
Dopamine and silane functionalized barium titanate with improved electromechanical properties for silicone dielectric elastomers
Dan Yangab,
Mengnan Ruanbc,
Shuo Huangbc,
Yibo Wuab,
Shuxin Liab,
Hao Wangab,
Xiang Aoa,
Yafei Lianga,
Wenli Guo*ab and
Liqun Zhang*c
aDepartment of Materials Science and Engineering, Beijing Institute of Petrochemical Technology, Beijing 102617, China. E-mail: gwenli@bipt.edu.cn
bBeijing Key Lab of Special Elastomeric Composite Materials, Beijing 102617, China
cDepartment of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: zhanglq@mail.buct.edu.cn
First published on 1st September 2016
Abstract
In this study, a combination of bio-inspired poly(dopamine) (PDA) deposition and γ-methacryloxypropyl trimethoxy silane (KH570) grafting is proposed to functionalize the surface of barium titanate (BT) with the aim to improve the electromechanical properties of silicone dielectric elastomers. Inspired by the composition of adhesive proteins in mussels, we used dopamine self-polymerization to form a thin adherent PDA film onto the surface of BT. KH570, which contains double bonds, was then grafted through reactions with the hydroxyl and indole groups of the PDA surface. The surface composition and microstructure of the modified BT particles were characterized by X-ray photoelectron spectroscopy (XPS), Fourier transform infrared (FTIR) spectroscopy, high resolution transmission electron microscopy (HR-TEM), and thermogravimetric analysis (TGA). The results show that PDA grafted with KH570 (denoted as PDA + KH570) was successfully coated on the BT particles. In addition, the PDA + KH570 layer containing double bonds not only improved the dispersion of the fillers in the polymer matrix but also helped in the formation of strong interfacial interactions between the BT particles and silicone elastomer matrix, which led to an improved dielectric constant and actuated strain. Finally, the tensile strength of all the silicone dielectric composites is larger than 5 MPa, which is much higher than commercial acrylic dielectric elastomers. This is a big advantage for the practical application of a dielectric elastomer.
1 Introduction
Dielectric elastomers (DEs), which are generally similar to mammalian skeletal muscle, represent a new and promising electromechanical transducer technology due to their capabilities to respond to electric stimuli.1,2 Dielectric elastomer actuators (DEA) consist of an insulating membrane sandwiched between two soft electrodes, which reduces its thickness and expands in area when subjected to a voltage. By virtue of their simple working principle and outstanding deformation strains, DEA could find a wide range of applications in biomimetic robots, power generators, large strain capacitive sensors, optical devices, health monitors, and energy harvesters.3,4 Various types of soft materials, including acrylic elastomers,5 polyurethanes,6,7 silicone elastomers,8,9 and thermoplastic elastomers such as polystyrene-co-ethylene-co-butylene-co-styrene triblock copolymers (SEBS),10,11 have been investigated for such applications. Among them, silicone elastomers are the most promising materials for use in electromechanical actuators, because they have a fast response rate and resilience, low viscoelastic loss, high coupling efficiency, low creep, excellent biocompatibility, long-term stability and reliability, and can operate in a wide temperature range.12,13 However, the dielectric constant of silicone elastomers is relatively low, which means a high operating voltage (several kilovolts) is needed to induce a large deformation, which causes security risks and increases the costs of devices.9The actuation strain of a dielectric elastomer actuator is determined by
| Sz = P/Y = −εrε0E2/Y | (1) |
During recent years, two main approaches, namely modification of polymers backbone and addition high dielectric constant filler, have been applied to increase the dielectric constant of dielectric elastomers to improve the actuated strain. Polar p-nitroaniline, nitrobenzene, and cyanopropyl have been used to modify silicone in order to increase its dielectric constant.15–19 However, polar groups significantly reduce the dielectric strength of the polymer, improve the glass transition temperature (Tg) impacting elastic properties, increase sensitivity to moisture, and increase cost due to additional synthetic steps.9,20,21 Adding conductive particles to a polymer matrix can increase the dielectric constant obviously if the concentration of the conductive particles approaches the percolation threshold.22–24 However, an unacceptably dramatic reduction in dielectric strength and obvious increase in dielectric loss are usually accompanied.25,26 The main method for increasing the dielectric constant of polymers is the introduction of high-dielectric-constant ceramic particles (e.g. BaTiO3) into the elastomer matrix.27 However, the improvement in electromechanical properties is limited by poor dispersion properties due to the large contrast in surface characteristics between the inorganic and the organic phase. Extensive studies have been performed and a variety of techniques have been utilized to enhance the interfacial compatibility of ceramic/polymer composites. These studies and techniques mainly focused on chemical etching and grafting, such as hydroxylation,28 coupling agents,29 surfactants,30,31 phosphoric acids,32,33 and other organic molecular.34 However, these approaches are limited in practical industrial use due to multistep procedures, high costing instruments, stringent reaction conditions, and toxic materials.
In 2007, Lee et al.35 introduced a facile surface modification approach inspired by the adhesion mechanism of mussels. In this method, the mussel adhesive proteins excreted by marine mussels have the ability of strong adhesion to all types of inorganic and organic surfaces by the self-polymerization of dopamine (DOPA) in an aqueous solution.36,37 Recently, inspired by adhesive proteins in mussels, poly(dopamine) (PDA) was used as a general building block for the surface treatments of dielectric fillers to improve the electromechanical properties of DE.38–40 The improved electromechanical properties were attributed to the fact that PDA exhibits super strong adhesion, not only to the surface of inorganic fillers but also to macromolecules. More importantly, the poly(dopamine) layer could be used as a versatile platform for secondary reactions.41,42 Chawla et al.43 modified poly(dimethylsiloxane) (PDMS) with polyethylene glycol(PEG)–DOPA–lysine to improve lubrication properties. Sa et al.44 modified meta-aramid (MPIA) fibers through the combination of bio-inspired dopamine self-oxidative polymerization and epoxy functionalized silane (KH560) grafting to improve the adhesion of MPIA fibers with a rubber matrix. However, to the best of our knowledge, the surface modification of BaTiO3 (BT) by dopamine oxidative self-polymerization and secondary functionalization to improve the electromechanical properties of silicone elastomers has not been reported.
Herein, we report a simple and practical approach to prepare core–shell structure particles with BT as the core and PDA grafted with γ-methacryloxypropyl trimethoxy silane (KH570) as the shell (denoted as BT-(PDA + KH570)) by the combination of bio-inspired dopamine self-oxidative polymerization and KH570 grafting. It is well known that KH570, which contains double bonds, is not only able to graft to BT particles through the reactions with hydroxyl and indole groups of PDA surface but also can participate in the vulcanization reaction with silicone rubber to supply molecular interactions. The molecular interactions (chemical bonds) can provide stronger forces between the filler and the matrix than that of the adhesion of PDA (hydrogen bonds or van der Waals forces), thus leading to a better dispersion, which is an advantage for a DEA since it can be possibly used for a long time. The filler was mechanically blended into a silicone elastomer matrix to form dielectric composites. The effects of the PDA + KH570 shell layer on the mechanical and electromechanical performances of composites have been thoroughly investigated.
2 Experimental
2.1 Materials
Commercial grade methyl vinyl silicone rubber (SR) filled with 20 phr (parts per hundred parts of rubber) SiO2 nano-particles (Dow Corning C6-235) was used as the raw material. BT particles with an average diameter of 100 nm were purchased from Beijing DK nano technology Co. LTD (China). Dopamine and tris(hydroxymethyl)-amino-methane (Tris) were purchased from Alfa Aesar Company, USA. The modifier, γ-methacryloxypropyl trimethoxy silane (CH2C(CH3)COOCH2CH2CH2Si(OCH3)3, KH570), was purchased from Beijing Chemicals Company, Beijing, China. Dimethylsilicone oil with a viscosity of 0.1 Pa s and crosslinking agent dicumyl peroxide (DCP) were purchased from Beijing Chemical Reagents Co., Ltd. (China). All chemicals were of analytical regent grade and used without further purification.2.2 Surface modification of BT particles by PDA deposition and KH570 grafting
First, a dopamine solution with a concentration of 2.0 g L−1 was prepared by dissolving dopamine in distilled water. The pH of the solution was buffered to 8.5 by adding Tris. 10 mM Tris was dissolved in 100 mL of dopamine solution with a concentration of 2 g L−1. Then, 120 g BT particles were immersed in 1 L dopamine solution for the deposition of poly(dopamine). After 12 hours of deposition under mild stirring at room temperature, 6 wt% KH570 was added to the above dopamine solution for the grafting reaction for 8 hours at 60 °C. Last, the BT particles modified by poly(dopamine) self-oxidative polymerization and KH570 grafting were taken out, washed with deionized water, and dried at 60 °C under vacuum.2.3 Preparation of dielectric composites
The dielectric composites consisting of 0, 10, 30, and 50 phr of BT or BT-(PDA + KH570) particles, 2 phr of DCP, 10 phr of silicone oil, and 100 phr of silicone elastomer were prepared through mechanical mixing on a 6-inch two-roll mill. Silicone oil is a good plasticizer for silicone elastomers to decrease their elastic modulus and increase their actuated strain. When a large amount of silicone oil is added to the silicone elastomer matrix, the mechanical properties deteriorate. Thus, a suitable content of silicone oil (10 phr) was added to the silicone elastomer matrix in our paper. The cured elastomers were obtained at a pressure of 25 MPa for their optimum cure time, as determined by a rotorless curemeter (GT-M2000-FA, Goteah Testing Machines Inc., Taiwan) at 160 °C.The compliant electrodes were prepared by dispersing 10 g of silicone rubber containing 1 g of curing agent (Sylgard 184, Dow Co., Ltd.), 10 g of silicone oil (200FL, Dow Co., Ltd.), 1 g of curing agent, and 3.133 g of conductive carbon black (EC-300J) into 350 mL n-heptane. The DE sample was put into an electric blast below at 60 °C until the compliant electrodes were cured.
