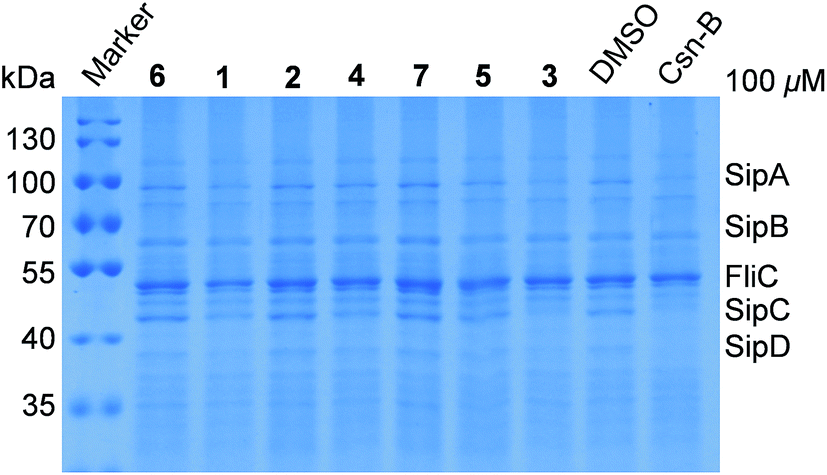 | ||
| Fig. 4 Ansatrieninols B–H (1–7) inhibited the secretion of invasion-associated SPI-1 effector proteins in vitro. Salmonella enterica serovar Typhimurium UK-1χ8956 (ΔP rpoS183:TT araC PBAD rpoS) was grown in a LB medium supplemented with 0.2% L-arabinose in the presence of solvent control or compounds 1–7 and Csn-B at the final concentration of 100 μM, respectively. The effector proteins of SPI-1 (Salmonella Pathogenicity Island-1) in the supernatant of culture were precipitated by 10% trichloroacetic acid and analyzed by 10% SDS-PAGE followed by staining with Coomassie Blue. SipA/B/C/D, SPI-1 effector proteins; FliC, flagellin of Salmonella. | ||
Ansatrienins with different chromophore moieties and side chains were isolated from different strains such as Streptomyces sp. MJ672-m3 (ref. 19 and 28) and Streptomyces rishiriensis T23.16 We collectively discovered ansatrienins with diverse post-PKS modifications from a single strain for the first time, supporting that deleting the N-cyclohexanoyl D-alanyl side chain facilitates an investigation of the post-PKS modifications in the biosynthesis of ansatrienins. These findings are interesting in that the post-PKS modifications not only contribute to the structure diversity of ansamycins but are also critical in determining their bioactivities.2,29,30 These post-PKS modifications should provide useful hints for the structural optimization of ansamycins as well. In particular, the formation of the oxazole ring in 1 and 2 would occur via a rare hetero-Diels–Alder reaction that has not been reported (Fig. 3),31–35 which might be catalyzed by a natural Diels–Alderase36 or generated by simultaneous reactions as observed in the formation of other ansamycins.8,10,12,37
Previously, we found that knockout of the astF2 gene led to the accumulation of ansatrienins without a C-19 hydroxyl group (unpublished data), indicating that AstF2 was responsible for the hydroxylation of C-19. Interestingly, compounds 6, 7, 9, and 10 were obtained without the C-19 hydroxyl group in the strain with an intact astF2 gene, suggesting that the hydroxylation of C-19 might occur during post-PKS modifications rather than the extension of polyketide chain.38
Experimental section
General experimental procedures
The NMR spectra were obtained on a Bruker DRX-600 MHz NMR spectrometer (Bruker Daltonics Inc., Billerica, Massachusetts) with tetramethylsilane (TMS) as the internal standard. HR-ESIMS were measured on an LTQ-Orbitrap XL. Sephadex LH-20 was obtained from the GE Amersham Biosciences (Piscataway, New Jersey). Reversed-phase (RP) C18 silica gel for column chromatography (CC) was obtained from Merck (Darmstadt, Germany). Silica gel GF254 for thin-layer chromatography (TLC) was purchased from Qingdao Marine Chemical Ltd. (Qingdao, China). Semipreparative HPLC was performed on a Waters 1525 Binary HPLC Pump and a Waters 996 Photodiode Array Detector, equipped with an Agilent Eclipse XDB-C18 column (5 μm, 9.4 × 250 mm). The compounds were visualized under UV light and sprayed with H2SO4/EtOH (1:9, v/v) and vanillic aldehyde, followed by heating.
