
Adsorption of asphaltenes on the calcite (10.4) surface by first-principles calculations
Raphael S. Alvimab,
Filipe C. D. A. Lima b,
Verónica M. Sánchezac,
Thomas F. Headend,
Edo S. Boeke and
Caetano R. Miranda*ab
b,
Verónica M. Sánchezac,
Thomas F. Headend,
Edo S. Boeke and
Caetano R. Miranda*ab
aCentro de Ciências Naturais e Humanas, Universidade Federal do ABC, Santo André, SP 09210-580, Brazil
bDepartamento de Física dos Materiais e Mecânica, Universidade de São Paulo, São Paulo, SP 05508-090, Brazil. E-mail: cmiranda@if.usp.br
cCSC, CONICET, Godoy Cruz 2390, 1425, Buenos Aires, Argentina
dISIS Neutron and Muon Source, Rutherford Appleton Laboratory, Harwell Campus, Didcot, UK
eDepartment of Chemistry, University of Cambridge, UK
First published on 20th September 2016
Abstract
Asphaltenes play a key role in oil production and its exploration from natural reservoirs. In carbonate reservoirs, the calcite (10.4) surface retains asphaltenes. However, its aggregate structure and deposition process are not fully understood. Using first-principles calculations based on density-functional theory (DFT) with van der Waals (vdW) dispersion, we studied the adsorption of asphaltene, resin and resin–asphaltene dimer molecular models on the CaCO3 surface in the presence of a dielectric water–toluene environment. These large molecules impose a challenging description at the electronic level. Our calculations indicate that there is a minor steric hindrance in the effective interaction of the aromatic region of asphaltene on the calcite surface. However, aliphatic chains with sulphide groups can play a significant role on the adsorption process and its availability to receive electronic charge density from the surface. Accordingly, the preferential LUMO localized in the aromatic region of asphaltene may also allow the adsorption on the calcite surface and π–π stacking interactions. Initially, the resin molecule tends to be trapped during dimer formation with the asphaltene, whereas a significant intramolecular charge rearrangement due to the heteroatoms is necessary to increase the π–π stacking interactions. For the dimer, the adsorbed form of asphaltene favors more available electronic states to increase the likelihood of nanoaggregation. Therefore, changes in the continuum dielectric constant only had a minor effect on the calculated adsorption energies. Experimental work related to the oil–water interface in the presence of toluene show similar behavior during asphaltene adsorption. Our studies indicate that nanoaggregates are grown through resin and the calcite (10.4) surface selectively adsorbs the less polar asphaltenes from oil.
1 Introduction
Asphaltenes are the macromolecular part of natural crude oil, defined by its insolubility in n-heptane and solubility in aromatic solvents such as toluene.1–4 The molecules aggregate in good solvents forming ∼10 nm-sized aggregates.1–3 Asphaltene molecules have a relatively high molecular weight and are heteroatom-rich. They are the densest part of oil with polar and non-polar groups giving some amphiphilic character. Furthermore, these compounds possess an ability to aggregate and adsorb to oil–water (hydrophobic–hydrophilic) interfaces to stabilize water-in-oil emulsions even at low concentrations.The presence of high concentrations of asphaltene aggregates at the bottom of a reservoir can provide more complex upgrading technologies of many types of by-products in the refining of heavy crude oils, such as the bitumen,5–7 or yield a lighter crude oil. Furthermore, some precipitating agents for asphaltenes, such as high-pressure reservoir gases, light paraffins and resins, may cause the partial or complete blockage of flow in the recovery and movement of petroleum through pipes, causing problems in the well and extraction facilities. For this reason, many structural features of these organic compounds have been extensively studied.4,8–11
Mainly due to their chemical composition, including polyaromatic and polycyclic ring containing heteroatoms (N, O, S and metals), asphaltene molecules are susceptible to intermolecular forces, which are responsible for their strong propensity to self-associate, form aggregates and to adsorb on the mineral surfaces of rocks.11,12 In this context, resin molecules may play an important role in bridging the gap between the solubility of asphaltenes in polar and non-polar oils.
The resins consist of molecules of intermediate polarity similar to asphaltenes, differing by a reduced number of aromatic structures. They may be regarded as an important component for stabilizing asphaltenes due to the solubility of asphaltenes with greater polarity and the non-polar moieties in the saturated oil matrix.13 These forces of attraction are mainly acid–base interactions, hydrogen bonds in the presence of water, coordination complexes, π–π stacking and van der Waals (vdW) interactions in hydrophobic regions.3,14 The π–π stacking interactions are the most dominant force for asphaltene aggregation. This type of interaction can be inhibited in the adsorption on the rock surface because of the significant steric hindrance.
Minerals are able to retain asphaltenes and therefore play an important role in the development of enhanced oil recovery (EOR) operations, mainly in natural reservoir rocks.15–18 Even in the absence of water, asphaltene adsorption on mineral surfaces is driven by polar interactions, surface precipitation and H-bond formation.17,18 The adsorption process produces marked changes in the wettability of the solid and reduces oil production from the reservoir.3,19–22
When compared to the bare mineral surface, González et al.19 observed an increase in the contact angle at the mineral–water–toluene solution interface for asphaltene-covered surfaces, such as calcite. On the other hand, for low asphaltene concentrations in toluene, there was a decrease in the contact angle despite the adsorption of asphaltene on the mineral surface and the consequent purification of the organic solvent. An earlier study20 showed an increase in the contact angle measured at the calcite–water–toluene interface from the greater calcite surface covered by asphaltene rather by resin molecules.
Asphaltenes are the primary species that adsorb on the surface of calcite (CaCO3),12 in which the most stable cleavage plane is denoted by the (10.4) surface. Accordingly, highly aromatic and polar asphaltenes are thought to be more prone to adsorb through the natural chemical composition in the asphaltene and resin molecules. It is believed that asphaltenes adsorb primarily by interactions between their aromatic and heteroatom groups, and the Ca2+ and CO32− sites present on the calcite (10.4) surface. This adsorption phenomenon may be associated with the colloidal interactions responsible for the adsorption of nanoaggregates.17,18 The adsorption processes may also be influenced by organic solvents in crude oil.15,23
Computer simulation techniques have been used to study the main molecular representations of the nanoaggregates of asphaltenes and resins from their adsorption on mineral surfaces.1,2 The adsorption of asphaltenes, resins and their aggregates onto solid surfaces is focused on the understanding of the process phenomenology that leads to the formation of these organic nanoaggregates. This aggregation process in solution could compete with adsorption.16 In order to simulate natural conditions, we used asphaltenes from water–toluene as the solvent and calcite as the adsorbent of the rock component.
Our aim is to study at the electronic level using first-principles calculations, with vdW forces included, the major oil interaction mechanisms between the asphaltene and resin molecules and the surface of CaCO3 as well as the adsorption process of the dimers formed. We are interested in evaluating the electrostatic arrangement of the adsorption processes of an isolated molecule and dimer of asphaltene on the calcite surface in the absence of the collective effects seen in asphaltene clusters. Therefore, the adsorption mechanisms of these molecules on the surface of minerals can be the basis for understanding a proper chemical mechanism for the different association nanoaggregates of asphaltene and resins molecules on the carbonate rock present in oil reservoirs.
2 Computational procedure
The calcite (10.4) surface with asphaltene and resin models was investigated at the density-functional theory (DFT)24 level with the generalized gradient approximation (GGA)25,26 implemented in the Quantum Espresso27–29 package. We used a plane wave basis set, periodic boundary conditions30 and Vanderbilt ultrasoft pseudopotentials.31 We used the exchange–correlation functional revised by Perdew, Burke and Ernzerhof (revPBE).32–34 In revPBE, it is allowed to properly apply vdW density functional (vdW-DF)35–37 including dispersion interactions to better describe the long-range vdW forces at the molecule–surface and dimer–surface interfaces.Calcite bulk is a trigonal crystal system with space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) C. The CaCO3 structure was built from a hexagonal unit cell with the lattice parameters calculated as a = b = 5.10 Å, c = 17.16 Å, α = β = 90.00° and γ = 120.00°. From the cleavage of this structure, the cubic unit cell of area (2 × 1) was replicated for the adsorption of the asphaltene molecules and the their dimer on the most stable plane (10.4). The lattice parameters were calculated as a = 29.94 Å, b = 23.47 Å and c = 38.63 Å. We used the tested vacuum layer with a little more than 30.00 Å to isolate the top of the asphaltene molecules from the bottom of the next calcite layer. The surface was thus represented as a three-layer super-cell (side view in Fig. 1A).
