
Fabrication of heterogeneous biocatalyst tethering artificial prosthetic groups to obtain omega-3-fatty acids by selective hydrolysis of fish oils†
S. Moreno-Pérezaf,
G. Fernández-Lorenteb,
O. Romeroac,
J. M. Guisán *a and
F. López-Gallego*de
*a and
F. López-Gallego*de
aEnzymatic Engineering Group, Instituto de Catálisis y Petroleoquímica, CSIC, C/Marie Curie 2, Madrid, Spain. E-mail: jmguisan@icp.csic.es
bFood Microbiology and Biocatalysis Group, Institute of Food Science Research, CSIC, C/Nicolás Cabrera 9, Madrid, Spain
cPontificia Universidad Católica de Valparaiso (PUCV), Facultad de Ingeniería, Valparaiso, Chile
dHeterogeneus Biocatalysis Group, CIC BiomaGUNE, Pase Miramon 182, San Sebastian-Donostia, Spain. E-mail: flopez@ikerbasque.cicbiomagune.es
eIkerbasque, Basque Foundation for Science, Bilbao, Spain
fPharmacy and Biotechnology Department, School of Biomedical Sciences, Universidad Europea, Madrid, Spain
First published on 6th October 2016
Abstract
The active site of lipase from Bacillus thermocathenolatus was selectively modified with allyl and naphthyl chains at different positions. Lipase immobilization and selective tethering of a naphthyl side chain to its position 320 improve both the hydrolysis rate of fish oils and the selectivity towards the eicosapentaenoic acid acyl chains.
Expanding the chemical diversity of protein structure by tethering non-natural molecules has proved to alter their properties for biotechnological purposes.1 This can be approached by random chemical modification exploiting the reactivity of some protein surface residues such as lysines, aspartic or glutamic acids.1,2 However, this approach lacks the selectivity to tether the synthetic group to a specific position of the protein.2,3 Advances in molecular biology have aided the site-selective modification of proteins by inserting unique and highly reactive amino acids into specific positions of the protein sequence.4 Such reactive amino acids can be either non-natural, by using AMBER codons, or natural, like cysteine which presents high selectivity and reactivity through thiol-exchange chemistry and rarely appears in the surface of native proteins.5
Lipases are one of the most exploited enzymes at the industrial level and have been shown to be excellent biocatalysts in synthetic chemistry since they can catalyze a great variety of chemical reactions with high enantioselectivity.6 The insertion of synthetic molecules into the lipase active centre can expand their catalytic properties and uses thereof. By using this approach, the active site of an immobilized lipase from Bacillus thermocatenulatus (BTL2) has been selectively modified with alkane chains and photochromic molecules. The insertion of alkene chain in the vicinity of the lipase binding pocket froze a hyperactivated conformation that resulted in a more activity and enantioselective enzyme.7 Likewise, the insertion of photochromic molecules into the active site of the same enzyme achieved the light-control of both activity and enantioselectivity of the lipase.8 Furthermore, these semi-synthetic and immobilized biocatalysts can be easily re-used, which is fundamental to apply these biocatalysts in industry.9
A valuable application of lipases in food technology is the obtaining of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) from their natural sources. These polyunsaturated fatty acids (PUFAs) present interesting biological activities in their pure form, and specific food enrichment with either EPA or DHA can have cardiovascular benefits for adults or development benefits for infant, respectively.10 However, the structures of EPA and DHA are similar and thus they are difficult to be separated by physical methods from their main natural source; fish oils.11 Acid hydrolysis fails to discriminate between both acids unlike lipases that are able to hydrolyze one PUFA better than the other from the acylglycerols forming the fish oil.12 Therefore, enzymatic hydrolysis is presented as an attractive alternative to obtain pure PUFAs, although the enzyme stability suffers under the reaction conditions (high concentration of organic co-solvents). Enzyme immobilization is one of the most successful approaches to address the stability issues under harsh conditions. Additionally, we can specifically tether different artificial prosthetic groups to the active site of the immobilized enzyme to discriminate between the EPA and DHA acyl chains, endowing the heterogeneous biocatalyst with a higher selectivity beside the improved stability given by the immobilization protocol. Nevertheless, the immobilization chemistry must promote the optimal enzyme orientation to ensure its stabilization and its successful modification with the synthetic prosthetic group at the target position. Consequently, we must find a complementarity between the immobilization protocol and the site-selective chemical modification aided by directed mutagenesis in order to develop more stable and selective heterogeneous biocatalysts for industrially relevant processes. In this work, we have firstly selected the optimal immobilization protocol to assure the highest activity and stability in the hydrolysis of fish oil and then tuned the active site of such immobilized BTL2 with both napthyl and allyl moieties to enhance the binding of polyunsaturated acyl chains. We have applied these heterogeneous biocatalysts to the selective release of omega-3 fatty acids (EPA and DHA) by hydrolysis of fish oil (Scheme 1).
