
Palladium-catalyzed cross-dehydrogenative coupling of maleimides with simple arenes: a fast track to highly substituted maleimides†
Farnaz Jafarpour* and
Mitra Shamsianpour
School of Chemistry, College of Science, University of Tehran, P. O. Box 14155-6619, Tehran, Iran. E-mail: Jafarpur@khayam.ut.ac.ir
First published on 25th October 2016
Abstract
Pd-catalyzed cross-dehydrogenative coupling of N-substituted maleimides with simple arenes is explored. The protocol provides a versatile and atom-economic method for construction of a range of highly functionalized maleimides. The study unveils that a combination of TFA/PivOH is critical for diarylation.
Direct C–H bond functionalization reactions, in lieu of pre-functionalizations, have found extensive applications since this strategy presents an atom-economic way to construct complex molecules from simple starting materials which increases the overall efficiency of multistep synthetic sequences.1 Likewise, palladium catalyzed cross-dehydrogenative coupling reactions have proven to be one of the most powerful methodologies in modern organic chemistry.2 In this regard, much effort has been made toward the palladium(II)-catalyzed oxidative cross-coupling of arenes and olefin bonds. Although these studies showcase the ability of palladium to perform direct arylations with simple unactivated arenes, however, there are several problems associated with direct arylation of disubstituted olefinic bonds to construct tetrasubstituted olefins.3 The difficulty of generating tetrasubstituted alkenes using Heck coupling may be rationalized to steric congestion around double bonds.
Bisaryl-substituted maleimides have received significant attention for two main reasons; (a) they belong to the group of organic photochromic compounds with potential applications for optoelectronic devices4 and (b) these compounds belong to an important class of substrates found in several natural products as well as pharmaceuticals with diverse biological activities such as cancer therapy or inhibitory effect on the growth of fungi and bacteria.5 With an array of interesting properties, several research groups have aimed at construction of these structural motifs. Aryl-substituted maleimides are typically synthesized via multistep ring closing reactions.6 In recent decade transition metal-catalyzed methods for synthesis of 3,4-disubstituted maleimides have been developed by several researches. However, in most synthetic approaches functionalization of maleimides or the arene coupling partner is a pre-requisite, and the more synthetically viable preparation of these privileged scaffolds via direct arylation of unfunctionalized maleimides with arenes continues to be problematic (Scheme 1).7
 | ||
| Scheme 1 Methods for bisarylation of maleimides. | ||
Based on these results and in a continuation of our efforts aimed at the development of palladium-catalyzed C–H functionalization reactions,8 herein we devise a new catalytic system which allows for palladium-catalyzed C–H functionalization of simple unactivated arenes followed by cascade vicinal diarylation of the olefinic bond of maleimides. The present study uncovers a straightforward derivatization at 3- and 4-positions of the maleimide core for one-pot construction of highly substituted maleimides precluding installment of any disposal functionalities.
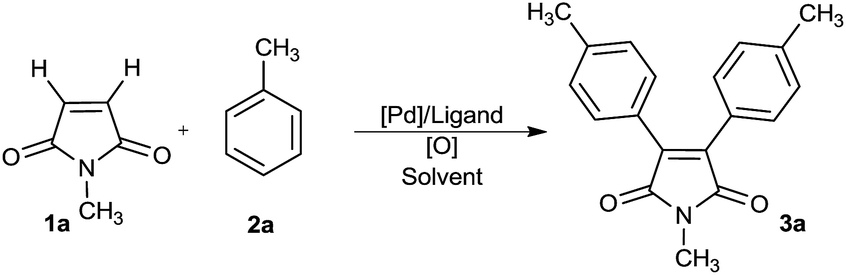
To test the hypothesis, we focused on oxidative arylation of N-methylmaleimide using toluene (Table 1). We were pleased to observe that the desired diarylated product was obtained employing a catalytic system comprising Pd(OAc)2, K2S2O8 and TFA, albeit in a 10% yield (entry 1). To our delight a combination of TFA/PivOH built up the yield fundamentally to 43% (entry 2). An oxidant screen revealed silver salts as the best choice of oxidants (entries 3–9). Accordingly, various silver salts were tested and the best result was obtained with AgOAC where, the desired product was gained in a 54% isolated yield (entries 6–9). In the absence of Ag salts however, the reactions did not proceed. Furthermore, the results showed that the presence of trifluoroacetic acid is essential to conduct the arylation process (entries 10 and 11). Among the Pd(II) catalysts tested, Pd(OAc)2 turned out to be the best, while no product could be detected in the presence of PdCl2 and Pd(dba)2 (entries 12–15). Ligands, such as phen, pph3, py or bpy, led to the comparable or lower yields of the arylated maleimide (entries 16 and 17). The best result was obtained from the reaction of maleimide 1a (0.1 mmol) with toluene 2a (4 mmol) using Pd(OAc)2 (20 mol%) and AgOAc (3 equiv.) in PivOH/TFA at 110 °C for 24 h.
