
A controlled tandem transformation of conjugated enynones with arenes under superelectrophilic activation leading to aryl-substituted dienones and indenes†
Steve Saulniera,
Alexander A. Golovanovb and
Aleksander V. Vasilyev *ac
*ac
aDepartment of Organic Chemistry, Institute of Chemistry, Saint Petersburg State University, Universitetskaya nab., 7/9, Saint Petersburg, 199034, Russia. E-mail: aleksvasil@mail.ru; a.vasilyev@spbu.ru
bDepartment of Chemistry, Chemical Processes and Technologies, Togliatti State University, Belorusskaya ul., 14, Togliatti, 445667, Russia
cDepartment of Chemistry, Saint Petersburg State Forest Technical University, Institutsky per., 5, Saint Petersburg, 194021, Russia
First published on 26th October 2016
Abstract
Under superelectrophilic activation in triflic acid (TfOH) and in the presence of arenes, 1,5-diarylpent-2-en-4-yn-1-ones lead to aryl-substituted indenes by addition of arene to the acetylenic bond followed by an intramolecular cyclization in a tandem process, with yields up to 97%. The regiochemistry of product formation is predicted and controlled by varying the aromatic substituents on the starting materials, which makes it possible to obtain diversely substituted indenes. The use of pyridine as a co-solvent of the superacid TfOH allows control of the extent of the tandem transformation and to isolate the aryl-conjugated dienone intermediates before cyclization with high E/Z diastereoselectivities and yields up to 95%. The reaction mechanism is highlighted and involves cationic species that have been observed and characterized by NMR.
Introduction
The synthesis of indene derivatives is of particular importance owing to their biological and pharmaceutical properties, as shown once again by two very recent papers on the anticholinesterase and amyloid beta inhibition activities of 1H-indene-2-carboxamides,1 and the discovery of the 1,3-dioxo-2,3-dihydro-1H-indene core as a promising scaffold for the protein kinase CK2 inhibition (a target for the treatment of cancer).2 Indenes are also among the most typical ligands in organometallic chemistry as they give transition metal complexes an enhanced reactivity, in particular complexes of group 4,3 8 (ref. 4) and 10 (ref. 5 and 6) metals. Among the classical approaches for the synthesis of indenes,7 cyclizations via intramolecular electrophilic aromatic substitutions (SEAr) have been widely used and involve unsaturated alcohols or carbonyl derivatives activated by Lewis or Brønsted acids. From this point of view, superelectrophilic activation may be an interesting method since it allows such Friedel–Crafts type reactions with very weak aromatic nucleophiles8 and thus might lead to structures otherwise difficult to achieve. Some examples have indeed been reported on intramolecular cyclizations into indenes under superelectrophilic activation of arylcyanopropionate,9 5-aryl-1-azapenta-1,4-dien-3-ones,10 α-acyl N-arylcinnamamides,11 dihydrocinnamoyl-substituted N-heterocycles12 and cinnamaldehydes.13Recently, we have shown that 1,5-diarylpent-2-en-4-yn-1-ones 1 undergo such electrophilic cyclization in trifluoromethanesulfonic acid (triflic acid, TfOH) to give the corresponding indan-1-ones 3 via the intermediate formation of dienyl triflates 2 (Scheme 1).14 More importantly, we observed that when the aryl ring conjugated with the acetylenic bond bore an electron donating group (for example with R1 = Me on Scheme 1), an intermolecular SEAr could take place before the ring formation, leading in that case to the indene derivatives 4 through two consecutive inter and intramolecular SEArs in a tandem reaction.
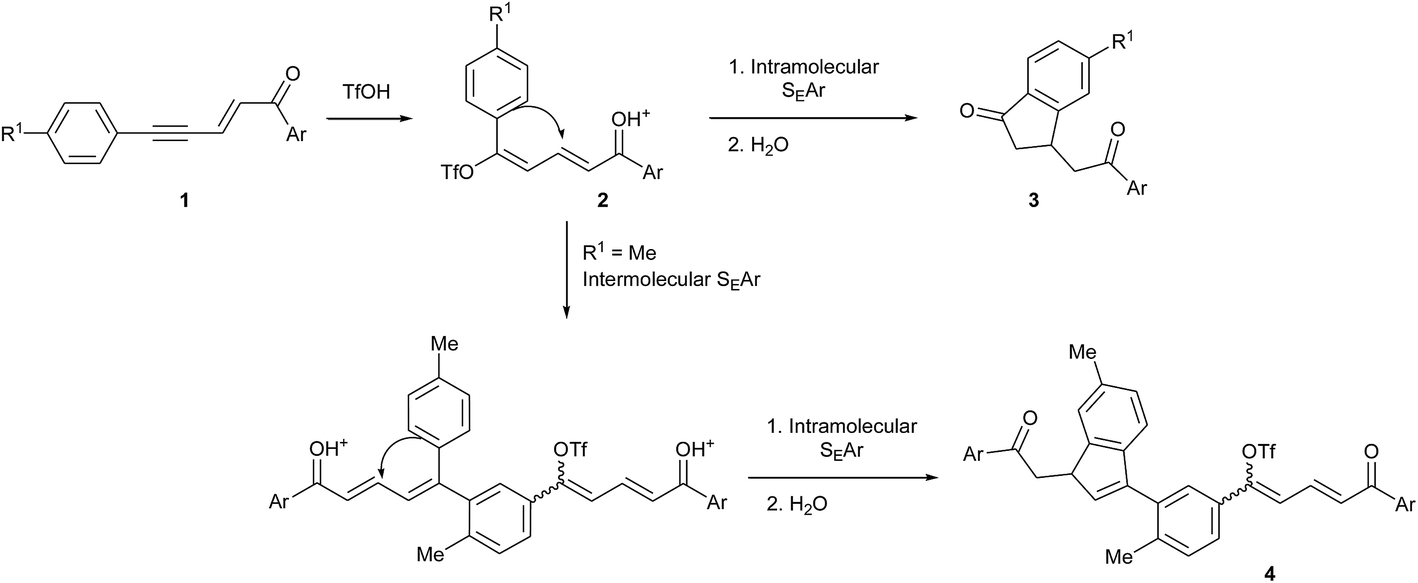 | ||
| Scheme 1 Reactivity of 1,5-diarylpent-2-en-4-yn-1-ones under superelectrophilic activation in TfOH showing their potential as precursors of indenes [ref. 14]. | ||
The present work aims at generalizing this observation by using benzene derivatives as external aromatic nucleophiles in order to obtain diversely functionalized indenes. The use of pyridine as a co-solvent, by modulating the acidity of the reaction medium, makes it possible to control the extent of the tandem reaction and to isolate the 1,5,5-triaryl dienone intermediates prior to cyclization. The reaction mechanisms as well as the regio- and stereo-selectivities of formation of the different products are discussed.