2.4 Characterization methods
X-ray photoelectron spectroscopy (XPS) was carried out on an ESCALAB 250 XPS system (Thermo Electron Corporation, USA) with an Al Kα X-ray source (1486.6 eV photons) to determine the chemical compositions of the surface of the BT and BT-(PDA + KH570) particles. Core-level signals were obtained at a photoelectron take-off angle of 45° with respect to the sample surface.The chemical structures of the BT and BT-(PDA + KH570) particles were characterized by Fourier transform infrared (FTIR) spectroscopy (Tensor 27, System 2000, Thermo Fisher Scientific, USA) under ambient conditions. Spectra were obtained in the mid infrared region of 800 cm−1 to 4000 cm−1. Typically, 32 scans at a resolution of 4 cm−1 were accumulated to obtain one spectrum.
A high resolution transmission electron microscope (HR-TEM) (Hitachi H9000, Japan) was used to characterize the morphologies of the BT and BT-(PDA + KH570) particles at a voltage of 300 kV.
Thermogravimetric analysis (TGA) was performed in the temperature range of 30–590 °C with a heating rate of 20 °C min−1 using a TA Q500 thermogravimetric analyzer under a nitrogen atmosphere.
Scanning electron microscopy (SEM, NanoSEM 430, FEI) was employed to observe the morphologies of the SR composites filled with different contents of BT and BT-(PDA + KH570) particles. The surface of the samples was sputtered by a thin layer of gold prior to each measurement.
Tensile tests of dumbbell-shaped samples (25 mm × 6 mm × 1 mm) of the vulcanizates were carried out by using a tensile apparatus (Instron 3366, Instron Corporation, American) at a crosshead speed of 50 mm min−1, according to ASTM D412. The elastic modulus of the samples was determined by the slope of the stress–strain curve at 10% strain. Every experimental data point of elastic modulus is the average of the results obtained from at least five samples.
Equilibrium swelling was used to obtain crosslink density by the Flory–Rehner expression.34 Cured samples of the SR composites were pre-weighed, placed in a Soxhlet extractor containing n-hexane, and allowed to swell for 48 hours at 30 °C. After extraction, excess solvent on the surfaces of the samples was removed and the weights of the swollen wet samples were recorded. Then, the samples were dried in an oven at 60 °C for 48 hours and the weights of the dry network were recorded again. The Flory–Rehner equation is given as follows:
| Ve = −(ln(1 − Vr) + Vr + χVr2)/(V1Vr1/3 − 1/2Vr) | (2) |
The dielectric properties of the samples at room temperature were measured by an impedance analyzer (Alpha-A, Novochtrol, Germany) over the frequency range of 10 to 106 Hz. The obtained experimental data point of the dielectric properties is the average of the results obtained from at least five samples.
The samples for actuated strain tests on a circular membrane actuator had a thickness of 1 mm and diameter of 5 cm. Prior to use, compliant graphite electrodes with a diameter of 11 mm were applied to each side of the dielectric elastomer film by an airbrush. Since it is difficult to accurately measure the change in thickness, we instead measured the change in planar area, Sp. During measurements, the dielectric elastomer films were laid flat between two circular frames without any pre-strains. To obtain the largest strain, the voltage loaded on the electrode area was increased from 0 kV mm−1 until an electric breakdown occurred. During actuation, video images of the biaxial extension of the electrode area were captured by a camera (Canon Ixus 210, Japan) fitted with a wide-angle lens, and the relative change in planar area was determined by analyzing the captured video images with Adobe Photoshop graphics editing software. The planar strain can be calculated according to
| Sp = (A − A0)/A0 × 100% | (3) |
3 Results and discussion
3.1 Surface chemical modification of BT particles
The procedure for the preparation of BT-(PDA + KH570) particles by dopamine polymerization and silane grafting is shown in Scheme 1. It consists of two main steps. First, the polymerization of PDA functional layers occured on the BT surfaces by oxidative self-polymerization. The mechanism for the in situ spontaneous oxidative polymerization of dopamine is elusive at the moment. A possible mechanism is that the dihydroxyl group protons in dopamine are deprotonated, becoming dopamine-quinone through intramolecular cyclization via 1,4-Michael addition, then oxidation to the more readily oxidizable leucodopaminechrome, formation of 5,6-dihydroxyindole, and further oxidation to 5,6-indolequinone, which can further undergo polymerization reactions to form PDA, which is a melanin-like polymer.38,41 Second, KH570 was added to the above dopamine solution, which reacts with the hydroxyl and indole groups in PDA on the surface of the BT particles. Then we obtained modified BT particles with BT as the core and PDA + KH570 as the shell, which allowed double bonds outside of the surface of the BT-(PDA + KH570) particles. A possible reaction mechanism for PDA grafting with KH570 is shown in Scheme 2.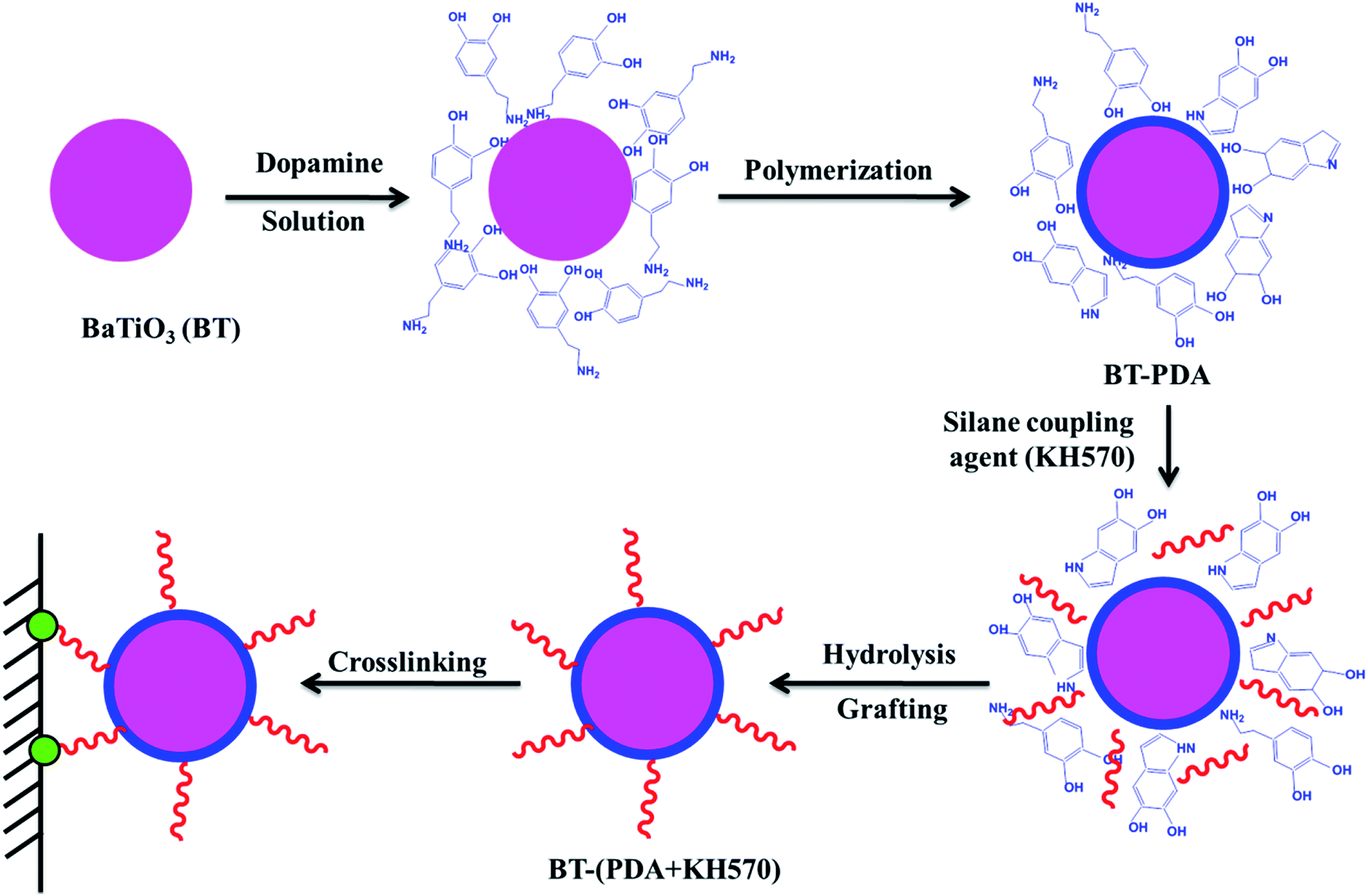 | ||
| Scheme 1 Illustration of the procedure for the preparation of BT-(PDA + KH570) particles by dopamine polymerization and silane grafting. | ||