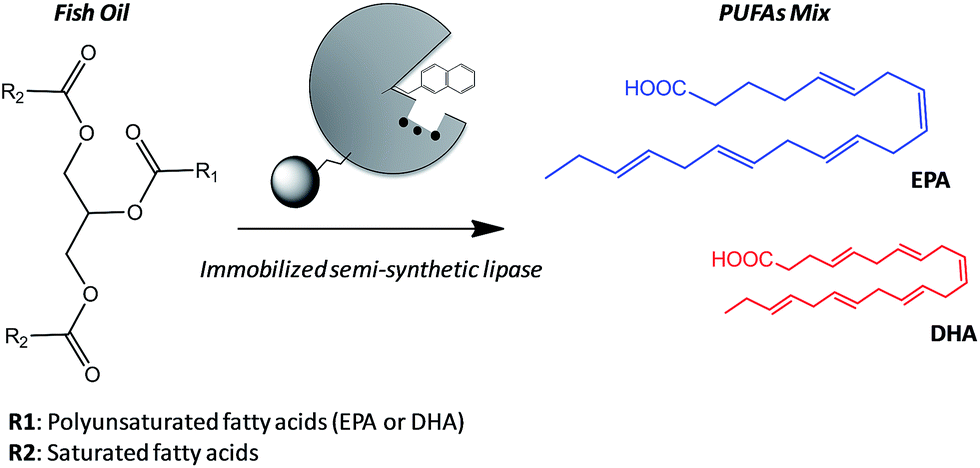 | ||
| Scheme 1 Selective hydrolysis of fish oil to obtain polyunsaturated fatty acids (PUFAs). The reaction is catalyzed by an immobilized semi-synthetic enzyme to yield a mix of PUFAs (eicosapentaenoic acid (EPA) or docosahexaenoic acid (DHA)). Some heterogeneous and semi-synthetic biocatalysts preferentially hydrolyze EPA acyl chains (represented as bigger structure in the scheme). | ||
BTL2 was immobilized on porous agarose beads activated with cyanogen bromide groups (Ag–CB), with octyl groups (Ag–O) and with glyoxyl groups (Ag–G). These immobilization protocol have been extensively exploited for many enzymes; BTL2 among them. Briefly, the immobilization on Ag–CB takes place through the N-terminus of the protein that efficiently reacts with the cyanogens bromide groups at the support surface. The immobilization on Ag–O is based on the interactial activation of the lipases between the “lid” domain and the octyl chain in the support surface. Finally, the immobilization on Ag–G involves the region containing the most reactive lysines that enable the nucleophilic attack to the aldehyde groups on the support under alkaline conditions forming reversible imine groups that are finally reduced to irreversible secondary amines under mild conditions. Therefore, we tested three immobilization strategies: irreversible and univalent immobilization on Ag–CB, the hydrophobic adsorption on Ag–O and the irreversible and multi-valent immobilization on Ag–G. We offered 600 international units (IU) of BTL2 (see ESI†) per gram of wet agarose for all cases, the immobilization yields ranged 43–66%, and the absolute recovered activities towards p-nitrophenol butyrate were 277 to 400 IU gwet agarose−1, which meant a relative recovered activity of 90–100% in all cases (Table S1, ESI†).