Results and discussion
A series of 1,5-diarylpent-2-en-4-yn-1-ones bearing various substituents on the aryl rings (Fig. 1) was prepared by cross-aldol condensation of the corresponding aryl propynals and aryl methyl ketones.15 Such conjugated enynones can give rise to electrophiles in the presence of different activating agents and we decided to explore TfOH and sulfuric acid (H2SO4), both used as reaction solvents, and aluminum chloride (AlCl3) used in excess in an appropriate anhydrous solvent. The acidic strength of the reaction medium is obviously an important aspect and can be modulated by using a non-nucleophilic organic base as a co-solvent. Pyridine is well-suited for this purpose as it is fully protonated in superacidic media and therefore unreactive. As a co-solvent of TfOH, we found that pyridine could be used up to 30% volume (i.e. ratio TfOH/pyridine 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v) for a homogeneous mixture.
:3 v/v) for a homogeneous mixture.
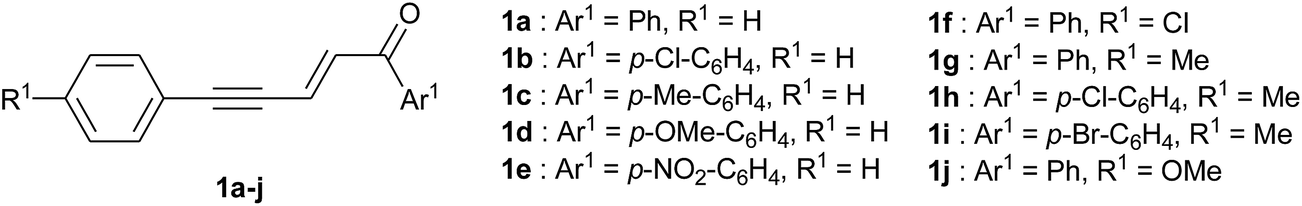 | ||
| Fig. 1 Starting 1,5-diarylpent-2-en-4-yn-1-ones used in this study. | ||
In the presence of benzene, enynone 1a gave the 3-phenyl indene 5a with the three activating agents tested (see Table S1 in ESI†) but the best result was obtained in TfOH with a yield of 97% after 12 hours (Table 1, entry 1). Indene 5a was also obtained from dienyl triflate 2a (entry 2), which was generated in situ from 1a in TfOH (see ref. 14) before the addition of benzene. In that case, the lower yield of 63% is due to the lower reactivity of 2a and the competitive formation of indanone 3 (see Scheme 1). Enynones 1b–d, with different substituents on the aromatic ring Ar1 conjugated with the carbonyl group, gave under the same conditions the corresponding indene derivatives 5b–d with rather good yields (86–96%, entries 3–6). In the case of enynone 1e (Ar1 = p-NO2-C6H4), the presence of the nitro substituent affects the stability of the carbocationic intermediates and makes the formation of indene 5e more difficult, which was isolated with only 48% yield (entry 6).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Starting enynone | Ar1 | R1 | Ar2 | Product | Yieldb, % | Mole fractionc |
| a Reactions carried out with 0.1 mol L−1 of starting enynone and 11 equivalents of benzene or 3.6 equivalents of mesitylene.b Yields of isolated products.c Amount of product 5 divided by the total amount of all regioisomers in mixture after separation by chromatography.d 2a was generated in situ from enynone 1a. | |||||||
| 1 | 1a | Ph | H | Ph | 5a | 97 | |
| 2 | 2ad | Ph | H | Ph | 5a | 63 | |
| 3 | 1b | p-Cl-C6H4 | H | Ph | 5b | 87 | |
| 4 | 1c | p-Me-C6H4 | H | Ph | 5c | 96 | |
| 5 | 1d | p-OMe-C6H4 | H | Ph | 5d | 86 | |
| 6 | 1e | p-NO2-C6H4 | H | Ph | 5e | 48 | |
| 7 | 1a | Ph | H | 2,4,6-Me3-C6H2 | 5f | 86 | 0.89 |
| 8 | 1f | Ph | Cl | 2,4,6-Me3-C6H2 | 5g | 77 | 0.89 |
| 9 | 1g | Ph | Me | 2,4,6-Me3-C6H2 | 5h | 49 | 0.99 |
| 10 | 1j | Ph | OMe | 2,4,6-Me3-C6H2 | 5i | 39 | 0.99 |
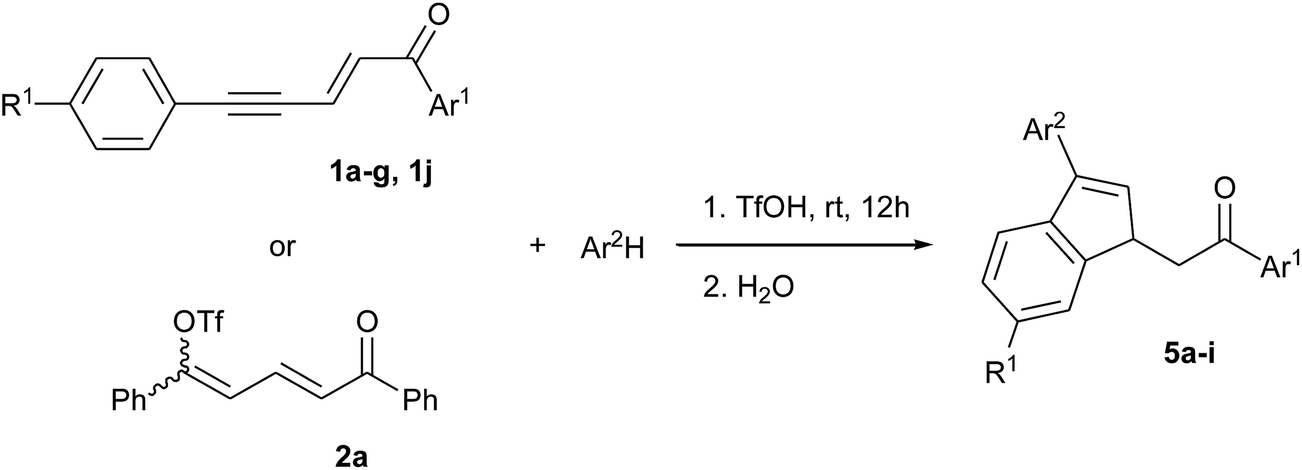
Under the same conditions and in the presence of 1,3,5-trimethylbenzene (mesitylene), more nucleophilic with the presence of three activating methyl groups, enynone 1a gave the corresponding 3-mesityl indene 5f together with a minor regioisomer which could not be isolated and clearly identified (entry 7). Thus, 5f was isolated in mixture with 86% yield and 0.89 mole fraction. Since both the intermolecular SEAr on the mesitylene ring and the intramolecular cyclization are supposed to proceed without any regiochemistry issue, we assume that the presence of a minor regioisomer is due to some methyl shift. Enynones 1f, 1g, 1j, with substituents R1 on the aromatic ring conjugated with the acetylenic bond, gave the corresponding 3-mesityl indenes 5g–i in the same way (entries 8–10). However, in the case of an electron donating substituent (enynones 1g, 1j, R1 = Me and OMe, entries 9–10), the higher nucleophilicity of the aromatic ring leads to the competitive formation of condensation products 4 (see Scheme 1 and ref. 14) and therefore to lower yields of indenes 5h, 5i. On the other hand, the presence of such electron-donating substituents stabilizes the carbocationic intermediates so that the formation of minor isomers decreases, resulting in higher mole fractions of the main products.