Strain and fermentation
Strain XZQH13 was isolated from a soil sample collected at Qinghai, China. It was identified as a Streptomyces species, according to the 16S rRNA sequence. The Streptomyces sp. XZQH13OEΔastC strain was cultured for 4 d on YMG (glucose 4 g, yeast extract 4 g, malt extract 10 g, agar 20 g, distilled water 1 L, and pH 7.2) agar plates at 28 °C.Extraction and isolation
The 30 L culture was diced and extracted three times overnight with EtOAc–MeOH (80:20, v/v) at room temperature. The organic solvents were collected through filtration and the filtrates were combined and concentrated under vacuum. The crude extract was partitioned between water and EtOAc (1:1) until the EtOAc layer was colorless. The EtOAc extract was partitioned between MeOH and petroleum ether. The MeOH layer was concentrated under vacuum to afford the defatted MeOH extract (4.5 g), which was subjected to column chromatography over Sephadex LH-20 eluted with MeOH to give Fr. A–H. Fr. C (1.9 g) was purified by medium pressure liquid chromatography (MPLC) over RP-18 silica gel (60 g) eluted with gradient aqueous acetonitrile (30%, 40%, 50%, 60%, 70%, and 100% CH3CN, 500 mL each) to afford 6 fractions Fr. C1–C6. Fr. C2 (174 mg) was subjected to column chromatography over Sephadex LH-20 eluted with MeOH and then were purified by HPLC (Sunfire Prep OBD C18, 19 × 150 mm, 5 μm, 15 mL min−1, UV 220 nm) eluted with 20% acetonitrile to afford 6 (tR 25 min, 7 mg), 7 (tR 13 min, 2.3 mg) and 10 (tR 18 min, 10 mg) (ESI Fig. S56†). Fr. C4 (73 mg) was subjected to column chromatography over Sephadex LH-20 eluted with MeOH to give Fr. C4a (26 mg) and Fr. C4b (25 mg); furthermore, they were purified by HPLC (Agilent Eclipse XDB-C18, 5 μm, 9.4 × 250 mm, 3.5 mL min−1, UV 274 nm) eluted with 34% acetonitrile and 35% acetonitrile to afford 9 (tR 9.5 min, 3 mg) and 1 (tR 6.8 min, 3.2 mg), respectively (ESI Fig. S56†). Fr. C5 (65 mg) was subjected to column chromatography over Sephadex LH-20 eluted with MeOH and were purified by HPLC (Agilent Eclipse XDB-C18, 5 μm, 9.4 × 250 mm, 3.5 mL min−1, UV 254 nm) eluted with 34% acetonitrile to afford 2 (tR 12.5 min, 3.2 mg) and 4 (tR 17.5 min, 3.5 mg), respectively (ESI Fig. S56†). Fr. D (490 mg), Fr. E (226 mg) and Fr. F (140 mg) were subjected to medium pressure liquid chromatography (MPLC) over RP-18 silica gel (30 g) eluted with a gradient of aqueous acetonitrile (20%, 30%, 40%, 50%, 60%, 70% and 100% CH3CN, 200 mL each) and were purified by HPLC (Agilent Eclipse XDB-C18, 5 μm, 9.4 × 250 mm, 3.5 mL min−1, UV 254 nm) eluted with 31%, 35% and 26% acetonitrile to afford 8 (tR 15.5 min, 4.2 mg), 11 (tR 9.5 min, 5.4 mg) and 5 (tR 13.5 min, 2 mg), respectively (ESI Fig. S56†). Fr. H (946 mg) was subjected to column chromatography over Sephadex LH-20 eluted with MeOH to give Fr. H6 (123 mg); it was then purified by medium pressure liquid chromatography (MPLC) over RP-18 silica gel (30 g) eluted with a gradient of aqueous acetonitrile (20%, 30%, 40%, 50%, 60%, 70% and 100% CH3CN, 200 mL each) and HPLC (Agilent Eclipse XDB-C18, 5 μm, 9.4 × 250 mm, 3.5 mL min−1, UV 254 nm) eluted with 70% acetonitrile to afford 3 (tR 5.8 min, 2.2 mg) (ESI Fig. S56†).