C. The CaCO3 structure was built from a hexagonal unit cell with the lattice parameters calculated as a = b = 5.10 Å, c = 17.16 Å, α = β = 90.00° and γ = 120.00°. From the cleavage of this structure, the cubic unit cell of area (2 × 1) was replicated for the adsorption of the asphaltene molecules and the their dimer on the most stable plane (10.4). The lattice parameters were calculated as a = 29.94 Å, b = 23.47 Å and c = 38.63 Å. We used the tested vacuum layer with a little more than 30.00 Å to isolate the top of the asphaltene molecules from the bottom of the next calcite layer. The surface was thus represented as a three-layer super-cell (side view in Fig. 1A).
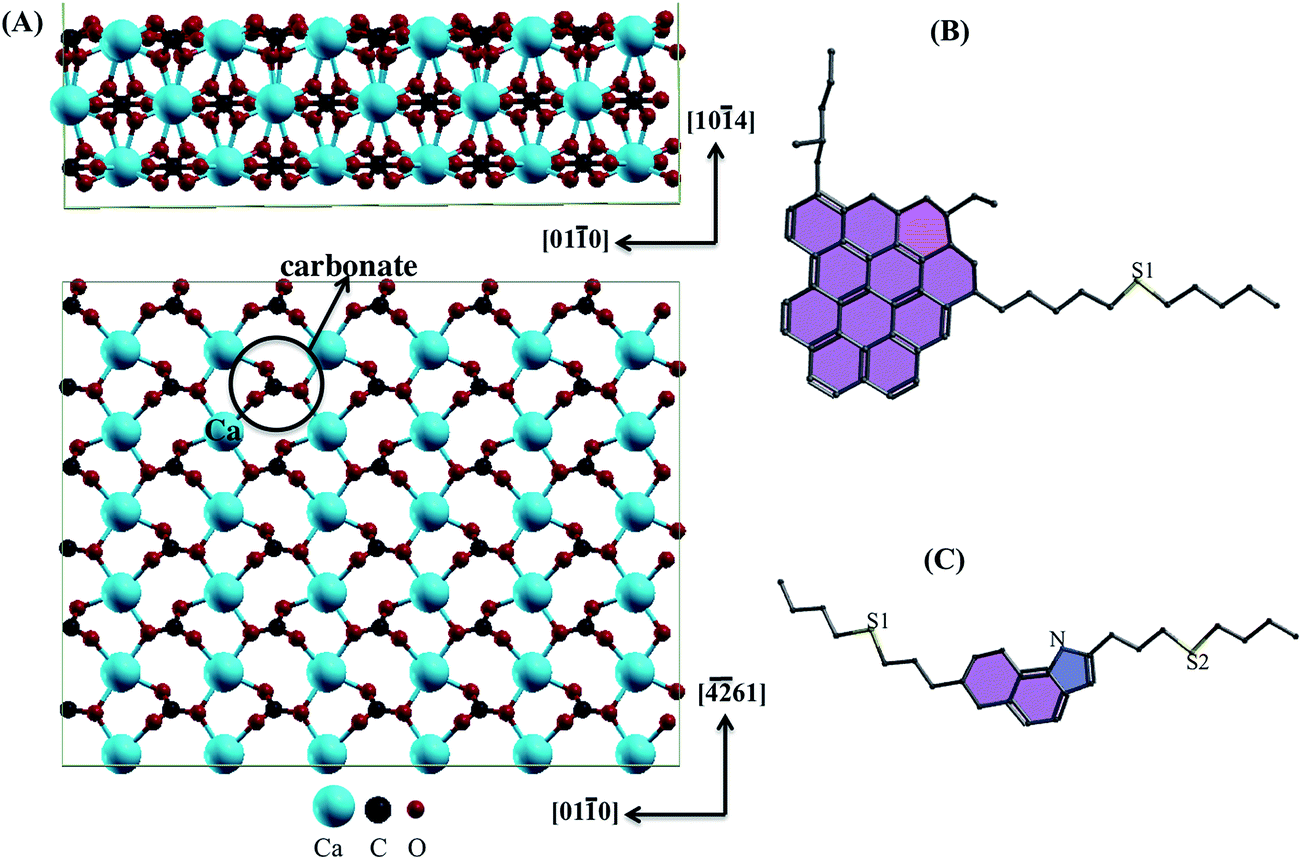 | ||
| Fig. 1 (A) Side- and top-views of the super-cell of calcite on the (10.4) surface. Projections along the of ring region of the molecular models parallel to the calcite (10.4) surface (in the absence of hydrogen atoms) of the (B) asphaltene and (C) resin models. The five and six-membered rings are light purple and pink, respectively. The sulfur sites are shown as S1 and S2. | ||
Asphaltenes are very complex mixtures. However, in order to make the problem tractable for simulation methods one must assume some representative structures. Asphaltene (Fig. 1B) and resin (Fig. 1C) models were obtained from the work of Boek et al.1 They used the quantitative molecular representation (QMR) method to build asphaltene molecules from aromatic, aliphatic groups and heteroatoms such as sulfur and nitrogen. Furthermore, these molecules were selected by best match with the experimental data, including mass spectroscopy, nuclear magnetic resonance and elemental analysis.38,39 This approach is widely used in other simulation papers and use the same or similar molecular structures for asphaltenes according to the Yen–Mullins model. In particular for the sulfur position, the sulphide group is of potential interest by interacting with the calcite surface to a greater extent than thiophene.
Our calculations were performed with a converged kinetic energy cut-off of 640 eV and charge density of 4400 eV at the Γ point. Based on the trust radius procedure for the Broyden–Fletcher–Goldfarb–Shanno (BFGS)40 algorithm, the sites were allowed to relax along the calculated Hellmann–Feynman41 forces until the residual force components were smaller than 0.05 eV Å−1. In calcite, just the first surface layer was allowed to relax, i.e., all the surface sites displayed in the top-view in Fig. 1A. Thus, the two bottom surface layers were fixed in the bulk positions. Regarding the asphaltene and resin molecules, all the sites were also allowed to relax.