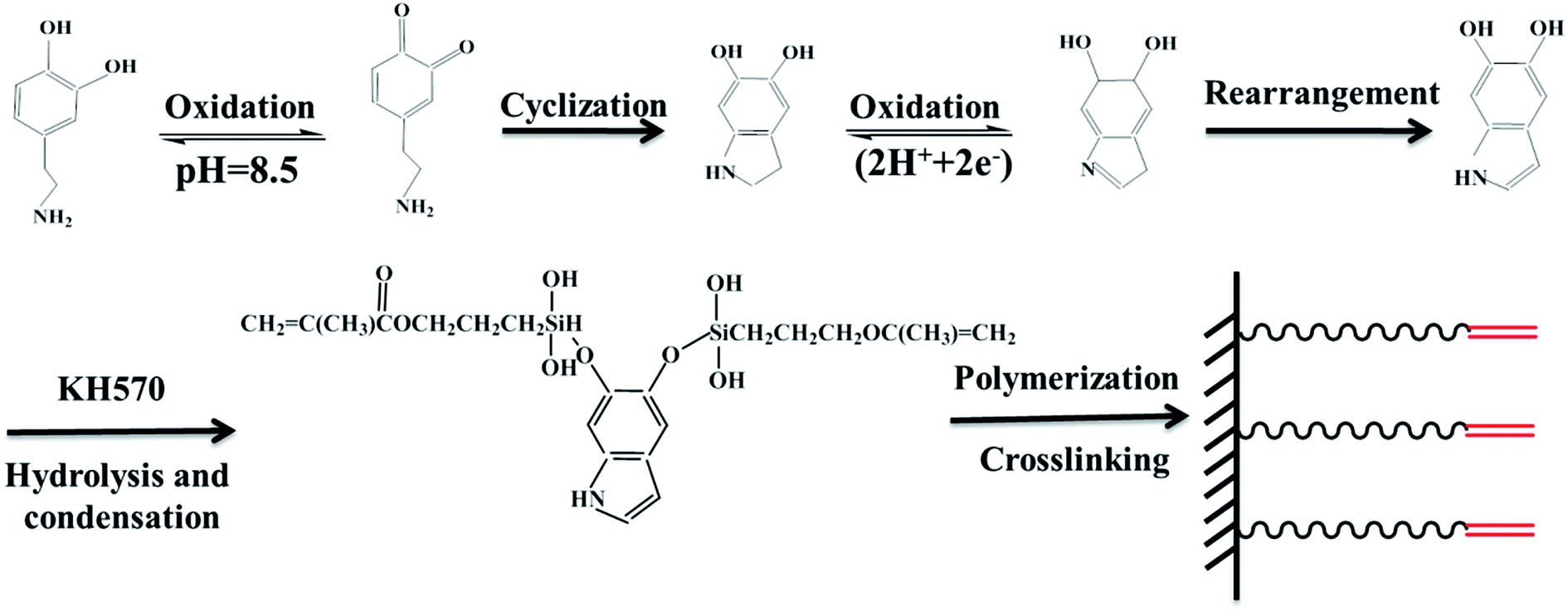 | ||
| Scheme 2 Illustration of the possible reaction mechanism for PDA grafting with KH570. | ||
The chemical compositions of the surfaces of the BT, BT-PDA, and BT-(PDA + KH570) particles were determined by XPS. Fig. 1 shows the wide scan, N 1s core-level spectra, and C 1s core-level spectra of the BT, BT-PDA, and BT-(PDA + KH570) particles. From Fig. 1(b) and (c), we can find there is a new peak component at the binding energy (BE) of about 400 eV in the BT-PDA particles and BT-(PDA + KH570) particles, which is attributed to the nitrogen-containing species in PDA. The N 1s spectra of the BT-PDA particles and BT-(PDA + KH570) particles can be curve-fitted into two typical peaks at BEs of about 398.5 and 399.5 eV, which are attributable to the imine (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) and amine (–N–H) species, respectively. On the basis of the conjectural polymerization mechanism of PDA, the N– species is attributed to the structure evolution during the self-polymerization of dopamine, whereas the –N–H species are formed by the indole groups. As shown in Fig. 1(a), the C 1s core-level spectrum of pristine BT particles can be curve fitted with three peak components at BEs of about 284.6, 286.4, and 288.5 eV, which are attributed to the C–C, C–O, and O–CO species, respectively. The appearance of an additional peak at 285.5 eV is attributed to the C–N species introduced by PDA in the BT-PDA particles and BT-(PDA + KH570) particles, as shown in Fig. 1(b) and (c). The above results confirm the in situ spontaneous oxidative polymerization of dopamine on the BT particles. In addition, the increased percentage peak of the CO and O–CO species of the BT-(PDA + KH570) particles compared with BT-PDA particles indicates that KH570 was successfully grafted on the surface of BT-PDA particles.
N–) and amine (–N–H) species, respectively. On the basis of the conjectural polymerization mechanism of PDA, the N– species is attributed to the structure evolution during the self-polymerization of dopamine, whereas the –N–H species are formed by the indole groups. As shown in Fig. 1(a), the C 1s core-level spectrum of pristine BT particles can be curve fitted with three peak components at BEs of about 284.6, 286.4, and 288.5 eV, which are attributed to the C–C, C–O, and O–CO species, respectively. The appearance of an additional peak at 285.5 eV is attributed to the C–N species introduced by PDA in the BT-PDA particles and BT-(PDA + KH570) particles, as shown in Fig. 1(b) and (c). The above results confirm the in situ spontaneous oxidative polymerization of dopamine on the BT particles. In addition, the increased percentage peak of the CO and O–CO species of the BT-(PDA + KH570) particles compared with BT-PDA particles indicates that KH570 was successfully grafted on the surface of BT-PDA particles.
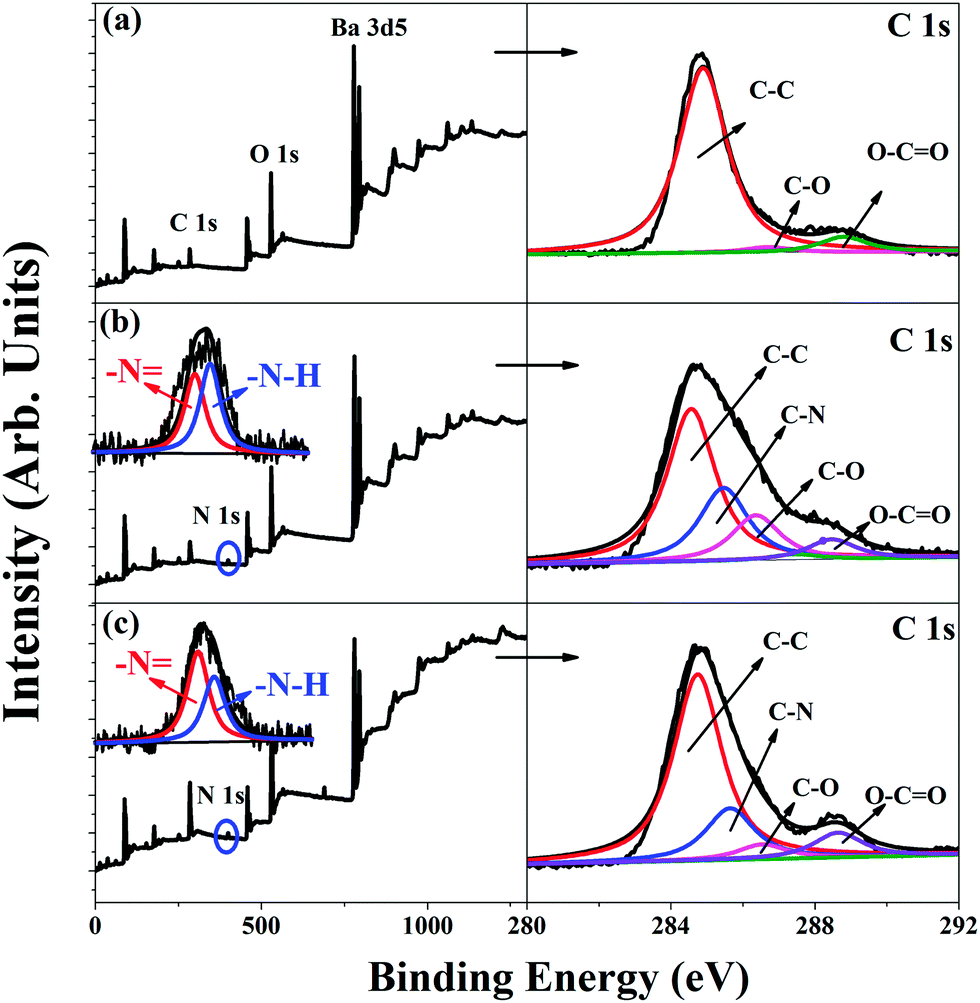 | ||
| Fig. 1 XPS wide-scan, N 1s core-level spectra, and C 1s core-level spectra of (a) BT, (b) BT-PDA, and (c) BT-(PDA + KH570) particles. | ||
The chemical structure of the BT particles before and after treatment was investigated further by FTIR measurements in the mid infrared region (4000–800 cm−1). Fig. 2 illustrates the FTIR spectra of the pristine BT particles, BT-PDA particles, and BT-(PDA + KH570) particles. The new absorption peak at 1500 cm−1 assigned to the benzene groups and absorption peak at 3200 cm−1 attributed N–H and O–H stretching vibration, indicate that PDA was successfully deposited on the surface of the BT particles.44 In addition, there are new absorption peaks at 940 cm−1, 1016 cm−1, 1168 cm−1, 1300 cm−1, and 1720 cm−1 attributed to the CC stretch vibration, Si–O stretch vibration, Si–O–R stretch vibration, C–O–C stretch vibration, and CO stretch vibration, respectively, which indicate that KH570 was grafted onto the BT surface by the chemical reaction between PDA and KH570.
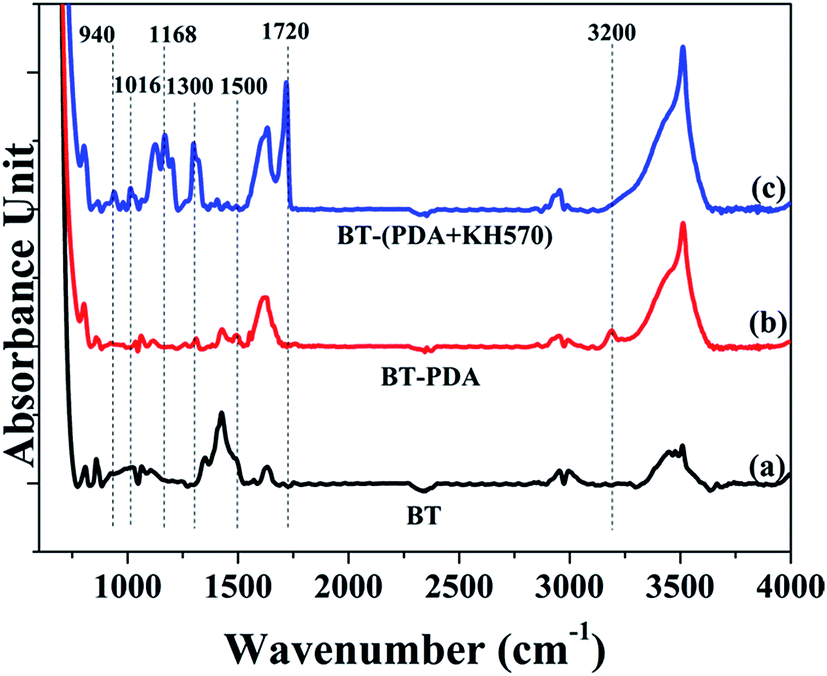 | ||
| Fig. 2 FTIR spectra of (a) BT, (b) BT-PDA, and (c) BT-(PDA + KH570) particles. | ||
The surface morphology of the BT particles before and after treatment was observed by HR-TEM. It can be seen from Fig. 3(a) that the pristine BT particles display a smooth surface. However, a distinct rough layer with a thickness of 1.8 nm is found on the surface of the BT particles (shown in Fig. 3(b)), which is attributed to the PDA layer formed by dopamine oxidative self-polymerization. In Fig. 3(c), the thickness layer of the BT-(PDA + KH570) particles (2.6 nm) is much larger than that of the BT-PDA particles, which indicates that KH570 is successfully grafted on the BT-PDA particles. In addition, the TGA curves of the BT, BT-PDA, BT-(PDA + KH570) particles, and pure PDA are shown in Fig. 4. When the samples are heated to 590 °C, the BT particles, BT-PDA particles, BT-(PDA + KH570) particles, and pure PDA show a weight loss of 0.33%, 3.26%, 4.96%, and 42.91%, respectively. The weight loss of BT-PDA is between the weight of pure PDA and pristine BT, which also demonstrates that the PDA was successfully deposited on the surface of the BT particles. In addition, the weight loss of BT-(PDA + KH570) is larger than that of BT-PDA particles, indicating the KH570 was successfully grafted to the BT-PDA particles again.