A plausible reaction mechanism for the formation of indenes is shown in Scheme 2 in the case of benzene as aromatic nucleophile. Under superacidic conditions, enynone 1 gives the dicationic species A (see DFT calculations of this cation in ref. 14) which undergoes an intermolecular SEAr on benzene to give cation C. The latter may also be generated from dienyl triflate 2 through its protonated form (cation B, see NMR data in ref. 14) by an addition–elimination mechanism. Two configurations (2E,4Z) and (2E,4E) are possible, corresponding to cations C and C′. They lead to the cyclic species D and D′ depending on whether the intramolecular SEAr involves the R1 substituted aryl ring or the phenyl ring. These cations D and D′ are protonated to give the stable dications E and E′, respectively, precursors of the corresponding indenes regioisomers 6 and 7 finally formed by aqueous quenching. When R1 = H, both pathways give the 3-phenyl indenes 5a–e. However, it should be mentioned that the 3-mesityl indenes 5f–i, obtained by using mesitylene instead of benzene, would be formed only via cation C and cyclization through the R1 substituted ring.
 | ||
| Scheme 2 Plausible reaction mechanism for the formation of indenes from enynones 1 or dienyl triflates 2 in the presence of benzene. | ||
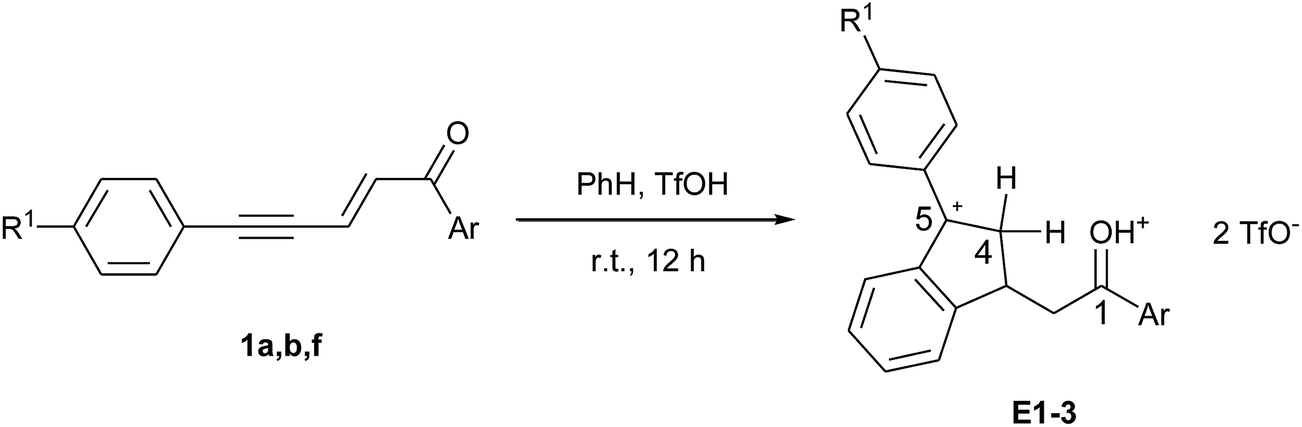
The formation of dication E by protonation of D was revealed by 1H and 13C NMR monitoring of the reactions between enynones 1a, 1b, 1f and benzene, which give cations E1–3, respectively, (see Table 2 and spectral figures in ESI†). For all three cations, the carbon cationic center C5 is shifted at around 220 ppm, slightly more deshielded than the protonated carbonyl C1. Carbon C4 appears as a secondary carbon at around 49 ppm in the DEPT-135 spectra and bears diastereotopic protons H4 with large germinal coupling constants 2J of 21–24 Hz due to hyperconjugation with the adjacent sp2 carbocation.16,17 These dications were relatively stable in superacidic media, even after extended reaction times. However, once isolated after aqueous quenching, the corresponding indene products were shown to be unstable and had to be stored at low temperature and under inert atmosphere.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Cation | Ar | R1 | 1H NMR, δH, ppm | 13C NMR, δC, ppm | |||
| H4 (doublet) | H4′ (multiplet) | C1 | C4 | C5 | ||||
| a 1H and 13C NMR spectra were recorded in TfOH at room temperature and referenced relative to the CH2Cl2 proton and carbon signals at δ 5.30 and 53.52 ppm, respectively. | ||||||||
| 1 | E1 | Ph | H | 4.09 (J 23.2 Hz) | 4.37–4.64 | 219.1 | 49.0 | 222.4 |
| 2 | E2 | p-Cl-C6H4 | H | 4.06 (J 21.7 Hz) | 4.44–4.62 | 216.9 | 48.9 | 222.1 |
| 3 | E3 | H | Cl | 4.04 (J 23.8 Hz) | 4.39–4.59 | 219.0 | 48.8 | 219.4 |
In order to study the formation of indenes 6 and 7, for which R1 ≠ H in Scheme 2, reactions of enynones 1f–j were performed in the presence of benzene and in different acidic media.‡ The results are presented in Table 3. In each case, both products 6 and 7 were observed in variable proportions and could not be separated by column chromatography. As expected, the cyclization takes place preferentially via the most nucleophilic ring: enynone 1f (R1 = Cl, entry 1) gave mostly the isomer 7 by cyclization via the phenyl ring rather than via the deactivated 4-chlorophenyl, while enynone 1g (R1 = Me, entries 2–5) gave mostly the isomer 6 by cyclization via the activated p-tolyl ring. Besides, minor regioisomers were formed together with 6b and 7b (R1 = Me) and could not be either isolated or identified. We assume again that they are products of methyl shifts since they were not observed for R1 = Cl or OMe. However, their formation was significantly reduced by the presence of pyridine in the reaction medium, as a consequence of the lowering of acidity. More importantly, the presence of pyridine prevents the competitive formations of indanone 3 and condensation product 4, as highlighted in our previous communication.14 Both factors result in a higher conversion to 6b (compare entries 2 and 5 in Table 3), which could finally be obtained with 0.79 mole fraction in mixture. Indenes 6c and 6d were formed in the same conditions from enynones 1h and 1i (R1 = Me, Ar1 ≠ Ph, entries 6 and 7) with a constant mole fraction of 0.79, showing that the presence of a substituent on Ar1 does not affect the regioselectivity of the cyclization step.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entrya | Enynone | Acidic medium | Product 6 | Product 7 | Isolated mixture | ||||
| No. | Conversionb, % | No. | Conversionb, % | Main product | Mole fractionc | Combined yieldd 6 + 7, % | |||