ε) 217 (4.26), 242 (4.24) nm; 1H and 13C NMR data, see Table 1; HRESIMS m/z 456.2383 [M + H]+ (calcd for C26H34NO6+, 456.2381), and 478.2199 [M + Na]+ (calcd for C26H33NO6Na+, 478.2200).ε) 215 (3.99), 245 (3.89) nm; 1H and 13C NMR data, see Table 1; HRESIMS m/z 456.2383 [M + H]+ (calcd for C26H34NO6+, 456.2381).ε) 247 (4.10), 272 (4.13) nm; 1H and 13C NMR data, see Table 2; HRESIMS m/z 486.1949 [M + H]+ (calcd for C26H32NO6S+, 486.1945).ε) 248 (4.36), 273 (4.30) nm; 1H and 13C NMR data, see Table 2; HRESIMS m/z 499.2267 [M + H]+ (calcd for C27H35N2O5S+, 499.2261), and 521.2079 [M + Na]+ (calcd for C27H34N2O5SNa+, 521.2081).ε) 222 (4.16), 260 (4.41) nm; 1H and 13C NMR data, see Table 2; HRESIMS m/z 537.2029 [M + Na]+ (calcd for C27H34N2O6SNa+, 537.2030), and 497.2107 [M + H − H2O]+ (calcd for C27H33N2O5S+, 497.2105).ε) 211 (4.20), 273 (4.29) nm; 1H and 13C NMR data, see Table 3; HRESIMS m/z 445.2698 [M + NH4]+ (calcd for C25H33NO5NH4+, 445.2697), and 450.2250 [M + Na]+ (calcd for C25H33NO5Na+, 450.2251).ε) 211 (4.01), 273 (4.20) nm; 1H and 13C NMR data, see Table 3; HRESIMS m/z 446.2540 [M + H]+ (calcd for C25H36NO6+, 446.2537), and 468.2355 [M + Na]+ (calcd for C25H35NO6Na+, 468.2357).Antimicrobial assay
The antimicrobial activities were tested against using the paper disc diffusion method.39 Kanamycin was used as a positive control. Compounds 1–11 were absorbed onto individual sterile filter paper disks (6 mm diameter) at 20 μg per paper disc and placed on the surface of the agar previously inoculated with Staphylococcus aureus ATCC 25923, Bacillus subtilis ACCC 11060 and Pseudomonas aeruginosa PA01 purchased from the China Center of Industrial Culture Collection (Beijing, China) and conserved in the laboratory of the authors. The assay plates were incubated for 24 h at 37 °C and examined for the presence of a zone of inhibition.Cytotoxicity assay
The in vitro antiproliferative activities were measured against human cancer H1299 and HeLa cell lines purchased from the Institute of Biochemistry and Cell Biology, SIBS, CAS using the SRB assay.40 Briefly, the cancer cells were seeded into 96-well tissue culture plates at a density between 3000 and 8000 cells per well/100 μL in Dulbecco's modified Eagle's medium (Sigma-Aldrich) containing 10% fetal bovine serum (Biological Industries). After 24 h of cultivation, the cells in triplicate with different concentrations of the tested compounds were incubated for 72 h in a humidified incubator (37 °C, 5% CO2). To remove the medium, 100 μL 10% TCA (w/v) per well was added and the plates were incubated overnight at 4 °C. The supernatant was discarded; the plates were washed 5 times with water and dried in air. After that, 100 μL of the SRB solution 0.4% (w/v) in 1% acetic acid was added to each well. Ten minutes later, the plate was rinsed 5 times with 1% (v/v) acetic acid and dried in air. Finally, 200 μL of 10 mM Tris base solution (pH 10.5) was added to each well and the plates were shaken for 5 min on a gyratory shaker. The OD values of each well were measured at 570 nm by a microplate reader (Bio-Rad680, USA). The growth inhibitory rate was calculated using the following formula.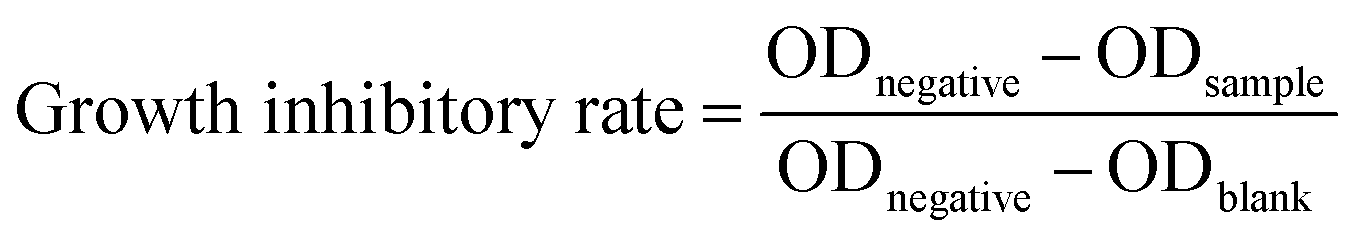