 | ||
| Fig. 3 HR-TEM images of (a) BT, (b) BT-PDA, and (c) BT-(PDA + KH570) particles. | ||
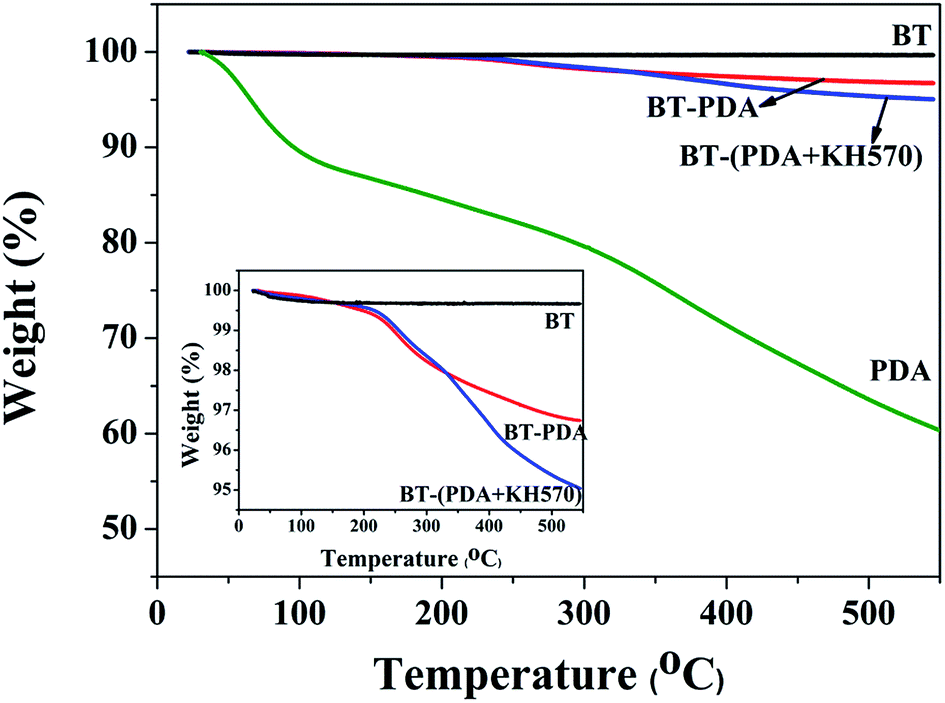 | ||
| Fig. 4 TGA curves of BT, BT-PDA, BT-(PDA + KH570) particles, and pure PDA. | ||
3.2 Microstructure of dielectric composites
Fig. 5 presents SEM images of the fractured surfaces of the SR composites filled with different amounts of BT and BT-(PDA + KH570) particles. From Fig. 5(b) and (c), we can see that almost every individual BT and BT-(PDA + KH570) particle is surrounded by a layer of polymer matrix without any contact with each other when a small amount of filler (10 phr) was added into the SR matrix. From Fig. 5(d) and (f), we can find that many particles are exposed on the fractured surfaces of BT/SR composites. Furthermore, a few agglomerates appear in the SR composite filled with 30 phr BT particles, and the size of the agglomeration increases and becomes large, which leads to the particles contacting with each other to form a filler network when the loading of BT increases to 50 phr. These phenomena are caused by the large difference in surface energy between the BT particles and SR matrix. However, we can find that the agglomerations in the SR composites filled with 30 phr BT-(PDA + KH570) and 50 phr BT-(PDA + KH570) particles are smaller than that of the SR composite filled with 30 phr BT and 50 phr BT particles, respectively. In addition, the interface between the BT-(PDA + KH570) particles and SR matrix is more indistinct than that of the composites filled with pristine BT particles. This is attributed to the fact that KH570, which contain double bonds, coated on the BT surface is able to participate in the vulcanization reaction with rubber to supply molecular interactions to improve the compatibility between the filler and the matrix. The probably mechanism of the double bonds in KH570 crosslinking with SR chains is showed in Scheme 3. During the curing process, the peroxide decomposes and generates oxy radicals. The oxy radicals not only have the potential to abstract hydrogen from the backbone of SR, but also can add to the double bonds of silane, and thus produce radicals. The free electrons may combine molecules in a similar fashion to propagate the free radical reaction. Accordingly, the silane molecules are ultimately grafted onto SR chains through radical reactions.45–47 | ||
| Fig. 5 SEM micrographs of SR composites filled with (a) 0 phr BT, (b) 10 phr BT, (c) 10 phr BT-(PDA + KH570), (d) 30 phr BT, (e) 30 phr BT-(PDA + KH570), (f) 50 phr BT, and (g) 50 phr BT-(PDA + KH570) particles. | ||
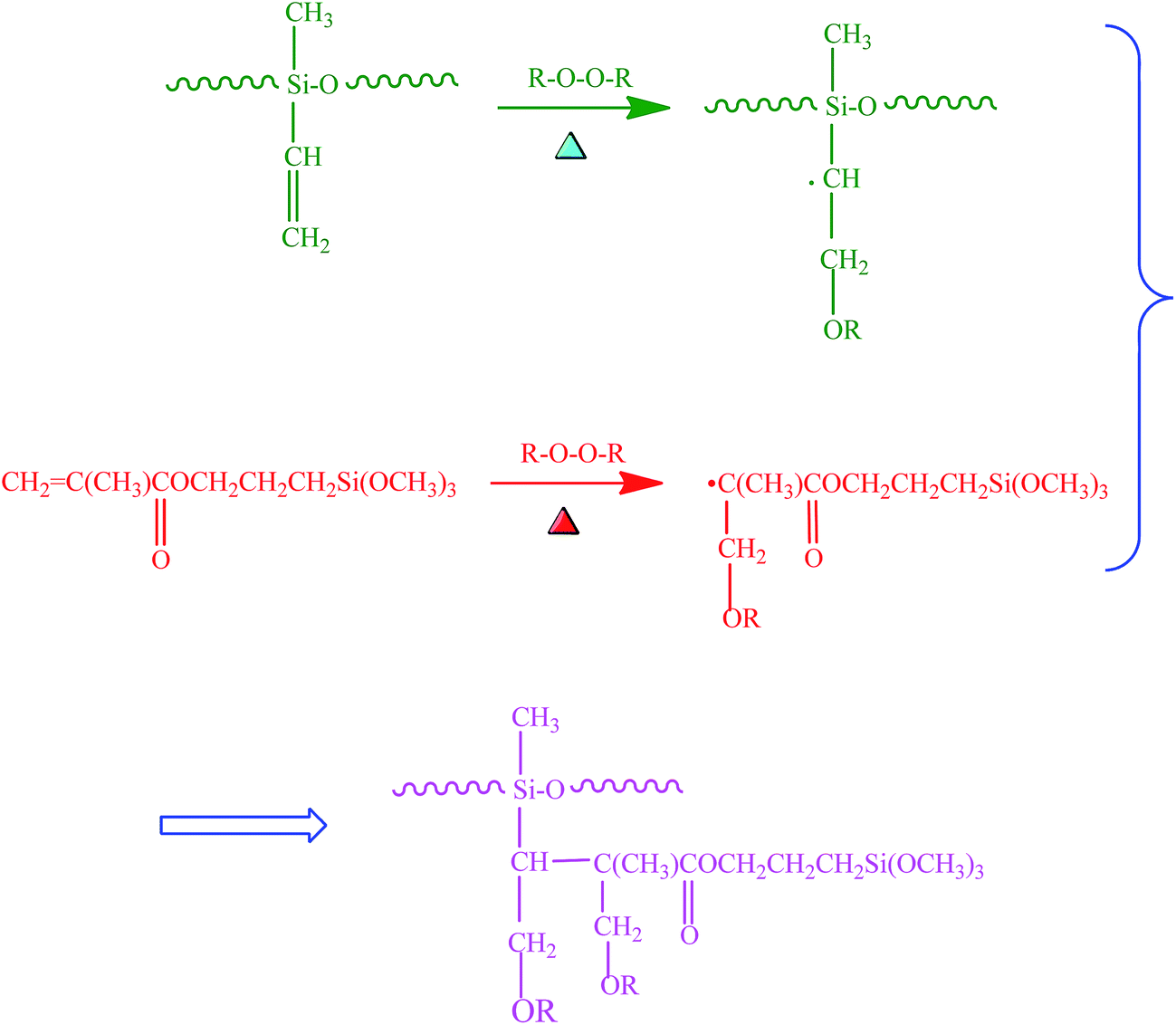 | ||
| Scheme 3 Possible mechanism of KH570 double bonds crosslinking with silicone rubber chains. | ||
3.3 Mechanical properties of dielectric composites
Fig. 6 shows the stress–strain curves of the SR composites filled with different contents of BT and BT-(PDA + KH570) particles. In addition, the stresses at 300% and 500% strains, elongation at break, and tensile strength of SR composites are shown in Table 1. From Fig. 6 and Table 1, we can find that the elongation at break of the SR composites filled with BT-(PDA + KH570) particles is smaller than that of the SR composites filled with pristine BT particles. This mainly ascribed to the strong interfacial interaction between thee BT-(PDA + KH570) particles and SR matrix. Usually, strong interfacial interaction between filler and matrix results in an increase in the number of organic macromolecules tightly bonded to the BT-(PDA + KH570) fillers, thus leading to a decrease in the mobility of the polymer chains and a decrease in elongation at break of the composites. The low elongation at break allows the SR composites filled with BT-(PDA + KH570) particles to display a low tensile