| a Reactions carried out at room temperature with 0.1 mol L−1 of starting enynone and 11 equivalents of benzene.b Conversions of 1 to products 6 and 7 determined by 1H NMR analysis of the crude reaction mixture.c Amount of the main product divided by the total amount of all regioisomers in the isolated mixture.d Yields of whole mixtures after separation by chromatography.e 5 equivalents of AlCl3 in solution in anhydrous dichloromethane as reaction solvent.f 20% volume of pyridine in TfOH as reaction solvent. | |||||||||
| 1 | 1f | TfOH | 6a | 6 | 7a | 88 | 7a | 0.94 | 90 |
| 2 | 1g | TfOH | 6b | 56 | 7b | 16 | |||
| 3 | 1g | H2SO4 | 6b | 38 | 7b | 11 | |||
| 4 | 1g | AlCl3 in CH2Cl2e | 6b | 21 | 7b | 7 | |||
| 5 | 1g | TfOH/pyridine 4:1f |
6b | 70 | 7b | 17 | 6b | 0.79 | 94 |
| 6 | 1h | TfOH/pyridine 4:1f |
6c | 7c | 6c | 0.79 | 67 | ||
| 7 | 1i | TfOH/pyridine 4:1f |
6d | 7d | 6d | 0.79 | 86 | ||
| 8 | 1j | TfOH | 6e | 34 | 7e | 46 | 7e | 0.57 | 85 |
| 9 | 1j | H2SO4 | 6e | 20 | 7e | 41 | |||
| 10 | 1j | TfOH/pyridine 4:1f |
6e | 77 | 7e | 15 | 6e | 0.83 | 83 |

Interestingly, in the case of enynone 1j (R = OMe, entries 8–10) the regioselectivity of the cyclization step depends on the reaction media. In the presence of pyridine, the product 6e of cyclization via the 4-methoxyphenyl ring was mostly formed with a mole fraction of 0.83, as rightly expected by the presence of the activating group OMe. In contrast, without the presence of pyridine, 7e was the main product in both TfOH and H2SO4, involving cyclization via the phenyl ring yet less nucleophilic. This can be explained by the ease of protonation of alkoxybenzenes in strong acidic media,18,19 including aqueous H2SO4,20 that deactivates the 4-methoxyphenyl ring. Seemingly, this does not happen in the presence of pyridine.
In order to achieve more complex structures, we explored the reactivity in the presence of disubstituted benzene derivatives. We first selected the reaction of enynone 1a (and the corresponding dienyl triflate 2a) with ortho-dimethylbenzene (o-xylene) as a model to understand and optimize the regioselectivity. The results are presented in Table 4. As we expected, the intermolecular SEAr takes place preferentially on the carbon C4 of o-xylene and the cyclization proceeds then preferentially via the activated dimethylphenyl ring, giving 7f as the main reaction product. In TfOH without any co-solvent, indene 7f was formed in similar yields of 61–67% regardless of starting material and temperature (entries 1–2 and 4–6). The yield decreased in the presence of pyridine (entry 3) with almost no conversion when dienyl triflate 2a was used as starting material (entry 7). This later observation is consistent with the high stability of dienyl triflates 2 in the TfOH/pyridine media already discussed.14
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Starting materialb | Acidic medium | Temp., °C | Time, h | Combined yieldc, % | Yield ofd 7f, % | Mole fraction of 7fe |
| a Concentration of starting material 0.1 mol L−1 and 4 equivalents of o-xylene for all reactions.b 2a was generated in situ from enynone 1a.c Yield of all regioisomers in the crude reaction mixture determined by 1H NMR.d Yield of 7f determined by 1H NMR analysis of the crude reaction mixture.e Ratio between the yield of product 7f and the combined yield of all regioisomers.f In parenthesis, combined yield and mole fraction after separation by chromatography. | |||||||
| 1 | 1a | TfOH | r.t. | 12 | 96 | 66 | 0.69 |
| 2 | 1a | TfOH | 0 | 6 | 93 | 67 | 0.72 |
| 3 | 1a | TfOH/pyridine 4:1 |
0 to r.t. | 12 | 55 | 38 | 0.69 |
| 4 | 2a | TfOH | r.t. | 12 | 89 | 63 | 0.71 |
| 5 | 2a | TfOH | 0 | 6 | 79 (74)f | 62 | 0.78 (0.86)f |
| 6 | 2a | TfOH | −38 to r.t. | 6 | 78 | 61 | 0.78 |
| 7 | 2a | TfOH/pyridine 5:1 |
r.t. | 12 | Not determined | 4 | Not determined |
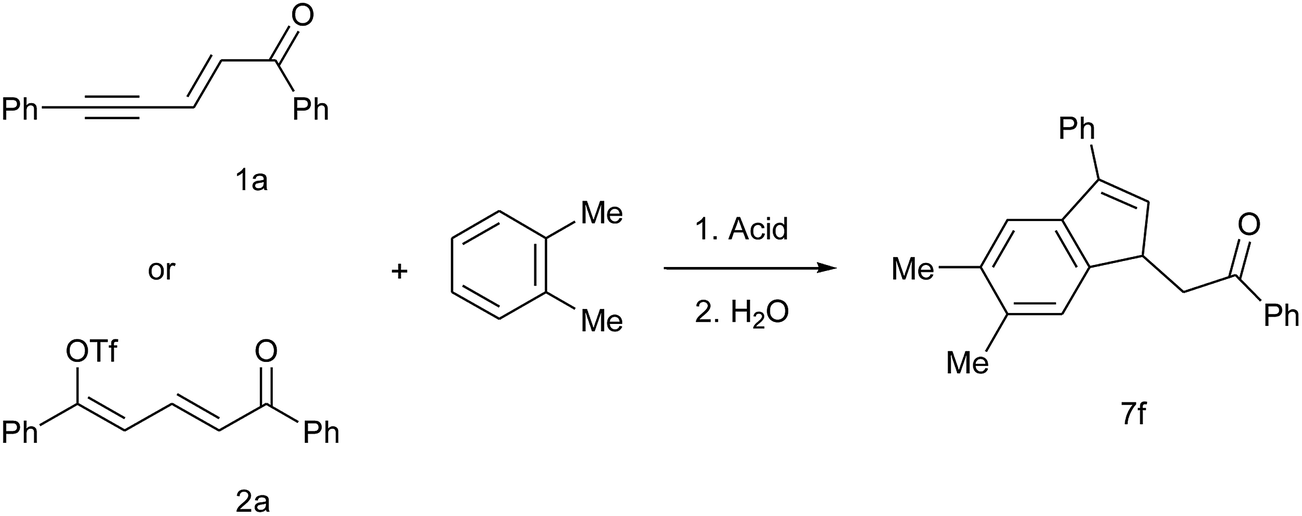
The mole fraction of 7f little varies when enynone 1a is used as starting material (0.69–0.72, entries 1–4), even in the presence of pyridine or at low temperature, but increases by starting from dienyl triflate 2 at 0 °C or −38 °C (0.78, entries 5–6). Since the intramolecular cyclization takes place through the same carbocationic intermediate C with both starting materials, the better result obtained from dienyl triflate 2 can be considered as an evidence of a higher regioselectivity concerning the intermolecular SEAr. This is linked with the difference of electrophilicity between cations A and B: the dication A, generated from enynone 1, has a more reactive sp carbocationic centre compared with the sp2 cation B (i.e. the protonated form of triflate 2). Since 7f could not be isolated from most of the minor regioisomers (mole fraction of 0.86 after separation, see entry 5), regioselectivity is an important aspect to take into consideration and makes dienyl triflates 2 useful starting materials for the synthesis of such polysubstituted indenes.