Anti-T3SS assay
The inhibitory activities of 1–7 on T3SS were assayed using Salmonella enterica serovar Typhimurium UK-1 χ8956 (ΔPrpoS183:TT araC PBAD rpoS) obtained from Roy Curtiss III (School of Life Sciences, Arizona State University),41 grown in LB broth (1% tryptone, 0.5% yeast extract, 1% NaCl, pH 7.4) supplemented with 0.2% L-arabinose at 37 °C or 25 °C in the presence of a solvent control or the tested compounds at the final concentration of 100 μM, respectively. The induction of SPI-1 to promote the secretion of effector proteins was achieved by changing the culture temperature. In particular, the bacterial culture was incubated overnight at 25 °C with agitation. On the next day, after the addition of compounds 1–7 and the positive control at 100 μM or solvent control into the 10× diluted cultures, the cultures continued to grow for 4 h at 37 °C with agitation. Cytosporone B (Csn-B) was used as a positive control.27 The proteins secreted in the supernatant of the culture were precipitated by 10% trichloroacetic acid and acetone. Sediments were dissolved with 1× sample buffer, boiled for 10 min at 95 °C to thoroughly denature the proteins, and detected by SDS-PAGE followed by staining with Coomassie Blue.Conclusions
In summary, we isolated and characterized 7 new ansatrienols, and 4 known ones from the Streptomyces sp. XZQH13OEΔastC strain. All eleven compounds showed no obvious cytotoxicities against the growth of the test bacteria S. aureus ATCC 25923, B. subtilis ACCC 11060 and P. aeruginosa PA01, and human cancer H1299 and HeLa cell lines. However, ansatrienol D (3) showed evident inhibitory activity against the secretion of SPI-1 effectors SipA/B/C/D and no effects on FliC. This study demonstrates that a deletion of the side chain contributes to the diverse post-PKS modifications during the biosynthesis of ansatrienins, which reveals not only new ansamycin scaffolds but also the anti-T3SS activity of ansamycins for the first time.Acknowledgements
This study was supported in part by the National Natural Science Foundation of China (81373304, 81530091, 81673317), the Independent Innovation Foundation of Shandong University (IIFSDU, 2014JC027) and the Program for Changjiang Scholars and Innovative Research Team in University (IRT13028).Notes and references
- N. Maggi, C. Pasqualucci, R. Ballotta and P. Sensi, Rifampicin: a new orally active rifamycin, Chemotherapy, 1966, 11, 285–292 CrossRef CAS PubMed.