The adsorption energies Eads1 and Eads2 were calculated from the electronic energy differences between the molecule/dimer and the surface 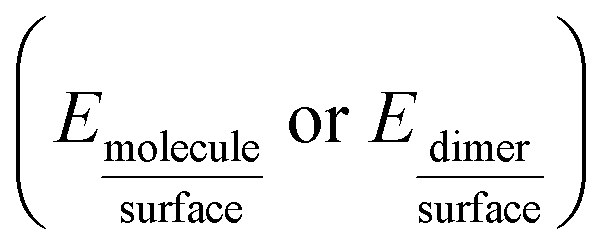 , and the pristine surface (Esurface) and the isolated molecule/dimer (Emolecule or Edimer) through:
, and the pristine surface (Esurface) and the isolated molecule/dimer (Emolecule or Edimer) through:
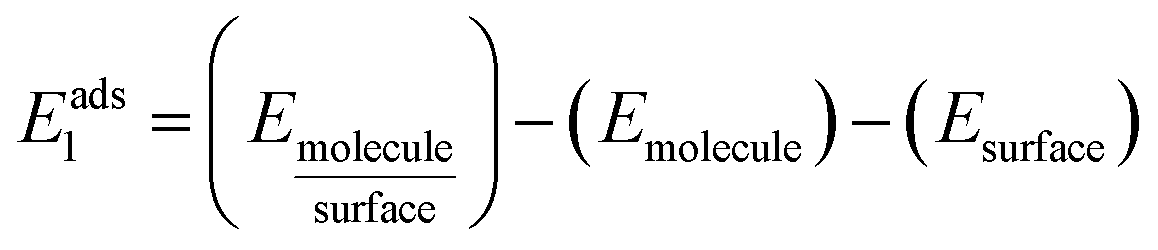
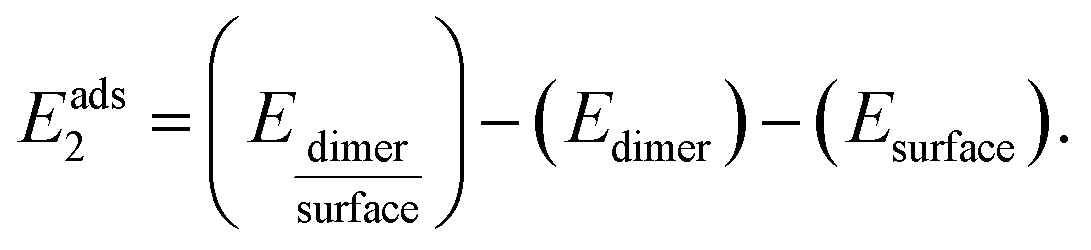
The charge density differences at the adsorption interfaces were calculated according to:
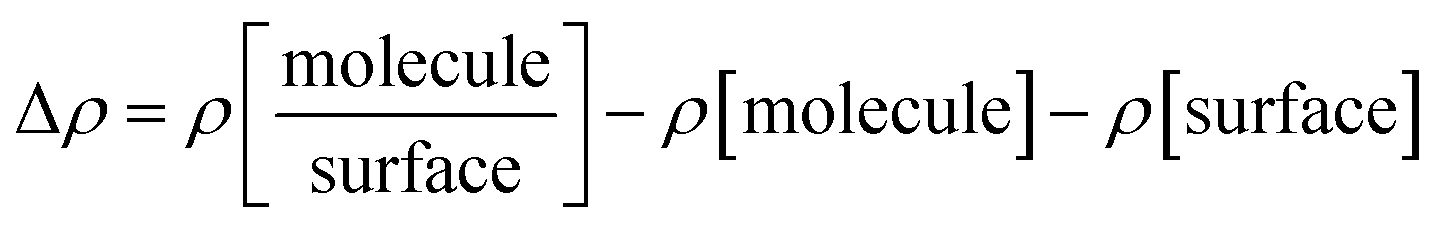
where
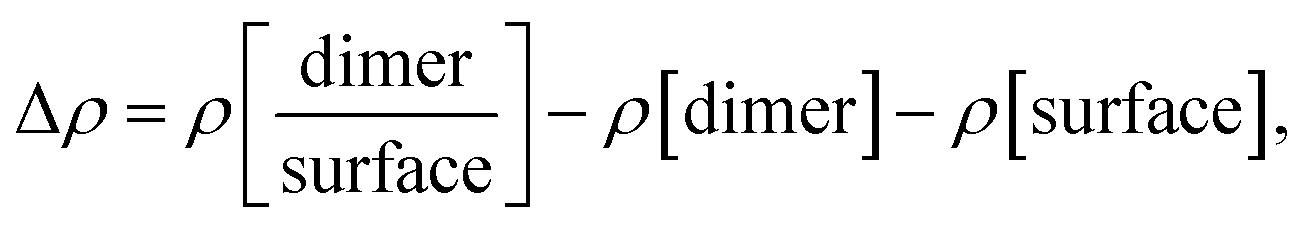
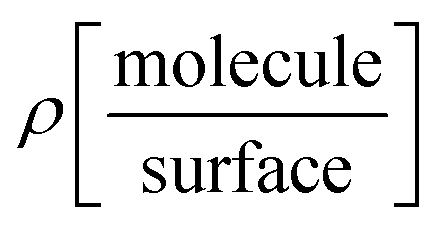 , ρ[molecule], ρ[surface],
, ρ[molecule], ρ[surface],  and ρ[dimer] are the charge densities of asphaltene or resin on calcite, the isolated asphaltene or resin, the isolated calcite surface, dimer on calcite and isolated dimer, respectively.
and ρ[dimer] are the charge densities of asphaltene or resin on calcite, the isolated asphaltene or resin, the isolated calcite surface, dimer on calcite and isolated dimer, respectively.
Charge analysis was carried out using Bader's definition available in the Bader charge analysis code,42–45 for the following valence electronic configurations at each pseudopotential: C 2s22p2, Ca 3s24s23p6, H 1s1, N 2s22p3, O 2s22p4 and S 3s23p4. Then, the charge for each element was obtained by the difference between the calculated Bader charge and number of electrons established at the valence electronic configuration of the pseudopotential cited above. Particularly for nitrogen in resin, it was compared to the charge of this element in pyridine with the same computational procedure to show the accuracy of the calculated charge.
Electrostatic effects due to the periodic boundary conditions in the first-principles quantum-mechanical simulations were calculated in vacuo and in combination with the continuum dielectric environment using the Environ46–48 package. From this, it was possible to obtain a better performance with the environment effects, in particular for applications in surface science and materials design from first-principles calculations.49,50 In these calculations, we used toluene and water with dielectric constants of 2.38 and 80.00, respectively. In these environment situations, the calculations were carried out without a new structural relaxation.
3 Results and discussion
Structural optimization was performed using the most stable coordinates of the asphaltene and resin molecules obtained from classical molecular dynamics.1 As shown in Fig. 2 in our first-principles calculations, the average distances for the aromatic and non-aromatic dC–C, dC–S and dC–N bonds and for the dmolecule–surface and dmolecule–molecule interaction distances, and the ∠C–S–C angles were obtained for isolated asphaltene, resin and their dimer, as well as both molecules and their dimer adsorbed on the calcite (10.4) surface, as shown in Table 1. Regarding the dmolecule–surface and dmolecule–molecule interaction distances, we used the center of mass of the ring island in the asphaltene and resin molecules.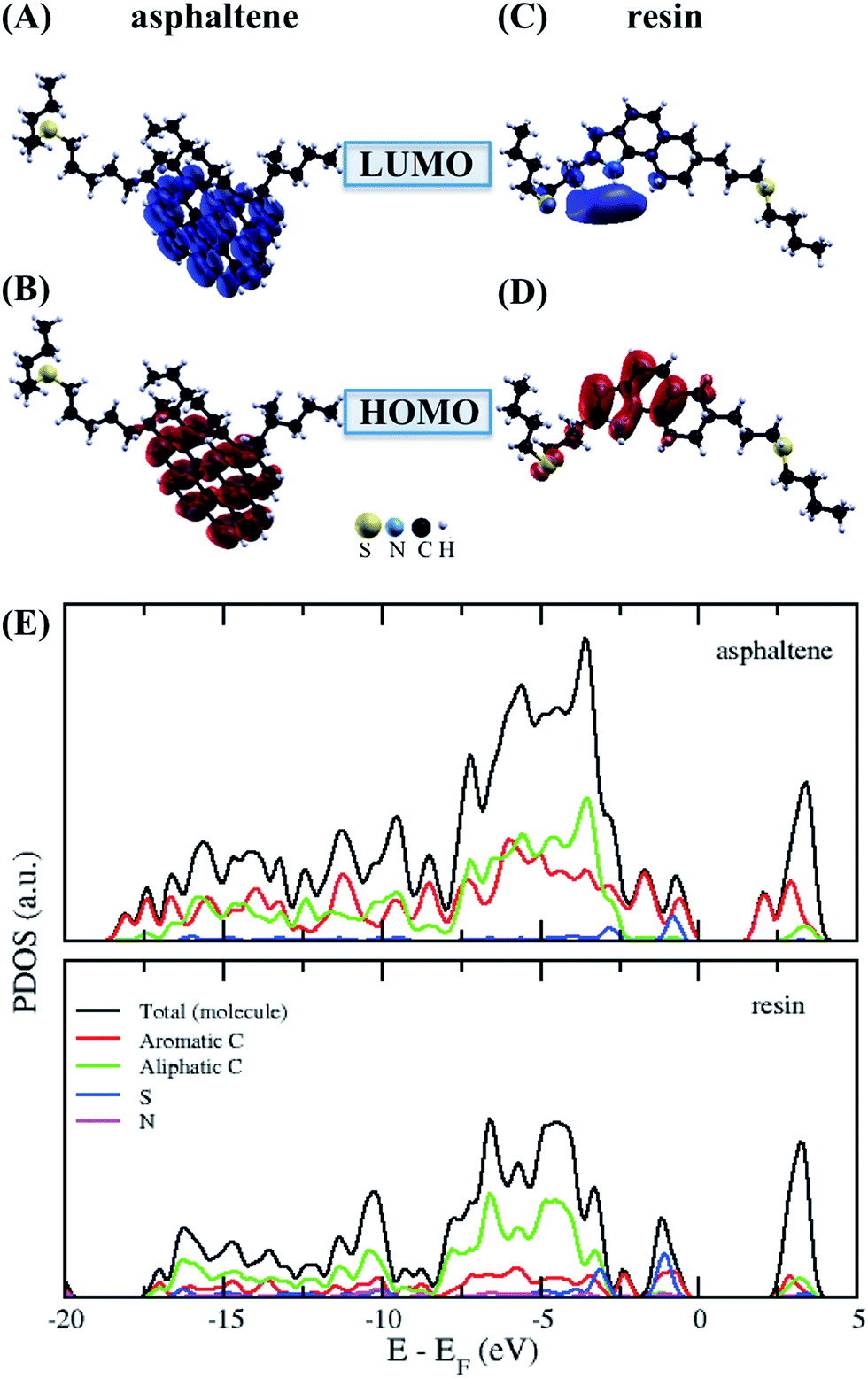 | ||