When these heterogenous biocatalysts were assayed in the hydrolysis of sardine oil, we observed that different immobilization chemistries conducted to different catalytic activities (Table 1). The multipoint and irreversible covalent attachment between the lipase and the Ag–G carrier increased the hydrolytic activity of wild type BTL2 by 72 and 8 times compared to the same enzyme immobilized via the one-point covalent attachment and through a hydrophobic adsorption on Ag–CB and Ag–O, respectively. A plausible explanation for these differences may rely on the orientation of the lipase on the carrier surface. As mentioned before, Ag–CB immobilizes the proteins through their N-terminus13 and the Ag–O promotes a hydrophobic adsorption of the lipases through their active sites,14 while the proteins are immobilized on Ag–G through their surface areas exposing the highest number of low-pKa lysine residues.13 Analysing the X-ray structure of BTL2 and calculating its surface electrostatics with Blues server,15 we have found that immobilization on Ag–G likely occurs through a region opposite to the active site which present at least four low-pKa lysine side chains clearly exposed to the media and ready to carry out the nucleophile attack to the aldehydes on the carrier surface, eventually forming the covalent bonds (Fig. 1A). We suggest that this orientation permits an unrestricted access of the oily substrates to the lipase catalytic pocket. On the contrary, the active site of BTL2 seems to be partially blocked by the carrier surface when the protein was immobilized on Ag–O carries through its most hydrophobic region (the lid). This orientation may drive to higher diffusional restrictions of the substrate to the active site which drives to lower hydrolytic activities than those immobilized enzymes on Ag–G, where the active site is clearly exposed to the reaction bulk. In fact, activity differences between the two immobilized preparations were less dramatic when smaller substrates were used (Table S1†). Unexpectedly, the immobilization on Ag–CB gave rise a poorly active heterogeneous biocatalysts although the orientation through the N-terminus seemed to ensure the free access of the bulky substrates to the active centre. Furthermore, the activity of BTL2 variants immobilized on Ag–CB towards the hydrolysis of the model substrate p-nitrophenyl butyrate under aqueous conditions were similar to the observed activity for the corresponding variants immobilized on Ag–G (Table S1†). This data point out that lipase immobilized on Ag–CB, although well oriented, presents a low stability under organic media, explaining the poor activity of such heterogeneous biocatalysts in the fish oil hydrolysis. Altogether, these results suggest that the BTL2 activity in the fish oil hydrolysis strongly relies on the orientation and the stability of the immobilized enzyme. In one hand, the protein orientation on the carrier surface is key to assure the access of esters of long and lyphophilic fatty acid chains (fish oil), while it is not as important for short-chain esters (p-nitrophenol butyrate) that can efficiently access the active site of lipase even when the binding pocket is facing the carrier surface. On the other hand, the protein stability guarantees the active conformation of the enzyme during the hydrolysis reaction under harsh conditions. As well as for other lipases,16 the immobilization of BTL2 on Ag–G carrier provides the suitable orientation with an excellent stability to efficiently catalyze the hydrolysis of fish oil in organic media.