- J. M. Cassady, K. K. Chan, H. G. Floss and E. Leistner, Recent developments in the maytansinoid antitumor agents, Chem. Pharm. Bull., 2004, 52, 1–26 CrossRef CAS PubMed.
- P. Kusari, S. Kusari, D. Eckelmann, S. Zuhlke, O. Kayser and M. Spiteller, Cross-species biosynthesis of maytansine in Maytenus serrata, RSC Adv., 2016, 6, 10011–10016 RSC.
- L. Whitesell, E. G. Mimnaugh, B. De Costa, C. E. Myers and L. M. Neckers, Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 8324–8328 CrossRef CAS.
- Q. Kang, Y. Shen and L. Bai, Biosynthesis of 3,5-AHBA-derived natural products, Nat. Prod. Rep., 2012, 29, 243–263 RSC.
- H. X. Wang, Y. Y. Chen, L. Ge, T. T. Fang, J. Meng, Z. Liu, X. Y. Fang, S. Ni, C. Lin, Y. Y. Wu, M. L. Wang, N. N. Shi, H. G. He, K. Hong and Y. M. Shen, PCR screening reveals considerable unexploited biosynthetic potential of ansamycins and a mysterious family of AHBA-containing natural products in actinomycetes, J. Appl. Microbiol., 2013, 115, 77–85 CrossRef CAS PubMed.
- W. Zhang, M. Wang, Y. Huang, S. K. Chea, Z. Zheng, X. Qian and Y. Shen, New and highly efficient methodology for screening high-yield strains of cytotoxic deacetylmycoepoxydiene (DAM), Lett. Appl. Microbiol., 2011, 52, 441–447 CrossRef CAS PubMed.
- S. Li, C. Lu, J. Ou, J. Deng and Y. Shen, Overexpression of hgc1 increases the production and diversity of hygrocins in Streptomyces sp. LZ35, RSC Adv., 2015, 5, 83843–83846 RSC.
- S. R. Li, G. S. Zhao, M. W. Sun, H. G. He, H. X. Wang, Y. Y. Li, C. H. Lu and Y. M. Shen, Identification and characterization of the biosynthetic gene cluster of divergolides from Streptomyces sp. W112, Gene, 2014, 544, 93–99 CrossRef CAS PubMed.
- G. Zhao, S. Li, Z. Guo, M. Sun and C. Lu, Overexpression of div8 increases the production and diversity of divergolides in Streptomyces sp. W112, RSC Adv., 2015, 5, 98209–98214 RSC.
- J. Zhang, Z. Qian, X. Wu, Y. Ding, J. Li, C. Lu and Y. Shen, Juanlimycins A and B, ansamycin macrodilactams from Streptomyces sp., Org. Lett., 2014, 16, 2752–2755 CrossRef CAS PubMed.
- S. Li, Y. Li, C. Lu, J. Zhang, J. Zhu, H. Wang and Y. Shen, Activating a Cryptic Ansamycin Biosynthetic Gene Cluster To Produce Three New Naphthalenic Octaketide Ansamycins with n-Pentyl and n-Butyl Side Chains, Org. Lett., 2015, 17, 3706–3709 CrossRef CAS PubMed.
- C. Xie, J. Deng and H. Wang, Identification of AstG1, a LAL family regulator that positively controls ansatrienins production in Streptomyces sp. XZQH13, Curr. Microbiol., 2015, 70, 859–864 CrossRef CAS PubMed.
- G. Shi, N. Shi, Y. Li, W. Chen, J. Deng, C. Liu, J. Zhu, H. Wang and Y. Shen, D-Alanylation in the Assembly of Ansatrienin Side Chain Is Catalyzed by a Modular NRPS, ACS Chem. Biol., 2016, 11, 876–881 CrossRef CAS PubMed.
- M. Sugita, T. Sasaki, K. Furihata, H. Seto and N. Otake, Studies on mycotrienin antibiotics, a novel class of ansamycins. II. Structure elucidation and biosynthesis of mycotrienins I and II, J. Antibiot., 1982, 35, 1467–1473 CrossRef CAS PubMed.