In the light of the previous observations, we prepared a series of substituted indenes 6 and 7 using different starting enynones and ortho-disubstituted benzene derivatives. The results are presented in Table 5. Regardless of the starting enynone used, the reactions with 1,2-dichlorobenzene gave indenes 6 as main products while indenes 7 were obtained with o-xylene and 1,2-dimethoxybenzene. This is consistent with their relative nucleophilicities.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entrya | Starting materialb | Ar1 | R1 | R2 | Temp., °C | Time, h | Combined yieldc, % | Main product | |
| No. | Mole fractiond | ||||||||
| a Concentration of starting material 0.1 mol L−1 (except for 2h, 0.05 mol L−1, entries 8–10) and 4 equivalents of the aromatic nucleophile.b 2a, 2c, 2f, 2h were generated in situ from the corresponding enynones.c Yields of all regioisomers in mixtures after separation by chromatography.d Mole fractions of the main products in mixtures determined by 1H NMR after separation.e Reactions carried out with 20% volume of pyridine in TfOH as reaction solvent. | |||||||||
| 1 | 2a | Ph | H | Cl | 0 | 6 | 73 | 6f | 0.76 |
| 2 | 2a | Ph | H | OMe | 0 | 6 | 85 | 7g | 0.90 |
| 3 | 2c | p-Me-C6H4 | H | OMe | 0 | 6 | 68 | 7h | 0.89 |
| 4 | 2f | Ph | Cl | Cl | r.t. | 16 | 74 | 6g | 0.78 |
| 5 | 2f | Ph | Cl | Me | r.t. | 12 | 81 | 7i | 0.88 |
| 6 | 2f | Ph | Cl | OMe | r.t. | 12 | 71 | 7j | 0.48 |
| 7 | 2f | Ph | Cl | OMe | 0 | 6 | 17 | 7j | 0.93 |
| 8 | 2h | p-Cl-C6H4 | Me | Cl | −38 to r.t. | 6 | 30 | 6h | 0.84 |
| 9 | 2h | p-Cl-C6H4 | Me | Me | −38 to r.t. | 6 | 46 | 7k | 0.69 |
| 10 | 2h | p-Cl-C6H4 | Me | OMe | −38 to r.t. | 6 | 45 | 7l | 0.85 |
| 11 e | 1j | Ph | OMe | Cl | r.t. | 12 | 23 | 6i | 0.60 |
| 12 e | 1j | Ph | OMe | Me | r.t. | 12 | 95 | 7m | 0.63 |
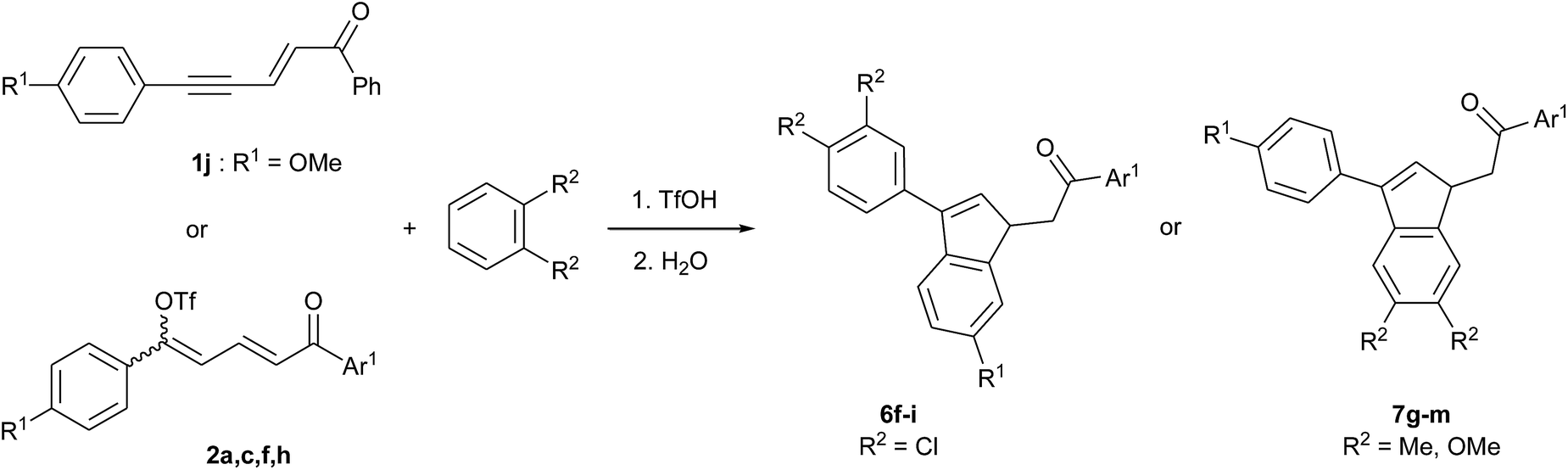
The reactions from enynones 1a, 1c, 1f, 1h were conducted through the corresponding dienyl triflate 2a, 2c, 2f, 2h, generated in situ. The reactions from 2f (R1 = Cl) were very slow at 0 °C and had to be run at room temperature. Nevertheless, the corresponding indenes 6g and 7i were isolated with rather high mole fractions of respectively 0.78 and 0.88 (entries 4 and 5). In the case of indene 7j, obtained from 2f and 1,2-dimethoxybenzene, the reaction at room temperature gave a poor regioselectivity (mole fraction 0.48) that was notably increased at 0 °C (0.93), at the expense of the yield (compare entries 6 and 7).
Lower yields were obtained with 2h (R1 = Me, entries 8–10) as a consequence of the higher nucleophilicity of the p-tolyl ring and the subsequent formation of condensation products 4 that could not be avoided, even by starting the reactions at −38 °C (see Scheme 1 and ref. 14). The reactions of 2h with 1,2-dichlorobenzene and 1,2-dimethoxybenzene gave the corresponding products 6h and 7l with similar mole fractions of around 0.85. However, with o-xylene, indene 7k was obtained with a mole fraction of only 0.69, probably due again to methyl shifts caused by the presence of the dimethylphenyl ring.