strength. However, the stresses at 300% and 500% strains of the SR composites filled with BT-(PDA + KH570) particles are larger than that of the SR composites filled with pristine BT particles at the same filler content (shown in Table 1), mainly because of the uniform dispersion of particles and strong interfacial interaction. Weak interfacial interaction will cause the adsorbed chains to slip easily, whereas good interfacial interaction could restrict the slippage of chains to form more orientated stretching straight chains, to uniformly distribute the external force.48 In addition, we can find that the tensile strength of all the composites is larger than 5 MPa, which is much higher than the 0.38 MPa of the commercial acrylic dielectric elastomer VHB 4910 (3M) at the elongation at break of 1500% according to our measurement. This is a big advantage for the output stress of a dielectric elastomer.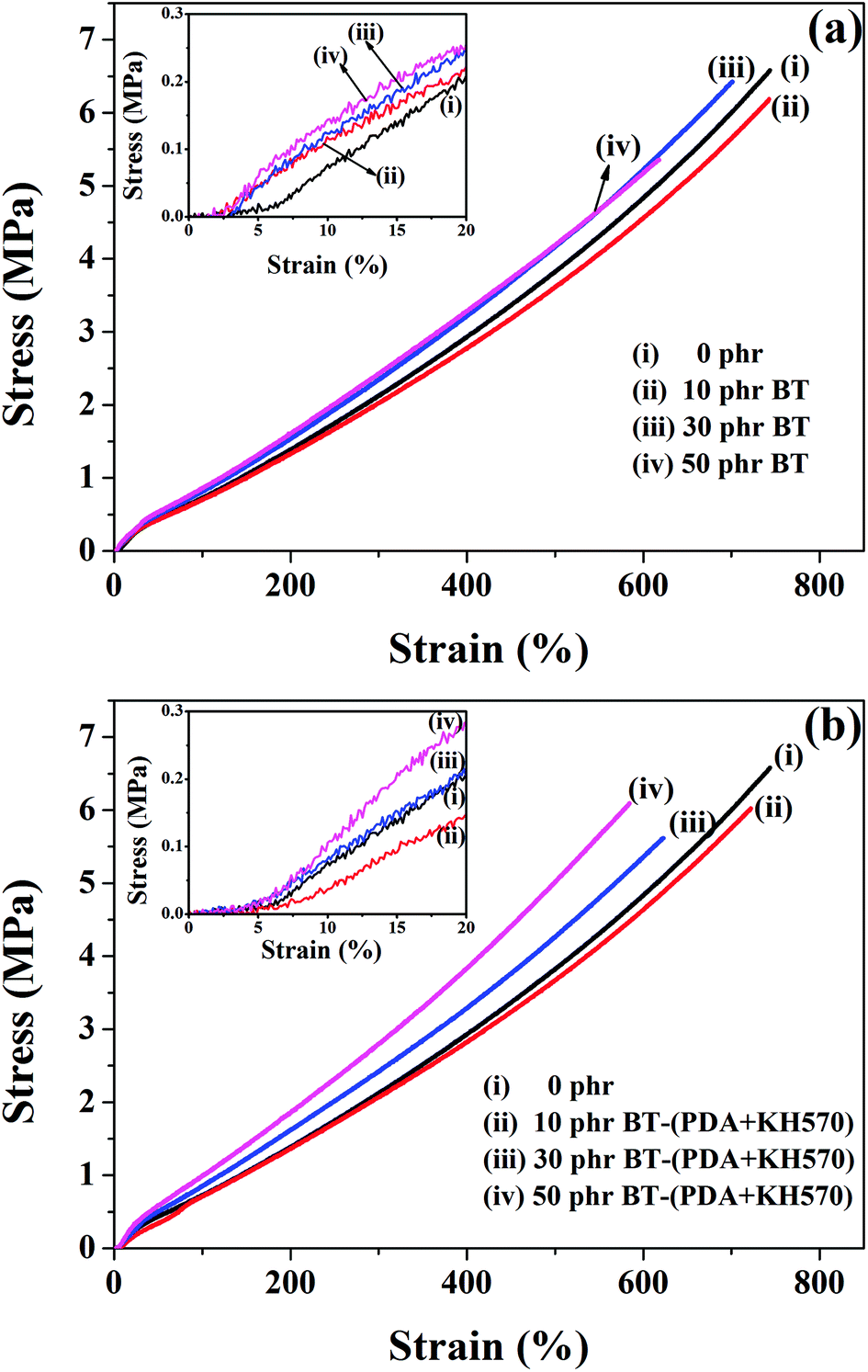 | ||
| Fig. 6 Stress–strain curves of the SR composites filled with different contents of (a) BT and (b) BT-(PDA + KH570) particles. | ||
| Sample | Stress at 100% strain | Stress at 300% strain | Elongation at break | Tensile strength | β | Maximum strain | Breakdown strength |
|---|---|---|---|---|---|---|---|
| (MPa) | (MPa) | (%) | (MPa) | (MPa−1) | (%) | (kV mm−1) | |
| 0 phr | 2.12 | 3.82 | 743.8 | 6.58 | 26.66 | 6.90 | 78 |
| 10 phr BT | 2.03 | 3.61 | 742.7 | 6.19 | 27.61 | 7.03 | 70 |
| 30 phr BT | 2.35 | 4.17 | 701.2 | 6.43 | 26.48 | 5.04 | 65 |
| 50 phr BT | 2.43 | 4.19 | 617.9 | 5.35 | 24.71 | 3.82 | 50 |
| 10 phr BT-(PDA + KH570) | 2.07 | 3.67 | 722.1 | 6.02 | 29.16 | 8.23 | 73 |
| 30 phr BT-(PDA + KH570) | 2.42 | 4.26 | 623.0 | 5.62 | 28.58 | 6.87 | 67 |
| 50 phr BT-(PDA + KH570) | 2.80 | 5.02 | 548.6 | 6.09 | 27.63 | 5.95 | 59 |
The elastic modulus of the composites filled with different contents of BT and BT-(PDA + KH570) particles are shown in Fig. 7. From Fig. 7, we can find that the elastic modulus of the SR composites filled with BT-(PDA + KH570) particles is lower than that of the SR composites filled with pristine BT particles at the same content of filler. In addition, we can find the difference ΔY between the elastic modulus of the SR composites filled with BT-(PDA + KH570) particles and pristine BT particles at the same content of filler increases with an increase in the content of filler. This might be due to the fact that the BT-(PDA + KH570) particles could partially display polymeric mechanical behavior when they are coated by a relatively thick (2.6 nm) PDA + KH570 layer with extremely lower modulus compared to the rigid BT particles.49,50 Schematic diagrams of the dispersion of BT particles and BT-(PDA + KH570) particles in the SR composites are shown in Fig. 8. The relatively soft PDA + KH570 layer coating on the BT particles increases the volume fraction of the whole polymeric phase in the composites, thus the elastic modulus of the composite filled with BT-(PDA + KH570) particles is lower than that of the composites filled with the original BT particles, even though the dispersion of BT-(PDA + KH570) particles is better than the original BT particles. Assuming that the BT particle was spherical and 50 g BT particles were incorporated into 100 g SR matrix, we can roughly calculate the volume of the relatively soft PDA + KH570 layer (VPDA+KH570) coated on the BT particles according to the following equations:
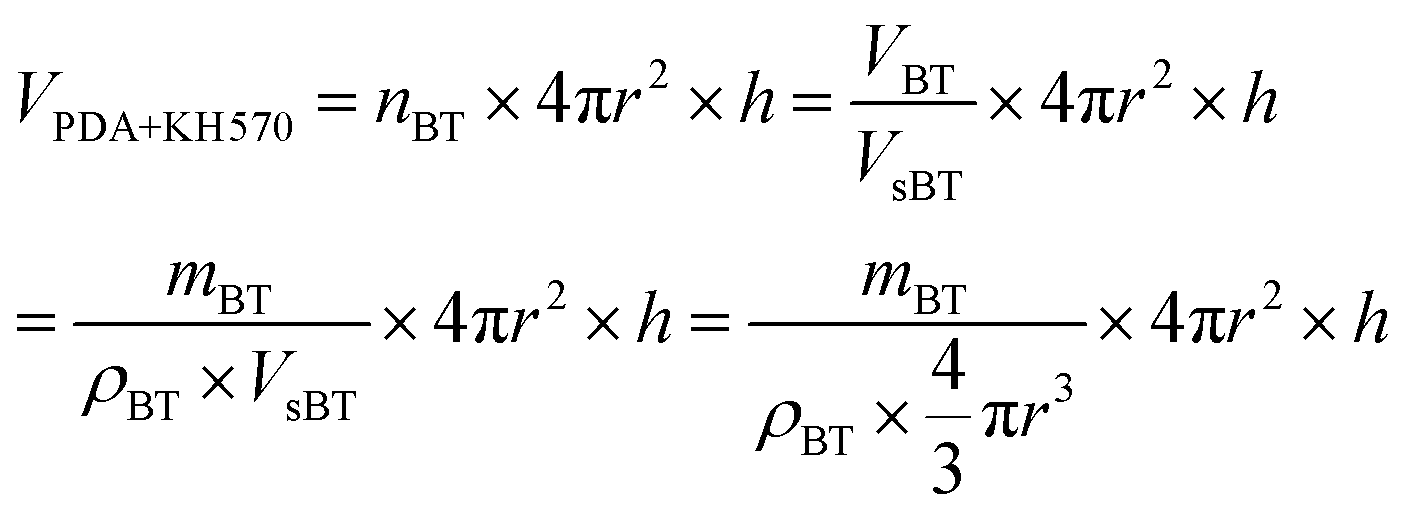 | (4) |
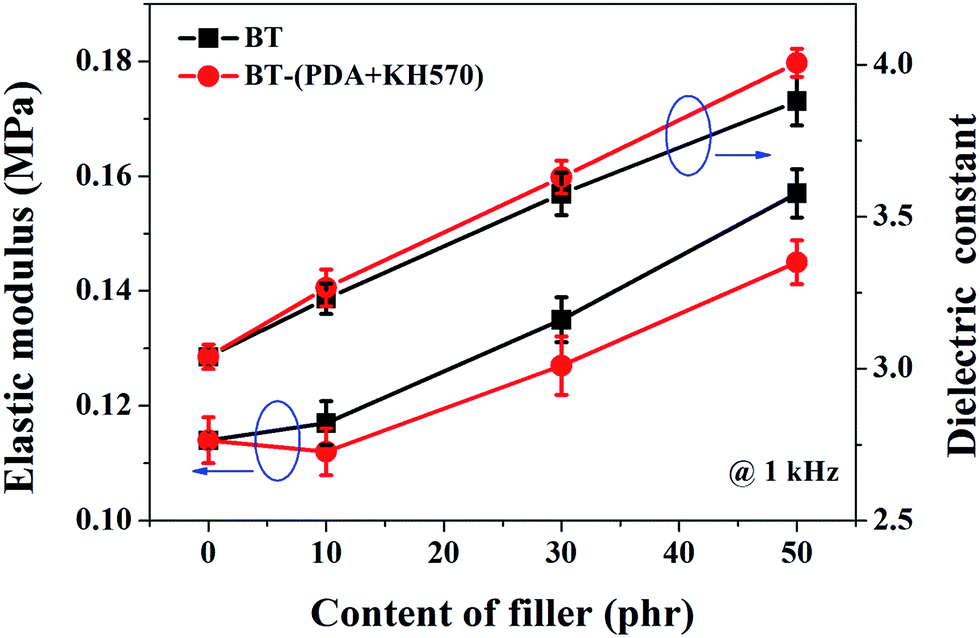 | ||
| Fig. 7 Elastic modulus and dielectric constant (1 kHz) of SR composites filled with different contents of BT and BT-(PDA + KH570) particles. | ||
 | ||
| Fig. 8 Schematic of dispersion of (a) BT particles and (b) BT-(PDA + KH570) particles in SR composites. | ||