| Fig. 2 The isolated asphaltene and resin molecules. The sum of the square module of the Kohn Sham states is at the bottom of the conduction band (LUMO) and at the top of the valence band (HOMO). The (A) LUMO and (B) HOMO of asphaltene and the (C) LUMO and (D) HOMO of resin. The isosurface of −0.0007 to 0.0007 electrons per Bohr3. (E) The projected density of states (PDOS) obtained for asphaltene and resin. | ||
| Structural parameters | Molecules | Dimer | ||||||
|---|---|---|---|---|---|---|---|---|
| Asphaltene |
|
Resin |
|
|
|
|||
| Asphaltene | Resin | Asphaltene | Resin | |||||
| Aromatic dC–C | 1.42 | 1.43 | 1.41 | 1.41 | 1.47 | 1.41 | 1.42 | 1.41 |
| Aliphatic dC–C | 1.55 | 1.54 | 1.54 | 1.54 | 1.54 | 1.54 | 1.55 | 1.54 |
| dC–S | 1.84 | 1.85 | 1.84 | 1.85 | 1.85 | 1.85 | 1.84 | 1.85 |
| dC–N | — | — | 1.40 | 1.40 | — | 1.40 | — | 1.40 |
| dmolecule–surface | — | 6.13 | — | 4.72 | — | — | 10.09 | — |
| dmolecule–molecule | — | — | — | — | 5.45 | 5.45 | 6.19 | 6.19 |
| ∠C–S1–C | 102.47 | 102.60 | 101.40 | 100.98 | 101.48 | 100.41 | 104.95 | 100.88 |
| ∠C–S2–C | — | — | 102.80 | 102.90 | — | 100.61 | — | 100.77 |
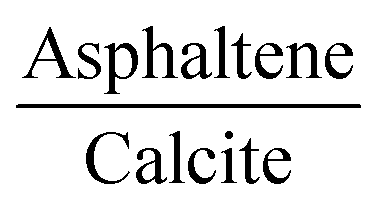
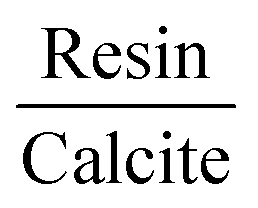
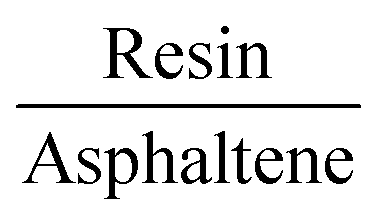

From the molecular dynamics calculations, the potential energy surface of the resin–asphaltene dimer led to a small number of energetically stable structures derived by the rotation of the resin on the asphaltene molecule. Accordingly, we chose the resin–asphaltene dimer structure shown in Fig. 3. The dimer was obtained from previous work with a potential energy surface survey with classical molecular mechanics using LAMMPS software.51 The force field employed was CHARMM.52,53 The resin was placed parallel to asphaltene at a distance of 5.00 Å. The resin was rotated from 10° to 350°. Then, the resin was translated by 0.50 Å. The same procedure was repeated until 14.00 Å. We have presented complete details regarding this and other sets of structures elsewhere.54
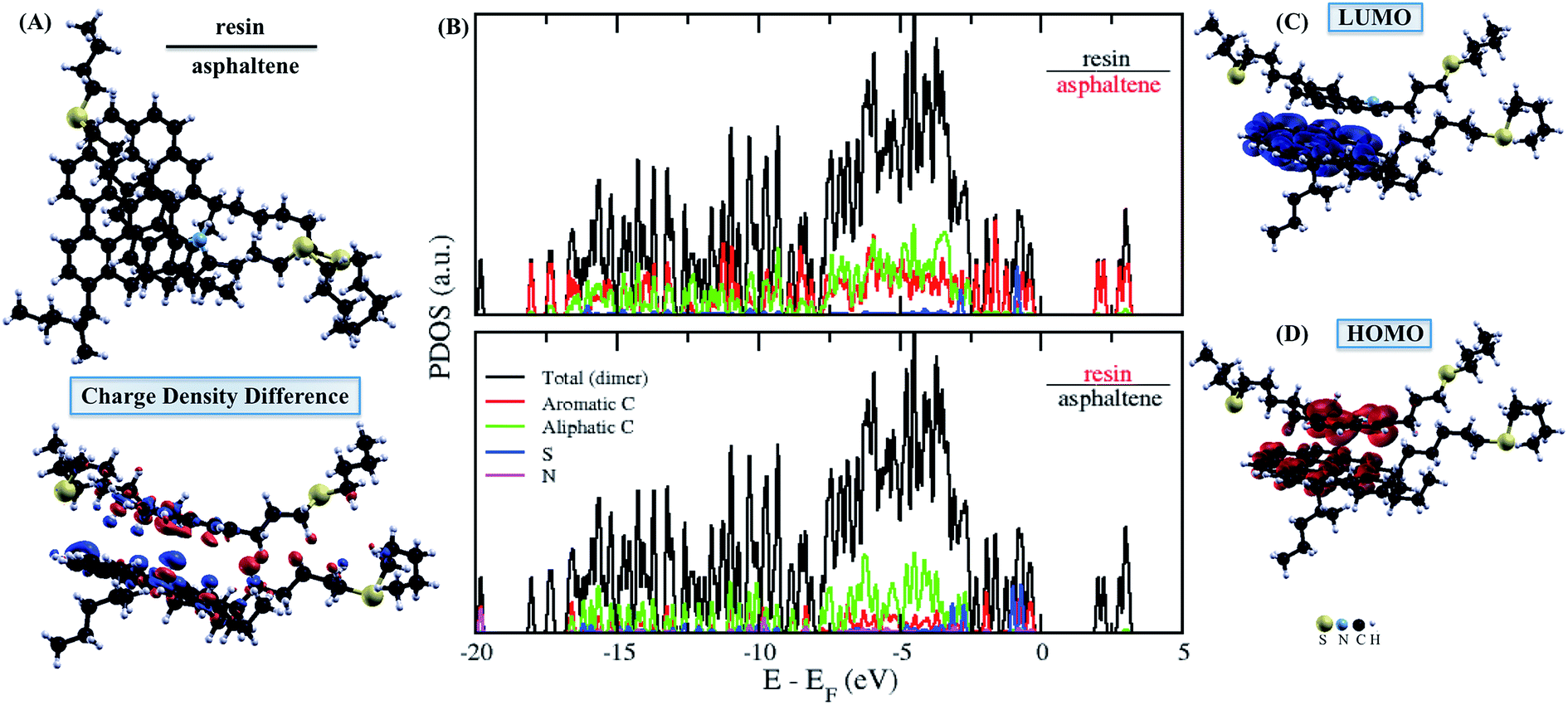 | ||
| Fig. 3 The isolated asphaltene and resin dimer molecules. (A) Top-view in up and charge density difference in down for resin–asphaltene. The red and blue colors indicate an increase and depletion in the charge density, respectively. (B) The projected density of states (PDOS) obtained for resin–asphaltene. The sum of the square module of the Kohn Sham states is (C) at the bottom of the conduction band (LUMO) and (D) at the top of the valence band (HOMO) for resin–asphaltene. The isosurface of −0.0007 to 0.0007 electrons per Bohr3. | ||
In this first analysis, we can observe that the analyzed structural parameters are rather similar in both the studied molecules even after molecular interaction and surface adsorption. Particularly, the dmolecule–surface distance was the parameter most affected due to the interaction influence onto the aromatic region in the asphaltene and resin molecules and in the dimer on the calcite surface. For the asphaltene and resin molecules, the molecule–surface distances were 6.17 and 4.72 Å, respectively. For the dimer, dmolecule–surface = 10.09 Å in the asphaltene–calcite interface. In turn, this increase in the molecule–surface distance was associated through resin–asphaltene interactions with the influence of the resin molecule in the dimer. Although not large, the variation in the ∠C–S–C angles may also show that the chains influence the molecule–molecule interface.