In the case of 1j (R1 = OMe), the reactions were run in the presence of pyridine in order to avoid the protonation of the methoxyphenyl ring previously discussed. This does not allow to use the corresponding dienyl triflate 2j and implies a loss of regioselectivity of the intermolecular SEAr. Thus, lower mole fractions of 0.60 and 0.63 were observed for the indene derivatives 6i and 7m (entries 11 and 12). The very low yield of 23% of the reaction of 1j with 1,2-dichlorobenzene (entry 11) is not surprising: in this case, the methoxyphenyl ring of 1j is the strongest nucleophile and leads to the formation of condensation products and oligomers.
More limited results were obtained with meta- and para-substituted benzene derivatives. In the presence of para-dimethylbenzene (p-xylene), complex mixtures of products were observed. We assume that despite the symmetry of p-xylene, which rules out regioselectivity issues, steric hindrance due to the SEArs on the nearest neighbour positions of the methyl groups may lead to the competitive formation of condensation products. In the case of meta-dimethylbenzene (m-xylene), two main indene isomers 8 and 9 were observed, which formation strongly depends on the reaction medium (Scheme 3). Alkylbenzenes are partially protonated in superacids and this occurs mostly in ortho/para positions, i.e. in positions 4 and 2 in the case of m-xylene.21 Thus, in TfOH the intermolecular SEAr takes place preferentially on position 5 of m-xylene and this leads to isomer 8. The degree of protonation varies with the acidity of the reaction medium and, consequently, in the less acidic medium TfOH/pyridine, the SEAr takes place preferentially on position 4 and leads to isomer 9. However, in this later case the yield was significantly decreased (38% versus 71% for the reaction without pyridine), probably due again to steric hindrance issues. It should also be mentioned that, in each case, several minor isomers were formed by either reaction on position 2 of m-xylene, cyclization via the phenyl ring of 1a, or methyl shifts, resulting in a reduction in mole fraction of products 8 and 9 in mixture (see xi values in Scheme 3). The structures of 8 and 9 were determined with absolute certainty by NOESY correlations (see spectral figures in ESI†).
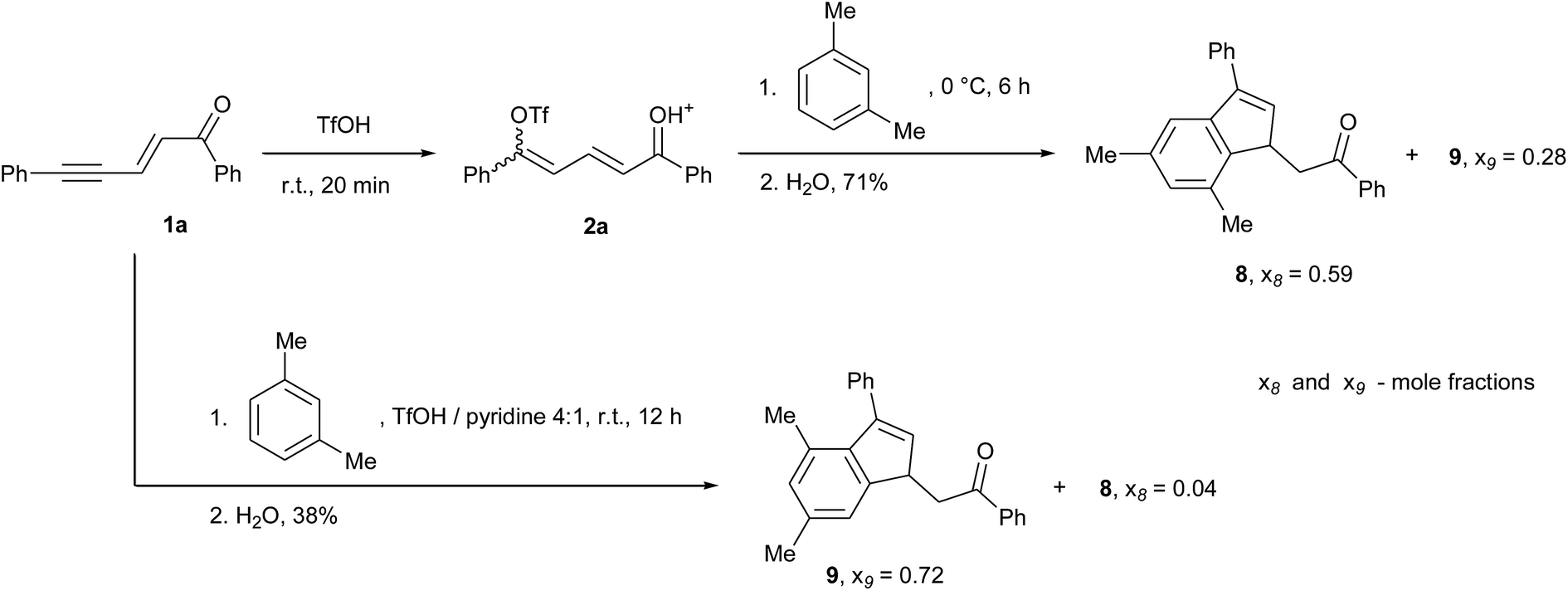 | ||
| Scheme 3 Reactions of enynone 1a and m-xylene leading to two products regioisomers depending on reaction conditions (x8 and x9 are the mole fractions of products 8 and 9 in mixture). | ||
Contrary to dienyl triflates 2 that can be isolated before cyclization into indan-1-ones 3 just by shortening the reaction time (Scheme 1 and ref. 14), the diaryl conjugated dienones 10, precursors of indenes 5, could never be observed in TfOH, even at low temperatures. But we showed in our previous communication14 that the presence of pyridine as a co-solvent of TfOH allowed more control on the cyclization step, as a result of the lowering in acidity (this was very useful for isolating the most reactive dienyl triflates 2g–j with R1 = Me and OMe). Based on this fact and because of the relative importance of dienones as intermediates in the synthesis of some pharmacologically active compounds,22 we explored different volume ratios of pyridine in TfOH and found out that a 1:6 ratio pyridine/TfOH decreases the cyclization rate enough to isolate the 5,5-diphenyl conjugated dienone 10a, already prepared and described in literature23 (see entry 1 in Tables 6 and S2 in ESI†). Higher volumes of pyridine lead to the competitive formation of dienyl triflate 2a as a consequence of the hydrogen bonding to pyridine which increases the nucleophilicity of triflate ions, in a similar way to fluoride ions in the Olah's reagent (i.e. 30% w/w pyridine in hydrogen fluoride).24 Actually, it was essential to limit as much as possible the formation of 2a since the (2E,4E)-isomer was found to be extremely difficult to separate from 10a.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Starting enynonea | Ar1 | R1 | Ar2 | Time, h | Product | Yieldb, % | Ratio (4Z)/(4E)c |