- M. Sugita, Y. Natori, N. Sueda, K. Furihata, H. Seto and N. Otake, Studies on mycotrienin antibiotics, a novel class of ansamycins. III. The isolation, characterization and structures of mycotrienols I and II, J. Antibiot., 1982, 35, 1474–1479 CrossRef CAS PubMed.
- S. Funayama, Y. Anraku, A. Mita, Z. B. Yang, K. Shibata, K. Komiyama, I. Umezawa and S. Omura, Structure–activity relationship of a novel antitumor ansamycin antibiotic trienomycin A and related compounds, J. Antibiot., 1988, 41, 1223–1230 CrossRef CAS PubMed.
- T. Kawamura, E. Tashiro, K. Shindo and M. Imoto, SAR study of a novel triene-ansamycin group compound, quinotrierixin, and related compounds, as inhibitors of ER stress-induced XBP1 activation, J. Antibiot., 2008, 61, 312–317 CrossRef CAS PubMed.
- N. Hosokawa, H. Naganawa, M. Hamada, H. Iinuma, T. Takeuchi, K. S. Tsuchiya and M. Hori, New triene-ansamycins, thiazinotrienomycins F and G and a diene-ansamycin, benzoxazomycin, J. Antibiot., 2000, 53, 886–894 CrossRef CAS PubMed.
- G. Lazar, H. Zahner, M. Damberg and A. Zeeck, Ansatrienin A2 and A3: minor components of the ansamycin complex produced by Streptomyces collinus, J. Antibiot., 1983, 36, 187–189 CrossRef CAS PubMed.
- A. B. Smith, J. L. Wood, W. Wong, A. E. Gould, C. J. Rizzo, J. Barbosa, K. Komiyama and S. Omura, (+)-Trienomycins A, B, C, and F and (+)-mycotrienins I and II: Relative and absolute stereochemistry, J. Am. Chem. Soc., 1996, 118, 8308–8315 CrossRef CAS.
- R. Nordfelth, A. M. Kauppi, H. A. Norberg, H. Wolf-Watz and M. Elofsson, Small-molecule inhibitors specifically targeting type III secretion, Infect. Immun., 2005, 73, 3104–3114 CrossRef CAS PubMed.
- E. Crabill, A. Joe, A. Block, J. M. van Rooyen and J. R. Alfano, Plant immunity directly or indirectly restricts the injection of type III effectors by the Pseudomonas syringae type III secretion system, Plant Physiol., 2010, 154, 233–244 CrossRef CAS PubMed.
- D. Aiello, J. D. Williams, H. Majgier-Baranowska, I. Patel, N. P. Peet, J. Huang, S. Lory, T. L. Bowlin and D. T. Moir, Discovery and characterization of inhibitors of Pseudomonas aeruginosa type III secretion, Antimicrob. Agents Chemother., 2010, 54, 1988–1999 CrossRef CAS PubMed.
- L. M. Stamm and M. B. Goldberg, Microbiology. Establishing the secretion hierarchy, VIP, 2011, vol. 331, pp. 1147–1148 Search PubMed.
- L. J. Worrall, E. Lameignere and N. C. Strynadka, Structural overview of the bacterial injectisome, Curr. Opin. Microbiol., 2011, 14, 3–8 CrossRef CAS PubMed.
- J. Li, C. Lv, W. Sun, Z. Li, X. Han, Y. Li and Y. Shen, Cytosporone B, an inhibitor of the type III secretion system of Salmonella enterica serovar Typhimurium, Antimicrob. Agents Chemother., 2013, 57, 2191–2198 CrossRef CAS PubMed.
- N. Hosokawa, H. Naganawa, H. Inuma, M. Hamada, T. Takeuchi, T. Kanbe and M. Hori, Thiazinotrienomycins, new ansamycin group antibiotics, J. Antibiot., 1995, 48, 471–478 CrossRef CAS PubMed.