3.4 Dielectric properties of dielectric composites
The frequency dependence of the dielectric constant of the SR composites filled with different contents of BT-(PDA + KH570) particles is shown in Fig. 9(a). From Fig. 9(a), we can find that the spectra of the dielectric constant of the composites are almost flat (the dielectric constant is frequency independent) in the entire observed frequency range, which indicate there is no flow of current caused by the accumulation of charges at the polymer/filler interface. Usually, the dielectric constant depends on frequency, especially at low frequencies, because of interfacial polarization (Maxwell–Wagner polarization), as shown in the literature.51,52 The PDA + KH570 layer surrounding the filler particles, which acts as an intermediate layer, prevents localized Maxwell–Wagner polarization between the matrix and the filler particles.53–55 In addition, the dielectric constant of SR composites filled with different contents of BT and BT-(PDA + KH570) particles at a frequency of 1 kHz are shown in Fig. 7. As expected, the dielectric constant increases with an increase in the content of BT particles, whether they are coated with PDA + KH570 layer or not. Due to the uniform dispersion of BT-(PDA + KH570) particles in the SR matrix, the dielectric constant of the composites filled with BT-(PDA + KH570) particles is higher than that of the composites filled with BT at the same filler content, which is an indication that BT-(PDA + KH570) particles are more effective in increasing the dielectric constant. The dielectric loss tangent spectra in Fig. 9(b) show that the dielectric loss tangent of all the samples is lower than 0.055 at all frequencies, which indicates that most of the electric energy can be converted into mechanical energy with just a little energy dissipation through dielectric loss during actuation. In addition, we can find that the dielectric loss tangent of the BT-(PDA + KH570)/SR composites increases with an increase in the content of BT-(PDA + KH570) particles. This may be due to the improvement of hydroxyl groups and indole groups resulting from PDA, which contributes to improve the dielectric loss tangent.29 However, we can find that the dielectric loss tangent of the composites decrease with an increase in frequency because the relaxation times of the polarization of the composites are not enough to catch up with the frequency change of the electric field.51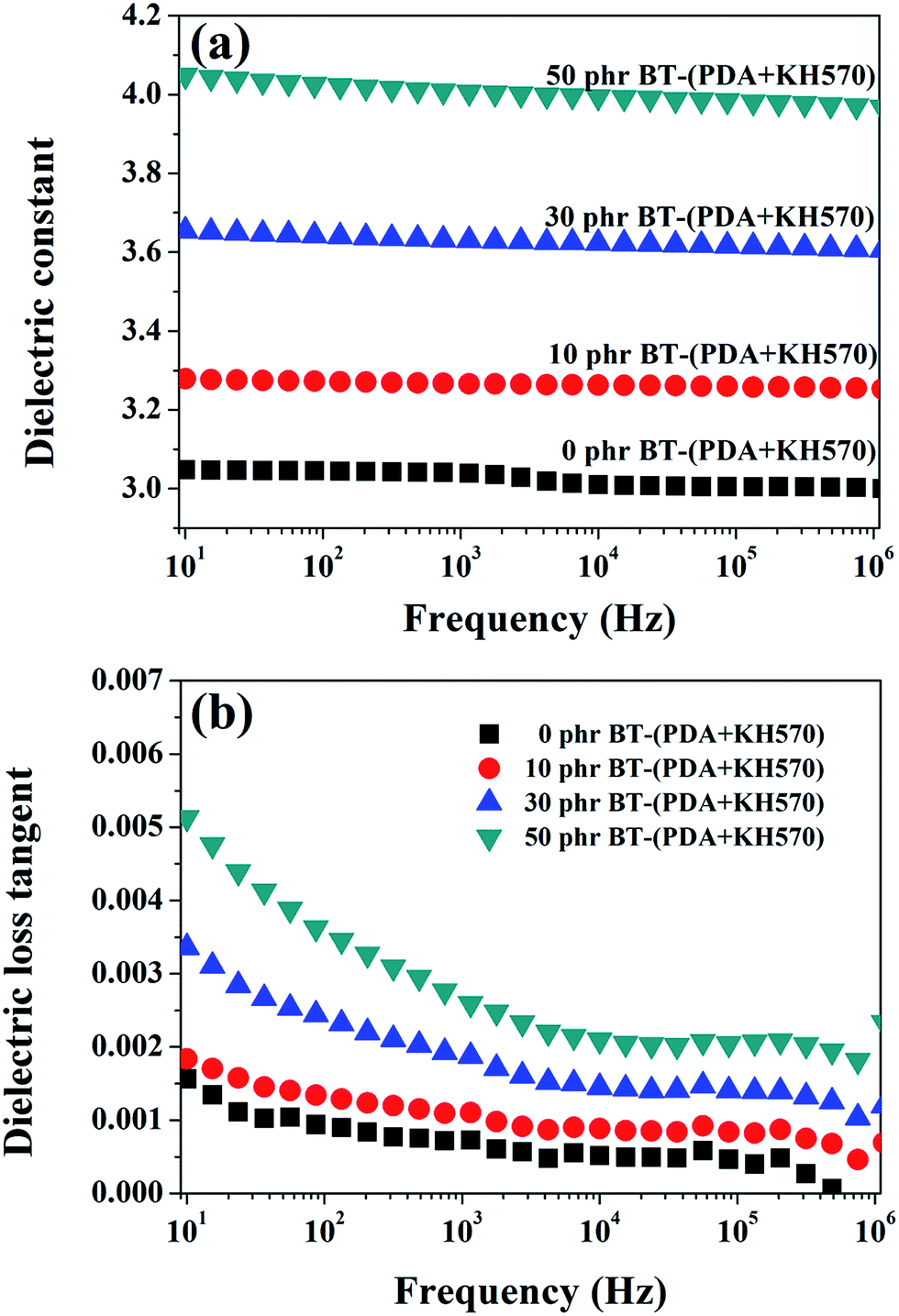 | ||
| Fig. 9 Dependence of (a) dielectric constant and (b) dielectric loss tangent of SR composites on frequency at different contents of BT-(PDA + KH570) particles. | ||
3.5 Actuated strain of dielectric composites
Fig. 10 dispalys the actuated strains of the SR composites filled with different contents of BT and BT-(PDA + KH570) particles against an applied electric field. From Fig. 10, we can easily see that the SR composites filled with 10 phr BT particles, whether they are modified or not, display the highest actuated strain at a given electric field, which is ascribed to the high electromechanical sensitivity, β (shown in Fig. 11). This can be explained by the fact that the dielectric constant of the SR composite increases whereas the elastic modulus changes a little with the addition of 10 phr dielectric filler because no obvious filler network exists in the system. In addition, we can find that the SR composites filled with BT-(PDA + KH570) particles exhibit higher actuated strain than the SR composites filled with pristine BT particles at the same electric field. This can be ascribed to the fact that the BT-(PDA + KH570)/SR composites exhibit a lower elastic modulus and higher dielectric constant compared with the BT/SR composites at the same filler content, thus leading to a larger electromechanical sensitivity β = εr/Y. Finally, the SR composites filled with 10 phr BT-(PDA + KH570) particles display the largest actuated strain of 8.23% at about 70 kV mm−1 without any pre-strains (see Table 1).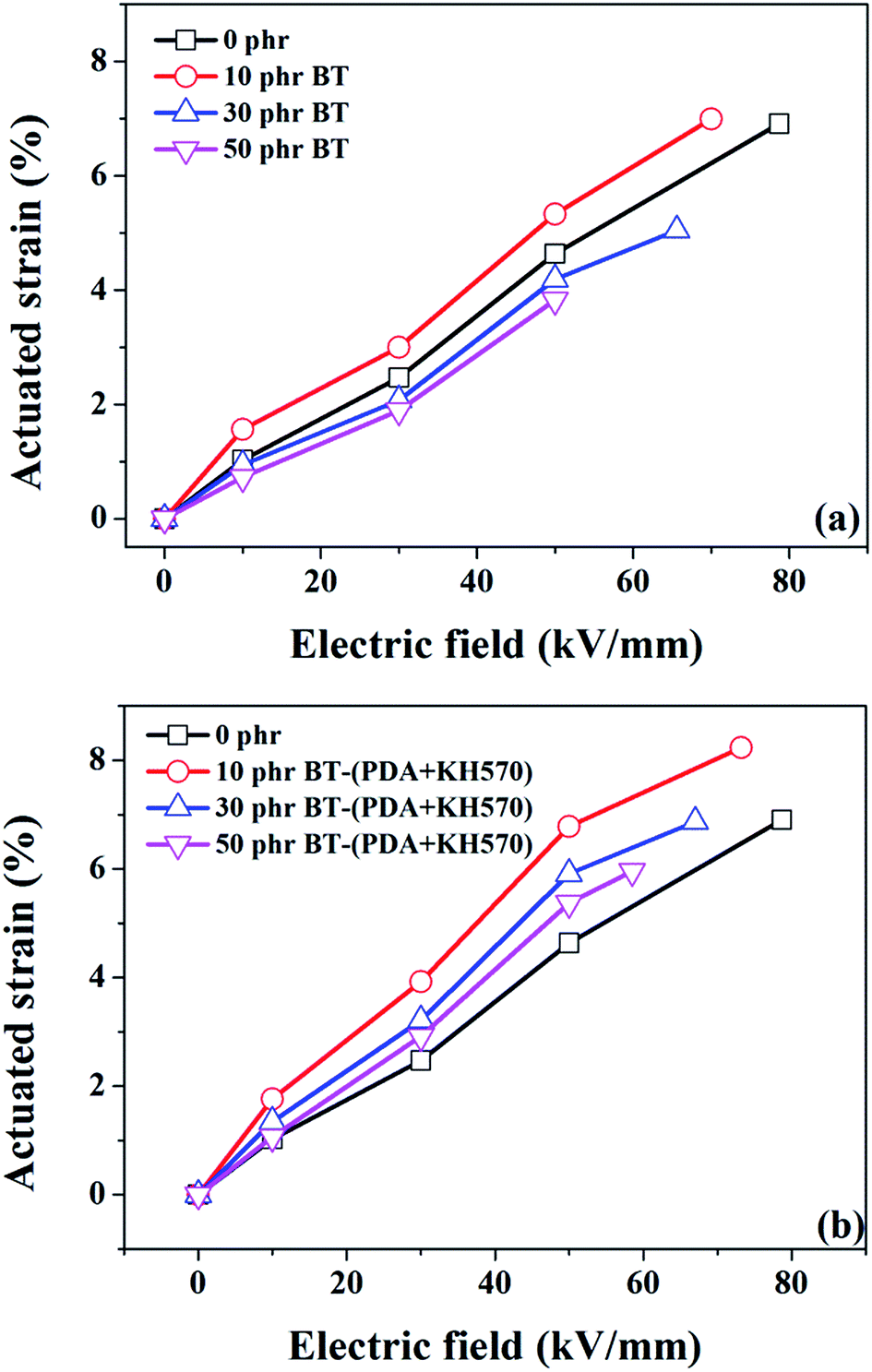 | ||