| a Concentration of starting enynone 0.2 mol L−1 and 10 equivalents of the aromatic nucleophile.b Yields of isolated products.c Ratios calculated by 1H NMR after separation by chromatography, when appropriate.d The cyclization product 5d was isolated as the main reaction product. | ||||||||
| 1 | 1a | Ph | H | Ph | 0.25 | 10a | 90 | |
| 2 | 1a | Ph | H | 2,5-Me2-C6H3 | 0.25 | 10b | 82 | 5.2:1 |
| 3 | 1a | Ph | H | 2,4,6-Me3-C6H2 | 0.25 | 10c | 74 | 3.8:1 |
| 4 | 1b | p-Cl-C6H4 | H | Ph | 0.25 | 10d | 89 | |
| 5 | 1c | p-Me-C6H4 | H | Ph | 0.25 | 10e | 57 | |
| 6 | 1d | p-OMe-C6H4 | H | Ph | 0.25 | 5dd | 89 | |
| 7 | 1e | p-NO2-C6H4 | H | Ph | 0.25 | 10f | 63 | |
| 8 | 1f | Ph | Cl | Ph | 0.25 | 10g | 47 | 1:1 |
| 9 | 1f | Ph | Cl | Ph | 1 | 10g | 91 | 1:1 |
| 10 | 1f | Ph | Cl | 2,5-Me2-C6H3 | 1 | 10h | 73 | 34:1 |
| 11 | 1f | Ph | Cl | 2,4,6-Me3-C6H2 | 1 | 10i | 83 | 34:1 |
| 12 | 1g | Ph | Me | Ph | 0.17 | 10j | 62 | 1:1 |
| 13 | 1g | Ph | Me | 2,5-Me2-C6H3 | 0.17 | 10k | 95 | 20:1 |
| 14 | 1g | Ph | Me | 2,4,6-Me3-C6H2 | 0.17 | 10l | 94 | 35:1 |
| 15 | 1j | Ph | OMe | 2,5-Me2-C6H3 | 0.17 | 10m | 87 | 17:1 |
| 16 | 1j | Ph | OMe | 2,4,6-Me3-C6H2 | 0.17 | 10n | 95 | 17:1 |
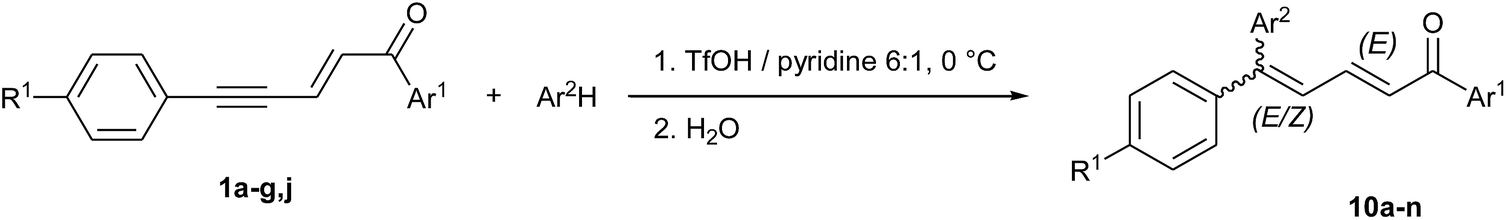
In the same reaction conditions, enynones 1b, 1c, 1e substituted on the aromatic ring Ar1 gave the corresponding 5,5-diphenyl dienones 10d–f as main reaction products (entries 4, 5 and 7). However, in the case of enynone 1d (Ar1 = p-OMe-C6H4) only the cyclization product 5d could be isolated (entry 6). Here, the presence of the stabilizing OMe substituent might favor the formation of a dicationic intermediate by further protonation of cation C (see Scheme 2), resulting in a faster intramolecular SEAr even in the presence of pyridine. Enynones 1f and 1g (with R1 ≠ H) reacted with benzene to give 1:1 mixtures of E/Z isomers of 10g and 10j which could not be separated by column chromatography (entries 9 and 12). Compound 1f (R1 = Cl) required an extended reaction time of 60 minutes for a full conversion while 10 minutes were sufficient in the case of 1g (R1 = Me), showing again the effect on reactivity caused by the presence of a substituent in this position.
In order to test the efficacy of the volume ratio TfOH/pyridine 6:1, reactions were conducted with the more nucleophilic derivatives p-xylene and mesitylene. Surprisingly, they gave predominantly the (2E/4Z)-isomers instead of 1:1 mixtures. Furthermore, the ratio 4Z/4E seems to increase with the presence of a substituent R1: for example, the 5-mesityl dienone 10l was obtained with a 35:1 ratio from the substituted enynone 1g (entry 14) against only 3.8:1 in the case of 10c, obtained from the non-substituted 1a (entry 3). The 4Z configuration of the major isomers was determined by NOESY correlations on the corresponding minor isomers in mixture (Fig. 2) and was confirmed by X-ray structure analysis of the compound (2E/4Z)-10l (Fig. 3).
 | ||
| Fig. 2 Selected NOESY correlations (arrows) showing the 4E configuration of the minor isomers (not observed on the major isomers). | ||
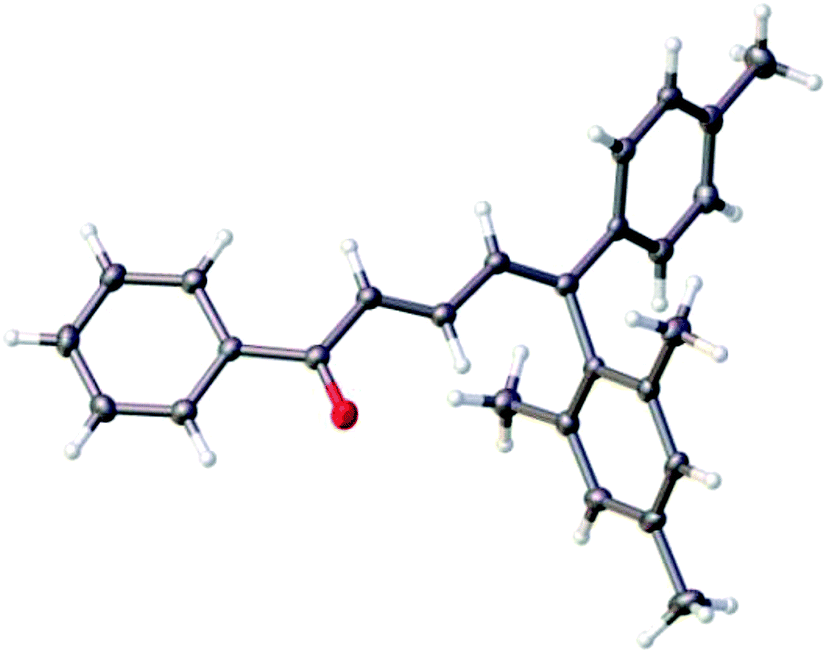 | ||
| Fig. 3 Molecular structure of (2E/4Z)-10l determined by X-ray structure analysis (ellipsoid contours of probability levels 50%). | ||
Finally, we could verify the cyclization of dienones 10 into the corresponding indene derivatives by dissolving them in TfOH (see Table S3 in ESI†) through cation C or C′ (see Scheme 2). In all cases, the cyclization products were obtained with the same regiochemistry as from the corresponding enynones and arenes but with slightly lower yields.