- S. J. Moss, L. Bai, S. Toelzer, B. J. Carroll, T. Mahmud, T. W. Yu and H. G. Floss, Identification of asm19 as an acyltransferase attaching the biologically essential ester side chain of ansamitocins using N-desmethyl-4,5-desepoxymaytansinol, not maytansinol, as its substrate, J. Am. Chem. Soc., 2002, 124, 6544–6545 CrossRef CAS PubMed.
- P. Spiteller, L. Bai, G. Shang, B. J. Carroll, T. W. Yu and H. G. Floss, The post-polyketide synthase modification steps in the biosynthesis of the antitumor agent ansamitocin by Actinosynnema pretiosum, J. Am. Chem. Soc., 2003, 125, 14236–14237 CrossRef CAS PubMed.
- S. Arumugam and V. V. Popik, Light-induced hetero-Diels–Alder cycloaddition: a facile and selective photoclick reaction, J. Am. Chem. Soc., 2011, 133, 5573–5579 CrossRef CAS PubMed.
- K. Takao, S. Noguchi, S. Sakamoto, M. Kimura, K. Yoshida and K. Tadano, Total Synthesis of (+)-Cytosporolide A via a Biomimetic Hetero-Diels–Alder Reaction, J. Am. Chem. Soc., 2015, 137, 15971–15977 CrossRef CAS PubMed.
- B. Liu, T. Y. Liu, S. W. Luo and L. Z. Gong, Asymmetric hetero-Diels–Alder reaction of diazenes catalyzed by chiral silver phosphate: water participates in the catalysis and stereocontrol, Org. Lett., 2014, 16, 6164–6167 CrossRef CAS PubMed.
- D. J. Sprague, B. M. Nugent, R. A. Yoder, B. A. Vara and J. N. Johnston, Adaptation of a small-molecule hydrogen-bond donor catalyst to an enantioselective hetero-Diels–Alder reaction hypothesized for brevianamide biosynthesis, Org. Lett., 2015, 17, 880–883 CrossRef CAS PubMed.
- M. P. Doyle, M. Valenzuela and P. Huang, Asymmetric hetero-Diels–Alder reaction catalyzed by dirhodium(II) carboxamidates, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5391–5395 CrossRef CAS PubMed.
- M. J. Byrne, N. R. Lees, L.-C. Han, M. W. van der Kamp, A. J. Mulholland, J. E. M. Stach, C. L. Willis and P. R. Race, The Catalytic Mechanism of a Natural Diels–Alderase Revealed in Molecular Detail, J. Am. Chem. Soc., 2016, 138, 6095–6098 CrossRef CAS PubMed.
- T. C. Le, I. Yang, Y. J. Yoon, S.-J. Nam and W. Fenical, Ansalactams B–D Illustrate Further Biosynthetic Plasticity within the Ansamycin Pathway, Org. Lett., 2016, 18, 2256–2259 CrossRef CAS PubMed.
- J. Xu, E. Wan, C. J. Kim, H. G. Floss and T. Mahmud, Identification of tailoring genes involved in the modification of the polyketide backbone of rifamycin B by Amycolatopsis mediterranei S699, Microbiology, 2005, 151, 2515–2528 CrossRef CAS PubMed.
- D. Raahave, Paper disk-agar diffusion assay of penicillin in the presence of streptomycin, Antimicrob. Agents Chemother., 1974, 6, 603–605 CrossRef CAS PubMed.
- V. Vichai and K. Kirtikara, Sulforhodamine B colorimetric assay for cytotoxicity screening, Nat. Protoc., 2006, 1, 1112–1116 CrossRef CAS PubMed.
- R. Curtiss, 3rd, S. Y. Wanda, B. M. Gunn, X. Zhang, S. A. Tinge, V. Ananthnarayan, H. Mo, S. Wang and W. Kong, Salmonella enterica serovar typhimurium strains with regulated delayed attenuation in vivo, Infect. Immun., 2009, 77, 1071–1082 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data and other relevant information for compounds 1–11. See DOI: 10.1039/c6ra19036g |
| This journal is © The Royal Society of Chemistry 2016 |