| Fig. 10 Dependence of actuated strain of SR composites filled with different contents of (a) BT and (b) BT-(PDA + KH570) particles on electric field. | ||
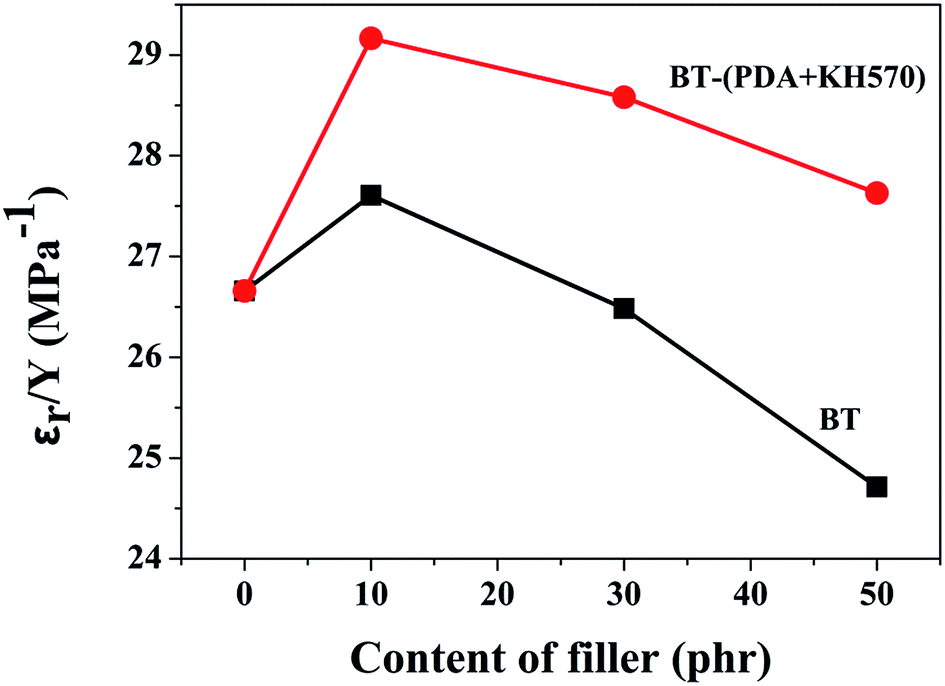 | ||
| Fig. 11 Electromechanical sensitivity of SR composites filled with different contents of BT and BT-(PDA + KH570) particles. | ||
Usually, the electrical breakdown field of composites decreases after they are filled with inorganic filler. In our work, the electrical breakdown field of the SR composites filled with BT, whether they are modified or not, decreases with an increase in the content of filler, which is in agreement with previous studies.29,56 Two reasons could be responsible for this phenomenon. First, with an increase in the content of filler in the system, more and more charged species are generated in the interface between the polymer matrix and filler, thus leading to the generation of interfacial charge to break down the system.6,57 Second, agglomerations of filler particles in the system will introduce defect centers which distort and enhance the local electric field.57,58 In addition, the BT-(PDA + KH570)/SR composites display a higher electrical breakdown field compared with the BT/SR composites at the same filler content, which is ascribed to the fact that the PDA + KH570 layer minimizes the concentration and mobility of charge carriers normally associated with the surface. This reduces leakage currents and sources of runaway conduction which can lead to high dielectric breakdown.29,59
As is known, history dependence, which is an important property of DEAs, such as pumps and sound generators, was also measured in our work. Fig. 12 shows the time dependence of the actuated strain of the 10 phr BT-(PDA + KH570)/SR composite actuator under an electric field of 30 kV mm−1 for 10 cycles without any pre-strains. Each application period of 10 seconds was followed by an off-interval of 3 seconds. We can find that the actuated strain is relatively stable with time, which is a desirable feature of the BT-(PDA + KH570)/SR composite.
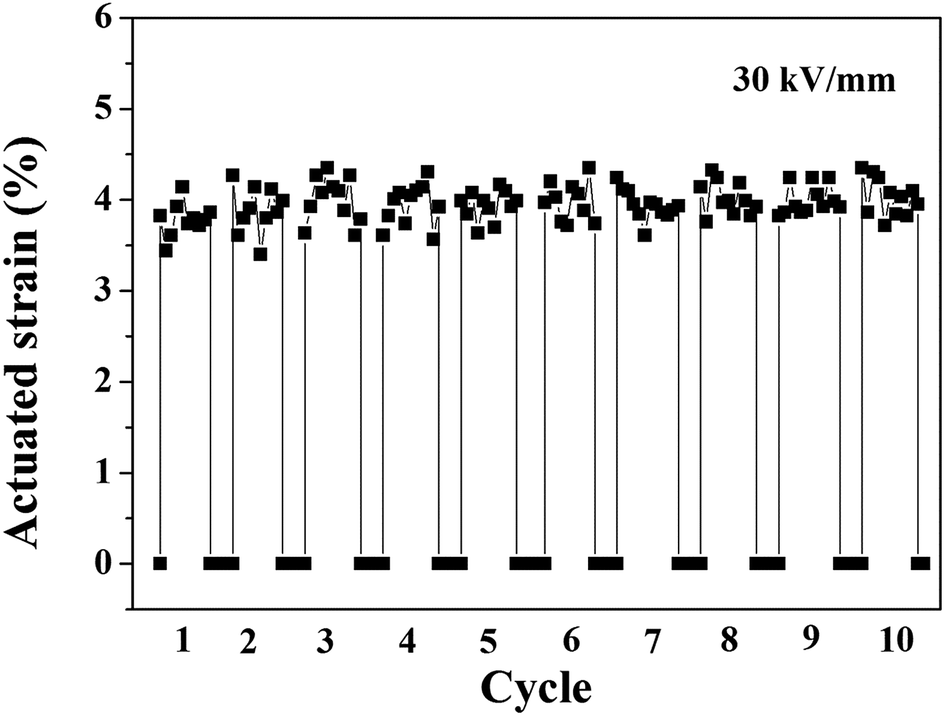 | ||
| Fig. 12 Time dependence of actuated strain of SR composite filled with 10 phr BT-(PDA + KH570) particles actuator under an electric field of 30 kV mm−1 for 10 cycles. | ||
4 Conclusion
BT particles coated with a continuous and uniform organic layer were successfully fabricated by bio-inspired dopamine deposition and KH570 grafting. The process is fast, simple, efficient, nontoxic, and controllable. The PDA layer grafted with KH570 not only improves the dispersion of fillers in the silicone elastomer matrix but also helps in the formation of strong interfacial interaction between the BT particles and silicone elastomer matrix, thus leading to an improved dielectric constant and actuated strain. This research might provide a new universal method for the modification of dielectric fillers to improve the electromechanical properties of dielectric elastomers.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51503019, No. 51373026, and No. 51573020), Beijing Natural Science Foundation (No. 2162014), Youth Backbone Personal Project of Beijing (No. 2014000020124G085), Undergraduate URT project (No. 2016J00137). Innovation Promotion Project of Beijing Municipal Commission of Education (TJSHG201310017034), Project of Petrochina (No. PRIKY14041).References
- C. Tugui, S. Vlad, M. Iacob, C. D. Varganici, L. Pricop and M. Cazacu, Polym. Chem., 2016, 7, 2709 RSC
.