The asphaltene molecule has more aromatic rings and fewer heteroatoms than the resin molecule. According to the calculated Bader charges, there is a charge difference mainly in the aromatic region between the asphaltene and resin molecules, as shown in Table 2. We calculated the sum of the square module of the Kohn–Sham states at the bottom of the conduction band, in the lowest unoccupied molecular orbital (LUMO), and at the top of the valence band, in the highest occupied molecular orbital (HOMO), of the studied molecules to analyze the regions related to the energetic levels more easily accessible to surface and molecular interactions. The aromatic carbon sites are in the more reactive region (Fig. 2A–D), being indeed responsible for the surface adsorption and the formation of the asphaltene and resin nanoaggregates on the natural reservoir rock surface. Although the energetic levels more easily accessible are due to the π–π stacking interactions, this does not eliminate the visible intramolecular contribution from the sulfur sites in the LUMO (Fig. 2C) and in the HOMO (Fig. 2D) of resin.
| Charge region | Molecules | Dimer | ||||||
|---|---|---|---|---|---|---|---|---|
| Asphaltene |
|
Resin |
|
|
|
|||
| Asphaltene | Resin | Asphaltene | Resin | |||||
| a The nitrogen charge in the resin is similar to the one calculated for pyridine (−2.83). | ||||||||
| Aromatic C | −0.01 | −0.01 | 0.20 | 0.19 | −0.01 | 0.21 | −0.01 | 0.22 |
| Aliphatic C | 0.07 | 0.06 | 0.07 | 0.05 | 0.06 | 0.05 | 0.06 | 0.05 |
| S | −0.10 | −0.10 | −0.06 | −0.05 | −0.06 | −0.06 | −0.10 | −0.06 |
| Na | — | — | −2.24 | −2.19 | — | −2.24 | — | −2.29 |
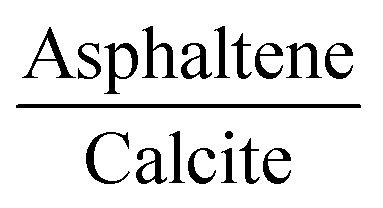
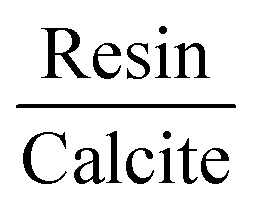
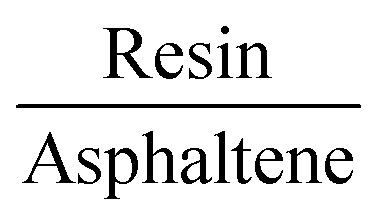

From the projected density of states (PDOS) analysis of asphaltene and resin (Fig. 2E), it is possible to confirm that the sulfur sites display a significant contribution, which are mainly in the valence band of the resin molecule. The aromatic carbon sites are present at the bottom of the conduction band of the asphaltene molecule; although the region of non-aromatic carbons is not adding electronic states at the top of the valence band in both molecules, the energy levels of these carbon sites contribute along with the unoccupied states in the resin molecule. Therefore, in addition to the contribution of the aromatic region, the S sites and the carbon chains where these heteroatoms are present can take part in the interaction processes including resin.
The more energetically stable faces of asphaltene and resin towards adsorption were determined. Then, after the interaction of isolated asphaltene and resin on the calcite surface with adsorption energies of −58.22 and −35.39 kcal mol−1, respectively there was no charge rearrangement in these molecules, as shown for the Bader charges in Table 2. For the charge density calculations in the molecule–surface interface, the aromatic rings take part in both the surface adsorption processes of asphaltene (Fig. 4A) and resin (Fig. 4B) molecules. Particularly in the case of asphaltene adsorption, even when displaying steric hindrance due the aliphatic chains, there is still an interaction between the region of the aromatic rings and the calcite (10.4) surface. This is energetically more favorably than in the resin–calcite interaction, even though the resin displays less steric hindrance and greater contribution of the S site in the interactions from the aliphatic chains.