| Enzyme variant | Ag–CB | Ag–O | Ag–G | |||
|---|---|---|---|---|---|---|
| Activityb (μU mg−1) | Selectivity EPA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DHAa :DHAa |
Activityb (μU mg−1) | Selectivity EPA:DHAa |
Activityb (μU mg−1) | Selectivity EPA:DHAa |
|
| a The selectivity was calculated as the ratio between the EPA and DHA releasing rates.b The activity was defined as the nmoles of PUFA (both EPA and DHA) released per minute by 1 mg of immobilized lipase during hydrolysis of fish oil (μU mg−1). The mean values are the average values of three independent experiments with standard deviation lower than 10% of the mean value. | ||||||
| WT | 13.8 | 1.34 | 119 | 1.6 | 999 | 1.68 |
| F17C | 7.77 | 1.09 | 75.8 | 1.15 | 1111 | 3.3 |
| L245C | 16.9 | 1.45 | 166 | 1.37 | 2670 | 1.28 |
| I320C | 35.1 | 1.71 | 164 | 1.55 | 333 | 2.42 |
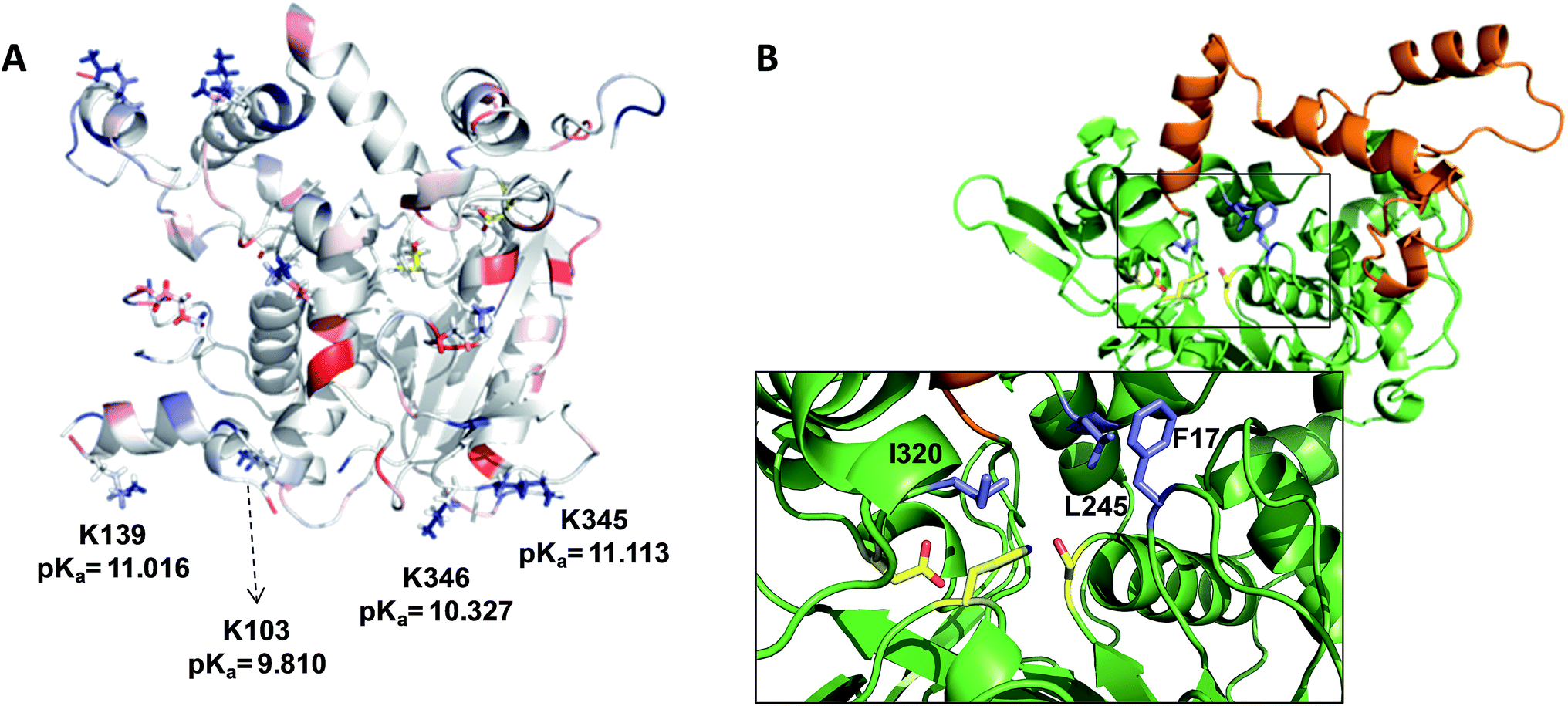 | ||
| Fig. 1 (A) Surface electrostatic potential of BTL2. Acid and basic residues are colored in red and blue respectively. The electrostatic potential of the enzyme surface was calculated with Blues server. (B) Cartoon representation of BTL2 structure (2W22). Lid is colored in orange while the protein core is colored in green. The inlet depicts the catalytic residues (yellow) and the selected position to be mutated by cysteines (purple). | ||
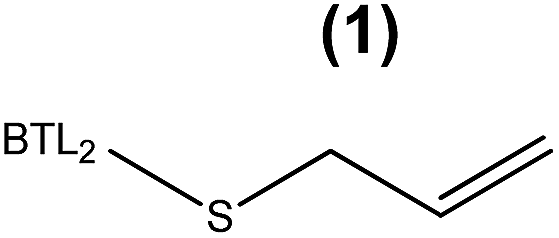
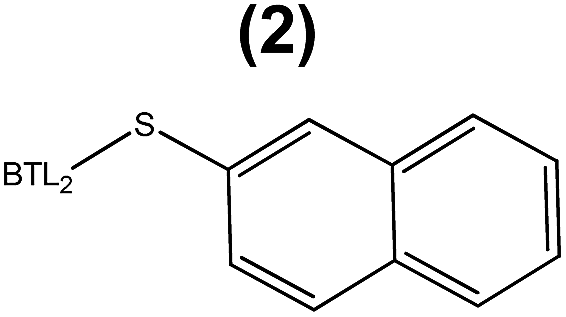
The enzymatic hydrolysis of sardine oil majorly yields two different free PUFAs; eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) (Fig. S1†). In this regard, all the immobilized enzyme variants showed a slight preference towards the EPA chains compared to the DHA ones, indicating that the effect of the immobilization chemistry on the enzyme selectivity was less pronounced than on the hydrolytic activity (Table 1). In order to improve the selectivity towards the EPA acyl chain, we used three different semi-synthetic variants of BTL2 containing non-natural side chains at certain positions to enhance the binding of the EPA acyl chain (20:5, n-3) that differs from the DHA chain (22:6, n-3) in the length, the number of unsaturated bonds and the position of the first unsaturated bond. From a small library of monocysteine BTL2 mutants existing in our laboratory, we selected the mutants bearing a unique cysteine at position 17 (BTL2-F17C), 245 (BTL2-L245C) and 320 (BTL2-I320C) to tether different artificial groups containing double bonds that favour π-stacking interactions between the doubles bonds from the PUFAs acylglycerols and