Conclusions
In conclusion, we have described a simple and efficient synthetic method towards diversely substituted indenes from conjugated 1,5-diarylpen-2-en-4-yn-1-ones and arenes via a tandem inter and intramolecular electrophilic aromatic substitutions in triflic acid. The regioselectivity of product formation is driven by the electronic properties of the aromatic substituents and has been optimized by transforming the starting enynones into the less reactive dienyl triflates, which give the indene products with higher mole fractions in a one-pot procedure. The use of pyridine as a co-solvent, by modulating the acidity of the reaction medium, allows at once to reduce the degree of protonation of the activated arenes and to control the extent of the tandem reaction. This way, the aryl-substituted dienone intermediates have been isolated before their cyclization into indenes and, in the case of substituted arenes, with high isomeric ratios in favour of the (2E,4Z) isomers.Experimental
Procedures for the transformation of enynones 1 into indenes 5, 6, 7, 8 and 9
General procedure for the transformation of enynones 1 into dienones 10
0.13 mL of benzene or 0.17 mL of p-xylene or 0.19 mL of mesitylene (1.4 mmol, 10 eq.) were dissolved in a solution of 0.1 mL of pyridine in 0.6 mL of TfOH. The resultant solution was stirred until homogeneous and cooled to 0 °C. Then, 0.14 mmol of enynone 1 were added portionwise and the reaction mixture was stirred at 0 °C during the indicated time (see details in ESI†). The mixture was then diluted with 3 mL of chloroform and quenched under vigorous stirring at 0 °C by dropwise addition of cold water (6 mL). The organic layer was separated and the aqueous layer was extracted again with chloroform. The combined organic layers were washed first with water and then with a diluted solution of NaHCO3, dried over Na2SO4 and concentrated under reduced pressure. The reaction products were isolated by flash column chromatography on silica gel. When appropriate, the isomeric ratios were determined by 1H NMR analysis.Acknowledgements
This work was supported by the Russian Scientific Foundation (grant no. 14-13-00448). Spectral studies were performed at the Centre for Magnetic Resonance, the Centre for Chemical Analysis and Materials Research, and the Research Centre for X-ray Diffraction Studies of the Saint Petersburg State University.Notes and references
- M. Koca, K. O. Yerdelen, B. Anil, Z. Kasap, H. Sevindik, I. Ozyurek, G. Gunesacar and K. Turkaydin, J. Enzyme Inhib. Med. Chem., 2016 DOI:10.1080/14756366.2016.1186019
.
- Z. Liu, R. Zhang, Q. Meng, X. Zhang and Y. Sun, Med. Chem. Commun., 2016, 7, 1352 RSC
- R. Leino, P. Lehmus and A. Lehtonen, Eur. J. Inorg. Chem., 2004, 2004, 3201 CrossRef
- V. Cadierno, J. Díez, M. P. Gamasa, J. Gimeno and E. Lastra, Coord. Chem. Rev., 1999, 193–195, 147 CrossRef CAS
- D. Zargarian, Chem. Rev., 2002, 233–234, 157 CAS
- C. Sui-Seng, A. Castonguay, Y. Chen, D. Gareau, L. F. Groux and D. Zargarian, Top. Catal., 2006, 37, 81 CrossRef CAS
- B. Gabriele, R. Mancuso and L. Veltri, Chem.–Eur. J., 2016, 22, 5056 CrossRef CAS PubMed
- G. A. Olah and D. A. Klumpp, Superelectrophiles and their chemistry, Wiley-Interscience, Hoboken, New Jersey, 2008 Search PubMed
- S. Nakamura, H. Sugimoto and T. Ohwada, J. Org. Chem., 2008, 73, 4219 CrossRef CAS PubMed
- N. Ghavtadze, R. Fröhlich, K. Bergander and E.-U. Würthwein, Synthesis, 2008, 21, 3397 Search PubMed
- X. Liu, Q. Zhang, D. Zhang, X. Xin, R. Zhang, F. Zhou and D. Dong, Org. Lett., 2013, 15, 776 CrossRef CAS PubMed
- K. N. Boblak and D. A. Klumpp, J. Org. Chem., 2014, 79, 5852 CrossRef CAS PubMed
- H. Kheira, P. Li and J. Xu, J. Mol. Catal. A: Chem., 2014, 391, 168 CrossRef CAS
- S. Saulnier, A. A. Golovanov, A. Y. Ivanov, I. A. Boyarskaya and A. V. Vasilyev, J. Org. Chem., 2016, 81, 1967 CrossRef CAS PubMed
- A. A. Golovanov, D. R. Latypova, V. V. Bekin, V. S. Pisareva, A. V. Vologzhanina and V. A. Dokichev, Russ. J. Org. Chem., 2013, 49, 1264 CrossRef CAS
- M. Barfield and D. M. Grant, J. Am. Chem. Soc., 1961, 83, 4726 CrossRef CAS
-
(a) R. C. Cookson, T. A. Crabb, J. J. Frankel and J. Hudec, Tetrahedron, 1966, 22(7), 355 CrossRef
- G. A. Olah and Y. K. Mo, J. Org. Chem., 1973, 38, 353 CrossRef CAS
- J. W. Larsen and M. Eckert-Maksic, J. Am. Chem. Soc., 1974, 96, 4311 CrossRef CAS
- G. A. Olah, A. M. White and D. H. O'Brien, Chem. Rev., 1970, 70, 561 CrossRef CAS
- D. Fărcaşiu, Acc. Chem. Res., 1982, 15, 46 CrossRef
- V. K. Tandon and K. A. Singh, Chem. Biol. Interface, 2012, 2, 76 CAS
- V. Cadierno, J. Díez, S. E. García-Garrido, J. Gimeno and N. Nebra, Adv. Synth. Catal., 2006, 348, 2125 CrossRef CAS
- G. A. Olah, J. T. Welch, Y. D. Vankar, M. Nojima, I. Kerekes and J. A. Olah, J. Org. Chem., 1979, 44, 3872 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization data and copies of NMR spectra of all products and cations, X-ray data. CCDC 1497472. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra21965a |
| ‡ According to the reaction mechanism, indenes 6b–e and 7b–e might also be obtained from enynone 1a by reaction with toluene or methoxybenzene. However, all attempts resulted in more complex reaction mixtures due to additional regioselectivity issues brought about by the first intermolecular SEAr on the substituted ring. |
| This journal is © The Royal Society of Chemistry 2016 |