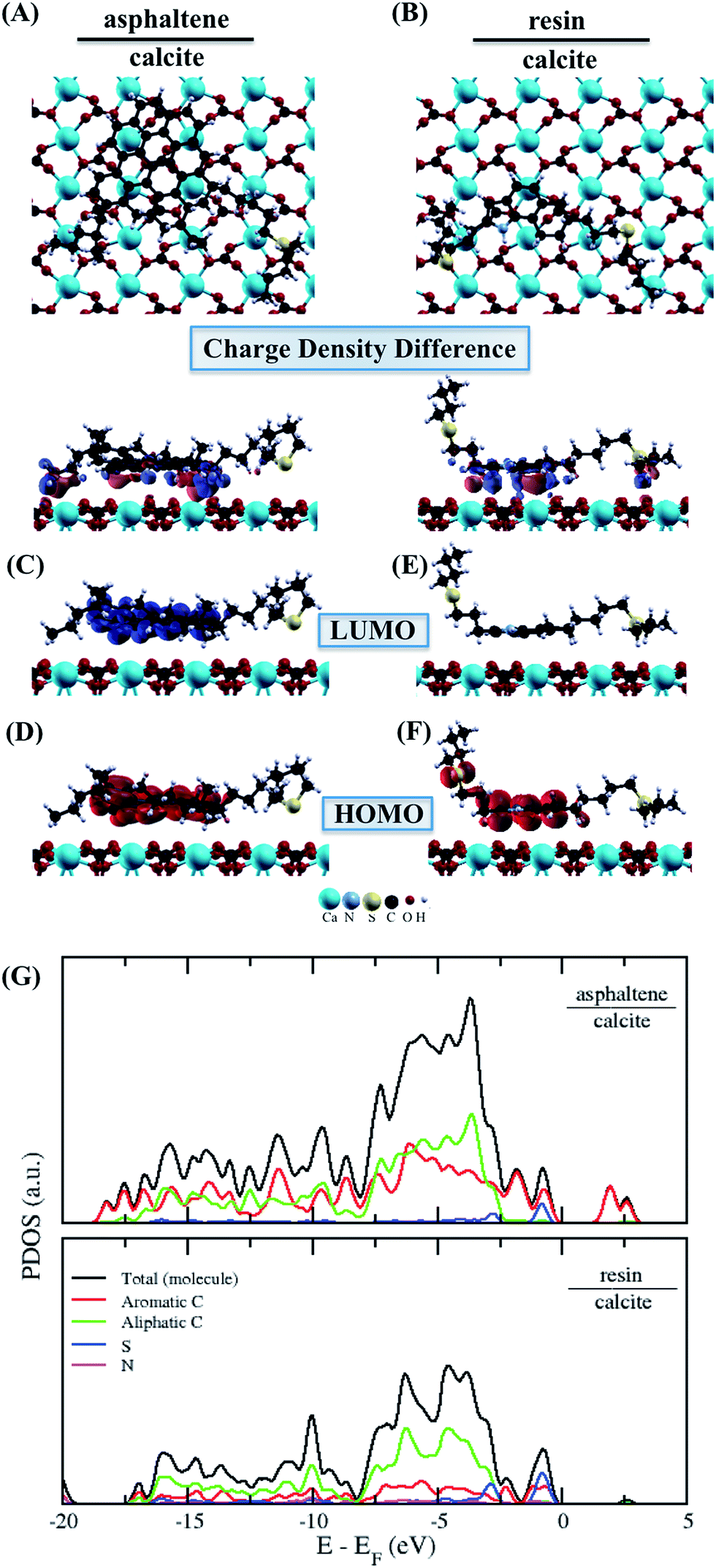 | ||
| Fig. 4 The adsorbed asphaltene and resin molecules. Top-view in up and charge density differences in down for (A) asphaltene–calcite and (B) resin–calcite. The red and blue colors indicate an increase and depletion in the charge density, respectively. The sum of the square module of the Kohn Sham states is at the bottom of the conduction band (LUMO) and at the top of the valence band (HOMO). The (C) LUMO and (D) HOMO of asphaltene–calcite. The (E) LUMO and (F) HOMO of resin–calcite. The isosurface of −0.0007 to 0.0007 electrons per Bohr3. The projected density of states (PDOS) obtained for (G) asphaltene–calcite and resin–calcite. | ||
The electrostatic effects with the inclusion of the implicit water (using the dielectric constant of water) did not show any significant influence on the adsorption energies, being −51.01 and −29.52 kcal mol−1 for the asphaltene–calcite and resin–calcite systems, respectively. In the case of the dielectric constant of toluene, this influence is even less marked when compared to water (−50.30 and −31.20 kcal mol−1, respectively). This indicates that the inclusion of a water or toluene dielectric environment has a minimal effect towards the surface adsorption processes from the interactions with both aromatic and non-aromatic regions in the asphaltene and resin molecules.
The electrostatic effects with a correction of dielectric effects in the absence of an explicit solvent, which is not so easy to calculate, afford an indication of the possible outside vacuum vulnerability towards the electrostatic arrangement appearing in the asphaltene–calcite interaction from a long variation of dielectric constants (toluene and water). Our results are thus important to show that solution electrostatic effects may be indeed somewhat a minor effect on the adsorption of asphaltene on the calcite surface. This proposition is consistent with the experimental work related to oil–water interfaces in the presence of toluene.55–57 Under these conditions, the interaction between asphaltene and water tends to be similar to our results found in the asphaltene–calcite interface, which is somewhat experimental difficult/limited to be analyzed due to the complex composition in the liquid–solid interfaces. Therefore, we indirectly validated our theoretical results due to the experimental adsorptive behavior of asphaltene on the water surface. Ruiz-Morales et al.58 also obtained similar results from dissipative particle dynamics calculations of asphaltene in the oil–water interface.
In general, the main experimental findings are: the interaction in a parallel way between the region of aromatic rings and the water surface, with aliphatic chains positioned towards the oil bulk where π–π stacking interactions are favored, and too high jamming at the oil–water interface by the asphaltene monomers that leads to asphaltene desorption. This last statement has been concluded from the experimental work of González et al.19,20 during their investigation of the mineral–water–toluene interface. Here, we showed in a similar way to calcite that the steric hindrance of asphaltene may be overcome through the strong electrostatic attraction between the aromatic region and the surface of the carbonate. Although the initial formation of the nanoaggregate (asphaltene–resin dimer) favored the π–π stacking interactions between the aromatic regions rather than with calcite, particular aliphatic chains with heteroatoms (S) may take place to keep the adsorption process on the surface possibly outside of the oil bulk with toluene. This would also explain the low influence of water molecules over asphaltene adsorption on the calcite surface due the lack of interference of water surface towards the aliphatic groups as seen experimentally.55–57
The results of the LUMO and HOMO of the asphaltene (Fig. 4C and D) and resin (Fig. 4E and F) molecules adsorbed on the surface of calcite ensure that the aromatic region remains accessible to the possible formation of resin–asphaltene agglomerates. From the PDOS results shown in Fig. 4G for asphaltene and for the resin, the electronic states are more affected in the conduction band region of the resin–calcite system, decreasing the available electronic states. Despite being energetically adsorbed on calcite, the resin adsorption process should not lead to further aggregation and therefore, no formation of nanoaggregates because of the delocalized LUMO of the resin molecule. This is according to the significant charge density mainly observed on the aliphatic chains and calcite interface after the charge redistribution seen in Fig. 4A. Indeed, the difference between the total Bader charges obtained in the aromatic region of the molecules leads preferentially to decreased resin adsorption with respect to asphaltene.
The next step of our study was to select the molecular dimer formed by asphaltene and resin, so that the adsorption process on the surface of calcite is started with the asphaltene molecule to form the resin–asphaltene–calcite system. The interaction energy in the dimer was found to be similar to the adsorption of the resin on the calcite surface. For the interface in the isolated dimer, the LUMO (Fig. 3C) and HOMO (Fig. 3D) indeed show that π–π stacking was favored in the molecular interactions between the aromatic regions of the resin and the asphaltene with no effective steric hindrance. This may also lead to resin adsorption on the calcite surface, as seen from the charge density mainly present on the resin–calcite interface (Fig. 4D). However, it was observed that a significant charge density change in the aliphatic regions occurs in both molecules, particularly after the their interactions in the dimer (Fig. 3A). This indicates that, although the interactions between the asphaltene and resin molecule tends to be mainly by π–π stacking, there is a possible charge rearrangement induced by the heteroatoms; these sites are not kept in the frontier of the Fermi level, according to the PDOS shown in Fig. 3B for the isolated dimer.