the protein active site (Fig. 1B). This mutant library was built using the double mutant BTL2-C64S/C295S as template. This cysteine-less variant presents the same catalytic properties and stability than the wild-type lipase and assures that the introduced cysteine is the unique cysteine17 that can be modified. We expressed and purified the variants BTL2-F17C, BTL2-L245C and BTL2-I320C and further immobilized them on Ag–G, Ag–CB and Ag–O. The immobilization of the mutants followed the same activity trend observed for the wild type lipase, being the most active preparations those ones immobilized on Ag–G (Table 1). Noteworthy, the immobilization of BTL2-F17C on Ag–G gave rise a heterogeneous biocatalyst 3 and 2 times more selective towards EPA side chains than the same mutant immobilized on Ag–CB and the wild type immobilized on Ag–G, respectively (Table 1). Furthermore, the variant BTL2-L245C immobilized on Ag–G presented a hydrolytic activity of the sardine oil in organic media almost three times higher than the wild type enzyme immobilized on the same carrier, although the activity of that mutant was slightly lower by using pNPB as substrate (Table S1†). In order to optimize the selectivity of the heterogeneous biocatalysts, the active sites of those three variants immobilized on Ag–G were selectively modified with either a allyl or a napthyl side chains through selective thiol chemistry. Hence, the selective chemical modification was carried out in solid-phase easing the work-up of the resulting semi-synthetic enzymes. The resulting disulfide bond tethering the synthetic group to the enzyme was converted into an irreversible thioether group by a phosphine-mediated desulfurization.18 The resulting thioether bond avoids the realising of the synthetic group during the operational process. The modification yields based on colorimetric titration of free cysteine residues varied from one synthetic group to the other; thiomethanesulfonic acid S-allyl ester (1) was efficiently tethered to positions 17 and 320, but tethering to position 245 only reached 60% yield, while the selective modification with 2-naphthalenethiol (2) reached >90% yield for all tested monocysteine mutants (see ESI, Table S2†). We evaluated the activity and the selectivity of the immobilized semi-synthetic enzymes towards the hydrolysis of sardine oil (Table 2). The lipases modified with compound (1) showed lower specific activity than those ones modified with compound (2), such activity reduction was even more dramatic in comparison to the immobilized wild type