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] | L | [O] | Solvent | Yield (%) |
| a Reaction conditions: N-methylmaleimide 1a (0.1 mmol), toluene 2a (4 mmol), catalyst (20 mol%), oxidant (0.3 mmol), ligand (20 mol%), TFA (1 mmol), PivOH (0.6 mmol), 110 °C, 24 h. | |||||
| 1 | Pd(OAc)2 | K2S2O8 | TFA | 10 | |
| 2 | Pd(OAc)2 | K2S2O8 | TFA/PivOH | 43 | |
| 3 | Pd(OAc)2 | Cu(OAc)2 | TFA/PivOH | 38 | |
| 4 | Pd(OAc)2 | TBP | TFA/PivOH | Trace | |
| 5 | Pd(OAc)2 | BP | TFA/PivOH | 16 | |
| 6 | Pd(OAc)2 | Ag2CO3 | TFA/PivOH | 50 | |
| 7 | Pd(OAc)2 | Ag2O | TFA/PivOH | 52 | |
| 8 | Pd(OAc)2 | AgNO3 | TFA/PivOH | 0 | |
| 9 | Pd(OAc)2 | AgOAc | TFA/PivOH | 54 | |
| 10 | Pd(OAc)2 | AgOAc | PivOH | Trace | |
| 11 | Pd(OAc)2 | AgOAc | TFA | 35 | |
| 12 | Pd(TFA)2 | AgOAc | TFA/PivOH | 50 | |
| 13 | PdCl2 | AgOAc | TFA/PivOH | 0 | |
| 14 | Pd(dba)2 | AgOAc | TFA/PivOH | 0 | |
| 15 | Pd(acac)2 | AgOAc | TFA/PivOH | 40 | |
| 16 | Pd(OAc)2 | Phen | AgOAc | TFA/PivOH | 52 |
| 17 | Pd(OAc)2 | PPh3 | AgOAc | TFA/PivOH | 50 |
Having identified the optimized conditions, we next investigated the scope of the proposed cross-dehydrogenative diarylation approach using various electron-rich and -deficient arenes (Table 2). We were pleased to find that a range of alkyl, alkoxy, aryl and halo substituted arenes smoothly underwent the diarylation process to give the desired multifunctionalized imides in moderate to good yields. To our delight, the catalytic system was also very effective toward construction of bis(3,4-dimethoxyphenyl)maleimide 3c, the key structural motif in permethyl ningalin B analogue which exhibits promising P-glycoprotein (P-gp)-modulating activity in Pgp-over-expressing breast cancer cell lines without causing any cytotoxicity toward normal cells (Scheme 2).9 Fluorobenzene was also tolerated in this reaction, leaving behind a good precursor for further functionalizations, albeit in low yield (3d). With bromobenzene however, bromide was engaged in the reaction course and replaced with imide unit to afford 3,4-diphenylmaleimide 3j as the sole product. Interestingly, employing biphenyl as coupling partner, gave rise to highly π-extended maleimide 3e which is proved to exhibit red-shifted photoluminescence bands.10


 | ||
| Scheme 2 P-glycoprotein inhibitor of permethyl ningalin B analogue. | ||
Furthermore, maleimides possessing N-aryl and -benzyl protecting groups also served as good coupling partners in this CDC reaction to afford diaryl maleimides in good to high yields (3f–3k). Cumene also led to relate adduct 3i, albeit in moderate yield. Unfortunately, no product was detected for the coupling reaction of highly electron-deficient arene and maleimide (3l). It should be mentioned that the treatment of N-methylmaleimide with methoxy substituted arenes, anisole and 2-methylanisole, provided a mixture of mono- and diaryl maleimides (Scheme 3). Possibly, introducing highly electron rich substituents on the olefinic bond, decreases its reactivity for the next arylation reaction and a mixture of products gains.