Finally, when compared to the adsorption of the asphaltene molecule (−58.22 kcal mol−1), the magnitude of the dimer adsorption energy shows small aggregates may take place on calcite before the growth of the resin–asphaltene interactions on the surface (Fig. 5). This can also be concluded from the similar interaction energies between resin–calcite (−35.39 kcal mol−1) and resin–asphaltene (−25.77 kcal mol−1), in which the resin molecule tends to agglomerate with asphaltene molecules in the absence of calcite. Besides increasing the available electronic states in the conduction band, the dimer allows for the formation of more electronic states occupied to the total system affording a possible increase in nanoaggregates on calcite. Taking into account the slightest influence of the correction of dielectric upon adsorption of the isolated molecules, we leave the assumption that it would be even more irrelevant to the variation of dielectric constants upon dimer adsorption, in which the π–π stacking is favored towards the oil bulk.58
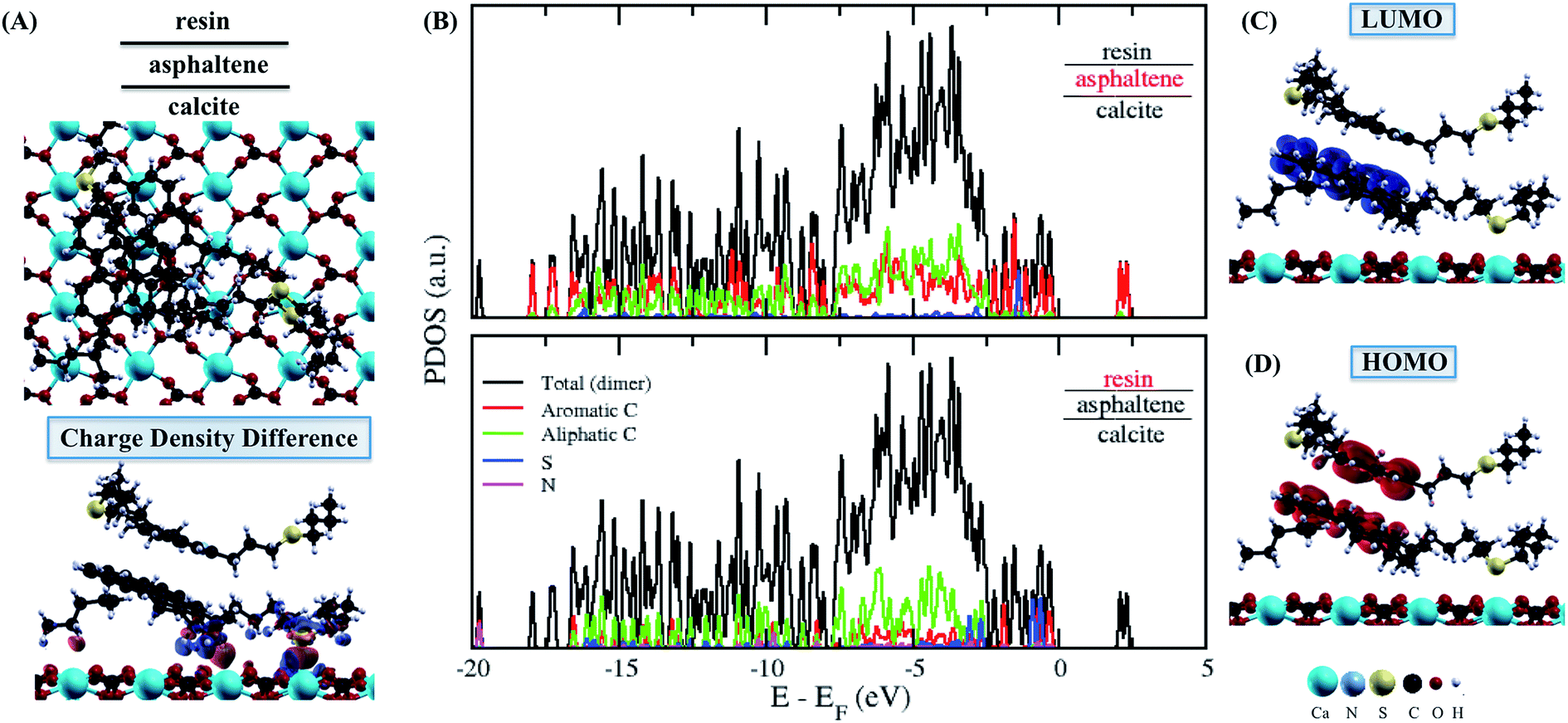 | ||
| Fig. 5 The adsorbed asphaltene and resin dimer molecules. (A) Top-view in up and charge density difference in down for resin–asphaltene–calcite. The red and blue colors indicate an increase and depletion in the charge density, respectively. (B) The projected density of states (PDOS) obtained for asphaltene and resin in resin–asphaltene–calcite. The sum of the square module of the Kohn Sham states is at the bottom of the conduction band (LUMO) and at the top of the valence band (HOMO). The (C) LUMO and (D) HOMO of resin–asphaltene–calcite. The isosurface of −0.0007 to 0.0007 electrons per Bohr3. | ||
The charge density at the interface between the resin–asphaltene dimer and the calcite surface was mainly related to the same interactions observed between the aromatic rings and the chain S sites in the asphaltene–calcite system (Fig. 5A). Particularly, the charge density difference can be seen from calcite to the aromatic rings, while the sulfur in the asphaltene molecule leads to the surface. Furthermore, due the influence of the resin molecule upon the asphaltene molecule previously discussed, the asphaltene molecule was also able to interact by π–π stacking of the adsorbed dimer, even in the absence of water. Despite the increase in the charge density seen in Fig. 5A, the LUMO (Fig. 5C) and HOMO (Fig. 5D) representations display the regions of electronic states related to asphaltene association and the further formation of hemi-micelles on the CaCO3 surface. In both the resin and asphaltene (Fig. 5B) molecules, the sulfur energetic levels are shifted considerably.
Our aim was to study the implicit solvent effect upon the asphaltenes adsorbed on the surface from the absence of water–calcite interactions. However, some considerations can be made in regard the possible effect of “real” solvent molecules on surface aggregation. Regarding the value of the adsorption enthalpy of water molecules on the calcite surface, this should be much lower when compared to that obtained for asphaltene adsorption. For example, the experimental adsorption enthalpies of ethanol show it is able to displace water from the pure calcite surface.59,60 Regarding the entropy for the release of solvent molecules upon asphaltene adsorption, Freeman et al.61 showed using molecular dynamics (MD) that the adsorption of mannose and methanoic acid molecules are favored by an increase in the entropy due to the displacement of water molecules from explicit solvent on the calcite (10.4) surface.
As a result of the adsorption of the asphaltene molecule and resin–asphaltene dimer, the hydrophilic character of the calcite surface may change to hydrophobic with implications on the system (e.g. reservoir) behavior. The charge of S on the aliphatic chains is different in the asphaltene and resin molecules, even before the adsorption. For this reason, we can say that there is a greater rearrangement between the aromatic region and the aliphatic chains (due to S) in resin, increasing the interaction region on this molecule in the dimer. This could favor the adsorption of more non-polar molecules with a minor influence of solvents, such as toluene. Therefore, small nanoaggregates can adsorb in a way similar to single asphaltene molecules until the CaCO3 surface is fully covered.
4 Conclusions
Our simulations are based on first-principles calculations with van der Waals (vdW) inclusion. We showed that the selectivity difference between more polar resin and less polar asphaltene on the CaCO3 (10.4) surface is present in natural reservoir rocks. Due to the self-association of nanoaggregates containing resin and asphaltene, the adsorption process of such dimers is important for understanding the description of the interactions on a molecular level for the asphaltene association on the calcite surface. Moreover, in order to approach the simulation of natural conditions, we used water and toluene as an implicit solvent, changing the dielectric constant.The formation of nanoaggregates may be enhanced on the surface when compared with the dimer. This leads to further laying of asphaltenes on the surface; higher electronic states available for π–π stacking interactions are induced by charge rearrangement and are therefore an important consideration for the asphaltene adsorption. Accordingly, the inclusion of a water–toluene dielectric environment does not influence the adsorption process, since the dimer is adsorbed on the calcite surface with less influence of the more polar molecules of resin. The influence of the solvent on the resin (mainly on the heteroatoms) may control the formation of the aggregates before interacting with the surface rather than of the growth of the agglomerates on the surface from the asphaltene molecule.
Although there is steric hindrance in the interaction of the aromatic ring region on the surface, our results indicate how asphaltene interacts at the particular calcite surface. Therefore, the calcite (10.4) surface can selectively adsorb the asphaltene molecules from oil or model solutions, as the first step in the formation of nanoaggregates. The aliphatic organic chains are also fundamental to the adsorption process, displaying the effect of vdW forces used in our calculations. In parallel, the increase in the polarity through the heteroatom content can activate specific portions of the resin molecules, in order to favor the interaction with asphaltene molecules in the agglomerates and the growth of the aggregates on the calcite surface. Future studies will look into a wider range of molecular structures for asphaltene and the specific effects of functional groups.
Acknowledgements
The authors acknowledge the financial support provided by PETROBRAS and the Brazilian agencies Fapesp and CNPq. The computational time for the calculations was provided by the Blue Gene/Q supercomputer support by Research Computing Support Group (Rice University) and High Performance Computing facilities at Universidade de São Paulo. We also acknowledge the computational support of CENAPAD-SP and UFABC supercomputer facilities.References
- T. F. Headen, E. S. Boek and N. T. Skipper, Energy Fuels, 2009, 23, 1220 CrossRef CAS.