enzyme. These results suggest that either the allyl side chains may adopt conformations that block the access of the triacylglycerols to the semi-synthetic active site, or the allyl side chain may unspecifically interact with other accessible aminoacids (i.e. histidine, tyrosine, lysine…) during the modification protocol. In this regard, a cysteine-less variant of BTL2 was inactivated when it was incubated with compound (1), suggesting that those unspecific modifications led to the enzyme inactivation (even with smaller substrates under fully aqueous conditions), explaining the low activity of such heterogeneous and semi-synthetic biocatalysts in ester hydrolysis (Fig. S2†).
| Enzyme variant |
|
|
||
|---|---|---|---|---|
| Activityb (μU mg−1) | Selectivity EPA:DHAa |
Activityb (μU mg−1) | Selectivity EPA:DHAa |
|
| a The selectivity was calculated as the ratio between the EPA and DHA yields.b The activity was defined as the nmoles of PUFA (both EPA and DHA) released per minute by 1 mg of immobilized lipase during hydrolysis of fish oil (μU mg−1). The mean values are the average values of three independent experiments with standard deviation lower than 10% of the mean value. | ||||
| WT | 999 | 1.68 | 999 | 1.68 |
| F17C | 1.51 | 2.09 | 278 | 2.03 |
| L245C | 0.92 | 1.49 | 255 | 1.58 |
| I320C | 2.97 | 2.23 | 2690 | 3.29 |
On the contrary, the incubation of the cysteine-less BTL2 with compound (2) negligible affected the enzyme activity, which means that the napthyl chains do not unspecifically interact with the enzyme surface; they only react with the unique cysteine located at the active site of the mutants. Interestingly, the tethering of compound (2) to the BTL2-I320C immobilized on Ag–G was the most active and most selective heterogeneous biocatalysts found in this study. This heterogeneous and semi-synthetic biocatalyst was 2.7 more active for the hydrolysis of fish oil and 2 times more selective towards EPA acyl chains than the wild type BTL2 also immobilized on Ag–G. The insertion of a napthyl group inside the catalytic pocket (position 320) of BTL2 may help the binding of PUFAs, and such synthetic group is even able to better bind EPA than DHA, resulting in more selective heterogeneous semi-synthetic biocatalysts towards the former one. Since the position of the napthyl chain is located above the catalytic serine at the entrance of the binding pocket, such group presumably creates new π-stacking interactions with the EPA acyl chain, positioning it in a more effective conformation within the active site than the DHA acyl chain.