 | ||
| Scheme 3 Coupling of N-methylmaleimide with anisole and 2-methylanisole. | ||
Further investigations on the aforementioned reaction revealed that replacing palladium(II) acetate with palladium(II) trifluoroacetate in the absence of pivalic acid resulted the monoarylation of N-methyl maleimide core where no diarylated product could be detected (Scheme 4).
 | ||
| Scheme 4 Monoarylation of N-methyl maleimide via oxidative cross-coupling reaction. | ||
The proposed mechanism for the CDC coupling involves a Pd(II)/Pd(0) catalytic cycle (Scheme 5). The first step comprises insertion of metal into the unactivated C–H bond through two most commonly invoked mechanisms: electrophilic addition or oxidative insertion. Next olefin insertion is followed by β-hydride elimination to afford the monoarylated adduct which in turn, inserts to the aryl palladium species. A second β-hydride elimination and reductive elimination/deprotonation generates diaryl maleimide and palladium(0) catalyst which on oxidation regenerates Pd(II) to render the C–H bond functionalization catalytic. For an evidence for conversion of monoaryl adducts to diaryl ones in the course of the reaction, samples of the reaction mixture of the oxidative Heck coupling of maleimide and toluene were taken after 6 and 12 h of the reaction progress and mono/diarylmaleimide ratios of 3.3 and 1.1 were obtained, respectively.
 | ||
| Scheme 5 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed a concise and efficient palladium-catalyzed method for direct diarylation of N-alkyl(aryl) maleimides in one step with simple unactivated arenes. It is the first report on significant waste free arylation of maleimides with diverse biological as well as photophysical activities. Furthermore, in the present approach direct diarylation of olefinic bond to construct a tetrasubstituted alkene through a CDC reaction which is rarely precedented is flourished.Acknowledgements
We acknowledge the financial support from the University of Tehran.Notes and references
- For some recent reviews on C–H functionalizations see: (a) H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343 CrossRef CAS PubMed; (b) J. J. Topczewski and M. S. Sanford, Chem. Sci., 2015, 6, 70 RSC; (c) I. A. Sanhueza, A. M. Wagner, M. S. Sanford and F. Schoenebeck, Chem. Sci., 2013, 4, 2767 RSC; (d) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (e) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed; (f) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (g) X. Chen, K. M. Engle, D. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed.
- For some recent reviews on cross-dehydrogenative couplings see: (a) Y. Wu, J. Wang, F. Mao and F. Y. Kwong, Chem.–Asian J., 2014, 9, 26 CrossRef CAS PubMed; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (c) C. J. Scheuermann, Chem.–Asian J., 2010, 5, 436 CrossRef CAS PubMed; (d) J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540 RSC; (e) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (f) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed.
- (a) N. Gigant, F. Quintin and J.-E. Bäckvall, J. Org. Chem., 2015, 80, 2796 CrossRef CAS PubMed; (b) Z. He, B. Wibbeling and A. Studer, Adv. Synth. Catal., 2013, 355, 3639 CrossRef CAS; (c) K. H. Kim, S. Lee, S. H. Kim, C. H. Lim and J. N. Kim, Tetrahedron Lett., 2012, 55, 5088 CrossRef; (d) H. S. Lee, K. H. Kim, S. H. Kim and J. N. Kim, Adv. Synth. Catal., 2012, 354, 2419 CrossRef CAS; (e) Z. He, S. Kirchberg, R. Fröhlich and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 3699 CrossRef CAS PubMed; (f) J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170 CrossRef CAS PubMed.
- (a) M. E. Jang, T. Yasuda, J. Lee, S. Y. Lee and C. Adachi, Chem. Lett., 2015, 44, 1248 CrossRef CAS; (b) H. Xie, L. A. Ho, M. S. Truelove, B. Corry and S. G. Stewart, J. Fluoresc., 2010, 20, 1077 CrossRef CAS PubMed; (c) H.-C. Yeh, W.-C. Wu and C.-T. Chen, Chem. Commun., 2003, 404 RSC; (d) W.-C. Wu, H.-C. Yeh, L.-H. Chan and C.-T. Chen, Adv. Mater., 2002, 14, 1072 CrossRef CAS; (e) J. C. Crano and R. J. Guglielmetti, Organic photochromic and thermochromic compounds, Kluwer Academic/Plenum, New York, NY, 1999 Search PubMed.