- T. F. Headen and E. S. Boek, Energy Fuels, 2011, 25, 499 CrossRef CAS.
- J. J. Adams, Energy Fuels, 2014, 28, 2831 CrossRef CAS.
- O. P. Strausz, T. W. Mojelsky, E. M. Lown, I. Kowalewski and F. Behar, Energy Fuels, 1999, 13, 228 CrossRef CAS.
- L. S. Kotlyar, B. D. Sparks, J. R. Woods and K. H. Chung, Energy Fuels, 1999, 13, 346 CrossRef CAS.
- O. P. Strausz, A. Morales-Izquierdo, N. Kazmi, D. S. Montgomery, J. D. Payzant, I. Safarik and J. Murgich, Energy Fuels, 2010, 24, 5053 CrossRef CAS.
- S. R. Kelemen, C. C. Walters, P. J. Kwiatek, H. Freund, M. Afeworki, M. Sansone, W. A. Lamberti, R. J. Pottorf, H. G. Machel, K. E. Peters and T. Bolin, Geochim. Cosmochim. Acta, 2010, 74, 5305 CrossRef CAS.
- T. W. Mojelsky, T. M. Ignasiak, Z. Frakman, D. D. McIntyre, E. M. Lown, D. S. Montgomery and O. P. Strausz, Energy Fuels, 1992, 6, 83 CrossRef CAS.
- M. A. Khadim and M. A. Sarbar, J. Pet. Sci. Eng., 1999, 23, 213 CrossRef CAS.
- E. Chouparova, A. Lanzirotti, H. Feng, K. W. Jones, N. Marinkovic, C. Whitson and P. Philp, Energy Fuels, 2004, 18, 1199 CrossRef CAS.
- A. K. Golovko, V. F. Kam'yanov and V. D. Ogorodnikov, Russ. Geol. Geophys., 2012, 53, 1374 CrossRef.
- M. Pons-Jiménez, R. Cartas-Rosado, J. M. Martínez-Magadán, R. Oviedo-Roa, R. Cisneros-Dévora, H. I. Beltrán and L. S. Zamudio-Rivera, Colloids Surf., A, 2014, 455, 76 CrossRef.
- M. A. Anisimov, Y. M. Ganeeva, E. E. Gorodetskii, V. A. Deshabo, V. I. Kosov, V. N. Kuryakov, D. I. Yudin and I. K. Yudin, Energy Fuels, 2014, 28, 6200 CrossRef CAS.
- J. Long, Z. Xu and J. H. Masliyah, Langmuir, 2007, 23, 6182 CrossRef CAS PubMed.
- T. Pernyeszi, A. Patzkó, O. Berkesi and I. Dékány, Colloids Surf., A, 1998, 137, 373 CrossRef CAS.
- S. Acevedo, M. A. Ranaudo, C. García, J. Castillo, A. Fernández, M. Caetano and S. Goncalvez, Colloids Surf., A, 2000, 166, 145 CrossRef CAS.
- D. Dudášová, S. Simon, P. V. Hemmingsen and J. Sjoblom, Colloids Surf., A, 2008, 317, 1 CrossRef.
- D. Dudášová, G. R. Flåten, J. Sjöblom and G. Øye, Colloids Surf., A, 2009, 335, 62 CrossRef.
- G. González and M. B. C. Moreira, Colloids Surf., 1991, 58, 293 CrossRef.
- G. González and A. Middea, Colloids Surf., 1988, 33, 217 CrossRef.
- J. S. Buckley and J. Wang, J. Pet. Sci. Eng., 2002, 33, 195 CrossRef CAS.
- D. L. Lord and J. S. Buckley, Colloids Surf., A, 2002, 206, 531 CrossRef CAS.
- A. W. Marczewski and M. Szymula, Colloids Surf., A, 2002, 208, 259 CrossRef CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244 CrossRef.
- Quantum-ESPRESSO is a community project for high-quality quantum-simulation software, based on density-functional theory, See http://www.quantum-espresso.org and http://www.pwscf.org.
- S. Scandolo, P. Giannozzi, C. Cavazzoni, S. Gironcoli, A. Pasquarello and S. Baroni, Z. Kristallogr., 2005, 220, 574 CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- G. Makov and M. C. Payne, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 4014 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533 CrossRef CAS.
- Y. Zhang and W. Yang, Phys. Rev. Lett., 1998, 80, 890 CrossRef CAS.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- T. Thonhauser, V. R. Cooper, S. Li, A. Puzder, P. Hyldgaard and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 125112 CrossRef.
- G. Roman-Perez and J. M. Soler, Phys. Rev. Lett., 2009, 103, 096102 CrossRef PubMed.
- E. S. Boek, D. S. Yakovleva and T. F. Headen, Energy Fuels, 2009, 23, 1209 CrossRef CAS.
- H. K. Al Halwachi, D. S. Yakovleva and E. S. Boek, Energy Fuels, 2012, 26, 6177 CrossRef CAS.
- S. R. Billeter, A. Curioni and W. Andreoni, Comput. Mater. Sci., 2003, 27, 437 CrossRef.
- R. P. Feynman, Phys. Rev., 1939, 56, 340 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899 CrossRef CAS PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 254 CrossRef.
- M. Yu and D. R. Trinkle, J. Chem. Phys., 2011, 134, 064111 CrossRef PubMed.
- O. Andreussi and N. Marzari, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 245101 CrossRef CAS.
- I. Timrov, O. Andreussi, A. Biancardi, N. Marzari and S. Baroni, J. Chem. Phys., 2015, 142, 034111 CrossRef PubMed.
- http://www.quantum-environment.org/home.html.
- G. La Penna, C. Hureau, O. Andreussi and P. Faller, J. Phys. Chem. B, 2013, 117, 16455 CrossRef CAS PubMed.
- A. Fortunelli, W. A. Goddard, Y. Sha, T. H. Yu, L. Sementa, G. Barcaro and O. Andreussi, Angew. Chem., Int. Ed., 2014, 53, 6669 CrossRef CAS PubMed.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1 CrossRef CAS.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes and I. Vorobyov, et al., J. Comput. Chem., 2009, 31, 671 Search PubMed.
- W. Yu, X. He, K. Vanommeslaeghe and A. D. MacKerell, J. Comput. Chem., 2012, 33, 2451 CrossRef CAS PubMed.
- F. C. D. A. Lima, R. S. Alvim and C. R. Miranda, Density-Functional Theory Calculations of the Asphaltene Nanoaggregation Mechanism Search PubMed, submitted for publication.
- J. P. Rane, D. Harbottle, V. Pauchard, A. Couzis and S. Banerjee, Langmuir, 2012, 28, 9986 CrossRef CAS PubMed.
- J. P. Rane, V. Pauchard, A. Couzis and S. Banerjee, Langmuir, 2013, 29, 4750 CrossRef CAS PubMed.
- V. Pauchard, J. P. Rane, S. Zarkar, A. Couzis and S. Banerjee, Langmuir, 2014, 30, 8381 CrossRef CAS PubMed.
- Y. Ruiz-Morales and O. C. Mullins, Energy Fuels, 2015, 29, 1597 CrossRef CAS.
- I. S. Pasarín, N. Bovet, M. Glyvradal, M. M. Nielsen, J. Bohr, R. Feidenhans'l and S. L. S. Stipp, J. Synchrotron Radiat., 2012, 19, 530 CrossRef PubMed.
- I. S. Pasarín, M. Yang, N. Bovet, M. Glyvradal, M. M. Nielsen, J. Bohr, R. Feidenhans'l and S. L. S. Stipp, Langmuir, 2012, 28, 2545 CrossRef PubMed.
- C. L. Freeman and J. H. Harding, J. Phys. Chem. C, 2014, 118, 1506 CAS.
| This journal is © The Royal Society of Chemistry 2016 |