Conclusions
In summary, we have demonstrated the potential of the immobilization coupled to the site-directed chemical modification of the enzyme active site to fabricate novel heterogeneous biocatalysts for more efficient and selective biotechnological transformations like the selective hydrolysis of sardine oil to obtain EPA, an interesting compound for the food, pharmaceutical and cosmetics industries. This is another example of how protein engineering, immobilization and bio-organic chemistry can be merged to advance in the development of the biocatalysis.Acknowledgements
We acknowledge COST action CM103-System biocatalysis. We would like to thank IKERBASQUE, Basque foundation for Science for the funding to PhD Fernando López Gallego. We gratefully recognize the European Commission for the “Lignofood project” contract for Dr Sonia Moreno-Perez and for the Ramón-Areces contract to Dr Gloria Fernández-Lorente.Notes and references
- B. G. Davis, Curr. Opin. Biotechnol., 2003, 14, 379 CrossRef CAS PubMed.
- E. Baslé, N. Joubert and M. Pucheault, Chem. Biol., 2010, 17, 213 CrossRef PubMed.
- R. C. Rodrigues, Á. Berenguer-Murcia and R. Fernandez-Lafuente, Adv. Synth. Catal., 2011, 353, 2216 CrossRef CAS.
- F. Liu, L. Wang, H. Wang, L. Yuan, J. Li, J. L. Brash and H. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 3717 CAS.
- S. M. Marino and V. N. Gladyshev, J. Mol. Biol., 2010, 404, 902 CrossRef CAS PubMed.
- F. Hasan, A. A. Shah and A. Hameed, Enzyme Microb. Technol., 2006, 39, 235 CrossRef CAS.
- F. López-Gallego, O. Abian and J. M. Guisán, Biochemistry, 2012, 51, 7028 CrossRef PubMed.
- A. Bautista-Barrufet, F. López-Gallego, V. Rojas-Cervellera, C. Rovira, M. A. Pericàs, J. M. Guisán and P. Gorostiza, ACS Catal., 2014, 4, 1004 CrossRef CAS.
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437 RSC.
- B. Ganesan, C. Brothersen and D. J. McMahon, Crit. Rev. Food Sci. Nutr., 2014, 54, 98 CrossRef CAS PubMed.
- A. Halldorsson, B. Kristinsson, C. Glynn and G. G. Haraldsson, J. Am. Oil Chem. Soc., 2003, 80, 915 CrossRef CAS.
- G. Fernández-Lorente, D. L.-V. Carolina Pizarro, L. Betancor, A. V. Carrascosa and J. M. G. Benevides Pessela, J. Am. Oil Chem. Soc., 2011, 88, 819 CrossRef.
- G. Fernandez-Lorente, F. Lopez-Gallego, J. M. Bolivar, J. Rocha-Martin, S. Moreno-Perez and J. M. Guisan, Curr. Org. Chem., 2015, 19, 1719 CrossRef CAS.
- A. Bastida, P. Sabuquillo, P. Armisen, R. Fernández-Lafuente, J. Huguet and J. M. Guisán, Biotechnol. Bioeng., 1998, 58, 486 CrossRef CAS PubMed.
- I. Walsh, G. Minervini, A. Corazza, G. Esposito, S. C. Tosatto and F. Fogolari, Bioinformatics, 2012, 28, 2189 CrossRef CAS PubMed.
- C. Pizarro, M. C. Brañes, A. Markovits, G. Fernández-Lorente, J. M. Guisán, R. Chamy and L. Wilson, J. Mol. Catal. B: Enzym., 2012, 78, 111 CrossRef CAS.
- C. A. Godoy, B. d. l. Rivas, V. Grazú, T. Montes, J. M. Guisán and F. López-Gallego, Biomacromolecules, 2011, 12, 1800 CrossRef CAS PubMed.
- G. J. L. Bernardes, E. J. Grayson, S. Thompson, J. M. Chalker, J. C. Errey, F. El Oualid, T. D. W. Claridge and B. G. Davis, Angew. Chem., 2008, 120, 2276 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Methods and characterization, Fig. S1–S2 and Tables S1–S2. See DOI: 10.1039/c6ra21121f |
| This journal is © The Royal Society of Chemistry 2016 |