- (a) M. H. Lauer, R. L. Drekener, C. R. D. Correia and M. H. Gehlen, Photochem. Photobiol. Sci., 2014, 13, 859 RSC; (b) G. Tsuji, K. Kawakami and Sh. Sasaki, Bioorg. Med. Chem., 2013, 21, 6063 CrossRef CAS PubMed; (c) I. Doi, G. Tsuji, K. Kawakami, O. Nakagawa, Y. Taniguchi and S. Sasaki, Chem.–Eur. J., 2010, 16, 11993 CrossRef CAS PubMed; (d) P. Y. Zhang, I. L. K. Wong, C. S. W. Yan, X. Y. Zhang, T. Jiang, L. M. C. Chow and S. B. Wan, J. Med. Chem., 2010, 53, 5108 CrossRef CAS PubMed; (e) C.-F. Cheng, Z.-C. Lai and Y.-J. Lee, Tetrahedron, 2008, 64, 4347 CrossRef CAS; (f) C. Peifer, T. Stoiber, E. Unger, F. Totzke, C. Schächtele, D. Marmé, R. Brenk, G. Klebe, D. Schollmeyer and G. Dannhardt, J. Med. Chem., 2006, 49, 1271 CrossRef CAS PubMed.
- (a) Ch. Zhu, G. Xu, D. Ding, L. Qiu and J. Sun, Org. Lett., 2015, 17, 4244 CrossRef CAS PubMed; (b) S. Roy, S. Roy and G. W. Gribble, Org. Lett., 2006, 8, 4975 CrossRef CAS PubMed; (c) S. Inoue, Y. Fukumoto and N. Chatani, J. Org. Chem., 2007, 72, 6588 CrossRef CAS PubMed.
- (a) L. H. Lim and J. Zhou, Org. Chem. Front., 2015, 2, 775 RSC; (b) M. Ambrogi, A. Ciogli, M. Mancinelli, S. Ranieri and A. Mazzanti, J. Org. Chem., 2013, 78, 3709 CrossRef CAS PubMed; (c) E. Awuah and A. Capretta, J. Org. Chem., 2011, 76, 3122 CrossRef CAS PubMed; (d) L. Bouissane, J. P. Sestelo and L. A. Sarandeses, Org. Lett., 2009, 11, 1285 CrossRef CAS PubMed; (e) A. El Yahyaoui, G. Felix, A. Heynderickx, C. Moustrou and A. Samat, Tetrahedron, 2007, 63, 9482 CrossRef CAS.
- (a) M. Khoobi, F. Molaverdi, F. Jafarpour, M. Abbasnia, M. Kubicki and A. Shafiee, Chem. Commun., 2015, 51, 11713 RSC; (b) F. Jafarpour, N. Jalalimanesh, M. Teimouri and M. Shamsianpour, Chem. Commun., 2015, 51, 225 RSC; (c) F. Jafarpour, H. Hazrati, N. Mohasselyazdi, M. Khoobi and A. Shafiee, Chem. Commun., 2013, 49, 10935 RSC; (d) F. Jafarpour, M. B. A. Olia and H. Hazrati, Adv. Synth. Catal., 2013, 355, 3407 CrossRef CAS; (e) F. Jafarpour, S. Rahiminejadan and H. Hazrati, J. Org. Chem., 2010, 75, 3109 CrossRef CAS PubMed; (f) F. Jafarpour and H. Hazrati, Adv. Synth. Catal., 2010, 352, 363 CrossRef CAS.
- J. W. Bin, I. L. K. Wong, X. Hu, Z. X. Yu, L. F. Xing, T. Jiang, L. M. C. Chow and W. S. Biao, J. Med. Chem., 2013, 56, 9057 CrossRef CAS PubMed.
- K. Onimura, M. Matsushima, M. Nakamura, T. Tominaga, K. Yamabuki and T. Oishi, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3550 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data, experimental procedures and copies of spectra. See DOI: 10.1039/c6ra24420c |
| This journal is © The Royal Society of Chemistry 2016 |