- K. B. Subramani, E. Cakmak, R. J. Spontak and T. K. Ghosh, Adv. Mater., 2014, 26, 2949 CrossRef CAS PubMed
- J. Zhou, L. Jiang and R. E. Khayat, Soft Matter, 2015, 11, 2983 RSC
- H. Sun, C. Jiang, N. Ning, L. Zhang, M. Tian and S. Yuan, Polym. Chem., 2016, 7, 4072 RSC
- R. Pelrine, R. Kornbluh, Q. Pei and J. Joseph, Science, 2000, 287, 836 CrossRef CAS PubMed
- Y. Yao, N. Ning, L. Zhang, T. Nishi and M. Tian, RSC Adv., 2015, 5, 23719 RSC
- M. H. Jomaa, L. Seveyrat, L. Lebrun, K. Masenelli-Varlot and J. Y. Cavaille, Polymer, 2015, 63, 214 CrossRef CAS
- A. H. A. Razak, P. Szabo and A. L. Skov, RSC Adv., 2015, 5, 53054 RSC
- S. J. Dünki, Y. S. Ko, F. A. Nüesch and D. M. Opris, Adv. Funct. Mater., 2015, 25, 2467 CrossRef
- H. Stoyanov, M. Kollosche, D. N. McCarthy and G. Kofod, J. Mater. Chem., 2010, 20, 7558 RSC
- H. Stoyanov, M. Kollosche, S. Risse, R. Waché and K. Guggi, Adv. Mater., 2013, 25, 578 CrossRef CAS PubMed
- F. B. Madsen, A. E. Daugaard, S. Hvilsted and A. L. Skov, J. Mater. Chem. C, 2016, 37, 378 CAS
- L. Zhang, D. Wang, P. Hu, J. W. Zha, F. You, S. T. Li and Z. M. Dang, J. Mater. Chem. C, 2015, 3, 4883 RSC
- M. Tian, B. Yan, Y. Yao, L. Zhang, T. Nishi and N. Ning, J. Mater. Chem. C, 2014, 2, 8388 RSC
- S. Risse, B. Kussmaul, H. Krüger and G. Kofod, Adv. Funct. Mater., 2012, 22, 3958 CrossRef CAS
- B. Kussmaul, S. Risse, G. Kofod, R. Waché, M. Wegener, D. N. McCarthy, H. Krüger and R. Gerhard, Adv. Funct. Mater., 2011, 21, 4589 CrossRef CAS
- F. B. Madsen, L. Yu, A. E. Daugaard, S. Hvilsted and A. L. Skov, RSC Adv., 2015, 5, 10254 RSC
- C. Racles, M. Cazacu, B. Fischer and D. M. Opris, Smart Mater. Struct., 2013, 22, 104004 CrossRef
- F. B. Madsen, L. Yu, A. E. Daugaard, S. R. Hvilsted and A. L. Skov, Polymer, 2014, 55, 6212 CrossRef CAS
- T. Chen, J. Qiu, K. Zhu, J. Li, J. W. Wang, S. Q. Li and X. Wang, J. Phys. Chem. B, 2015, 119, 4521 CrossRef CAS PubMed
- D. Opris, S. Dünki, M. Tress, F. Kremer, Y. S. Ko, F. Nueesch, C. D. Varganici and C. Racles, RSC Adv., 2015, 5, 50054 RSC
- D. Yang, M. Tian, W. Wang, D. Li, R. Li, H. Liu and L. Zhang, Electrochim. Acta, 2013, 87, 9 CrossRef CAS
- Y. Zhang, P. Huo, J. Wang, X. Liu, C. Rong and G. Wang, J. Mater. Chem. C, 2013, 1, 4035 RSC
- W. Hu, S. N. Zhang, X. Niu, C. Liu and Q. Pei, J. Mater. Chem. C, 2014, 2, 1658 RSC
- J. Lu, K. S. Moon, J. Xu and C. Wong, J. Mater. Chem., 2006, 16, 1543 RSC
- J. K. Yuan, Z. M. Dang, S. H. Yao, J. W. Zha, T. Zhou, S. T. Li and J. Bai, J. Mater. Chem., 2010, 20, 2441 RSC
- B. H. Fan, J. W. Zha, D. Wang, J. Zhao and Z. M. Dang, Appl. Phys. Lett., 2012, 100, 012903 CrossRef
- J. Li, S. I. Seok, B. Chu, F. Dogan, Q. Zhang and Q. Wang, Adv. Mater., 2009, 21, 217 CrossRef CAS
- Z. M. Dang, Y. J. Xia, J. W. Zha, J. K. Yuan and J. B. Bai, Mater. Lett., 2011, 65, 3430 CrossRef CAS
- H. Tang, Y. Lin and H. A. Sodano, Adv. Energy Mater., 2013, 3, 451 CrossRef CAS
- J. Li, P. Khanchaitit, K. Han and Q. Wang, Chem. Mater., 2010, 22, 5350 CrossRef CAS
- P. Kim, S. C. Jones, P. J. Hotchkiss, J. N. Haddock, B. Kippelen, S. R. Marder and J. W. Perry, Adv. Mater., 2007, 19, 1001 CrossRef CAS
- P. Kim, N. M. Doss, J. P. Tillotson, P. J. Hotchkiss, M. J. Pan, S. R. Marder, J. Li, J. P. Calame and J. W. Perry, ACS Nano, 2009, 3, 2581 CrossRef CAS PubMed
- P. Hu, Y. Shen, Y. Guan, X. Zhang, Y. Lin, Q. Zhang and C. W. Nan, Adv. Funct. Mater., 2014, 24, 3172 CrossRef CAS
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426 CrossRef CAS PubMed
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338 CrossRef CAS PubMed
- L. Q. Xu, W. J. Yang, K. G. Neoh, E. T. Kang and G. D. Fu, Macromolecules, 2010, 32, 684 Search PubMed
- N. Ning, Q. Ma, S. Liu, M. Tian, L. Zhang and T. Nishi, ACS Appl. Mater. Interfaces, 2015, 7, 10755 CAS
- M. F. Lin, V. K. Thakur, E. J. Tan and P. S. Lee, RSC Adv., 2011, 1, 576 RSC
- J. Liang, A. Betts, D. Kennedy and S. Jerrams, Mater. Des., 2015, 85, 733 CrossRef
- Y. Fu, L. Liu, L. Zhang and W. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 5105 CAS
- D. Yang, M. Ruan, S. Huang, Y. Wu, S. Li, H. Wang, Y. Shang, B. Li, W. Guo and L. Zhang, J. Mater. Chem. C, 2014, 4, 7724 RSC
- K. Chawla, S. Lee, B. P. Lee, J. L. Dalsin, P. B. Messersmith and N. D. Spencer, J. Biomed. Mater. Res., Part A, 2009, 90, 742 CrossRef PubMed
- R. Sa, Z. Wei, Y. Yan, L. Wang, W. Wang, L. Zhang, N. Ning and M. Tian, Compos. Sci. Technol., 2015, 45, 54 CrossRef
- G. B. Shah, M. Fuzail and J. Anwar, J. Appl. Polym. Sci., 2004, 92, 3796 CrossRef CAS
- D. Maldas, B. V. Kokta and C. Daneault, J. Appl. Polym. Sci., 1989, 37, 751 CrossRef CAS
- Y. Xie, C. A. S. Hill, Z. Xiao, H. Militz and C. Mai, Composites, Part A, 2010, 41, 806 CrossRef
- Z. Wang, J. Liu, S. Wu, W. Wang and L. Zhang, Phys. Chem. Chem. Phys., 2010, 12, 3014 RSC
- D. Yang, M. Tian, D. Li, W. Wang, F. Ge and L. Zhang, J. Mater. Chem. A, 2013, 1, 12276 CAS
- D. Yang, S. Huang, Y. Wu, M. Ruan, S. Li, Y. Shang, X. Cui, Y. Wang and W. Guo, RSC Adv., 2015, 5, 65385 RSC
- Z. M. Dang, H. P. Xu and H. Y. Wang, Appl. Phys. Lett., 2007, 90, 012901 CrossRef
- H. Liu, L. Zhang, D. Yang, Y. Yu, L. Yao and M. Tian, Soft Mater., 2013, 11, 363 CrossRef CAS
- H. Stoyanov, M. Kollosche, S. Risse, D. N. McCarthy and G. Kofod, Soft Matter, 2011, 7, 194 RSC
- G. Gallone, F. Carpi, D. De Rossi, G. Levita and A. Marchetti, Mater. Sci. Eng., C, 2007, 27, 110 CrossRef CAS
- D. Mc Carthy, S. Risse, P. Katekomol and G. Kofod, J. Phys. D: Appl. Phys., 2009, 42, 145406 CrossRef
- T. Zhou, J. W. Zha, R. Y. Cui, B. H. Fan, J. K. Yuan and Z. M. Dang, ACS Appl. Mater. Interfaces, 2011, 3, 2184 CAS
- S. Nayak, B. Sahoo, T. K. Chaki and D. Khastgir, RSC Adv., 2013, 3, 2620 RSC
- D. Yang, M. Tian, Y. Dong, H. Liu, Y. Yu and L. Zhang, Smart Mater. Struct., 2012, 21, 035017 CrossRef
- X. Dou, X. Liu, Y. Zhang, H. Feng, J. F. Chen and S. Du, Appl. Phys. Lett., 2009, 95, 132904 CrossRef
| This journal is © The Royal Society of Chemistry 2016 |