DOI:
10.1039/C6RA24546C
(Paper)
RSC Adv., 2016,
6, 109354-109360
Selective nitrogen bonding states in nitrogen-doped carbon via a solution plasma process for advanced oxygen reduction reaction†
Received
2nd October 2016
, Accepted 4th November 2016
First published on 10th November 2016
Abstract
Nitrogen-containing carbon matrix is a promising candidate for the oxygen reduction reaction (ORR) in metal–air batteries. However, there are still remaining challenges in the modification of desired carbon–nitrogen bonding for further enhancement of ORR catalytic activity. Herein, we designed a simple and effective method to have selective C–N bonding in synthesized carbon nanoparticles via a liquid plasma process. The nitrogen-containing carbon was synthesized by pyrazine (an aromatic structure precursor) and acrylonitrile (a linear structure precursor). From elemental analysis, the atomic percentage of nitrogen content in carbon synthesized from pyrazine and acrylonitrile was 8.58 and 2.97 at%, respectively. The structural analyses from scanning electron microscopy (SEM), X-ray diffraction (XRD) and Raman spectroscopy showed slight differences in the carbon matrices. X-ray photoelectron spectroscopy (XPS) demonstrated that the chemical bonding states of nitrogen were different and dependent on the structure of the original precursor. The relative percentage of amino-N of nitrogen was significantly higher when the linear structured acrylonitrile was applied as a precursor. In contrast, pyridinic-type nitrogen bonding was more preferred in carbon synthesized from heterocyclic structural pyrazine. From electrochemical analyses, the key factor for high ORR performance was not related to the total nitrogen content. Instead, a more positive ORR onset and peak potential was observed when the ratio of amino-N and quaternary-N increased, where the content of pyridinic-N had a significant effect on current density. This work opens new directions in designing and synthesizing selective C–N bonding within a N-doped carbon matrix through a simple solution process operated at room temperature.
1. Introduction
Metal–air batteries generate electricity through a redox reaction between the metal and oxygen in air. They feature an open cell structure, which admits a continuous supply of the cathode active material, oxygen, from the atmosphere (air).1–3 Since the cathode oxygen can be provided through the surrounding environment infinitely, the metal–air battery has a notably higher theoretical energy density compared to that of other traditional batteries, including metal-ion batteries.4 Among various types of metal–air family, the primary aluminium–air or rechargeable zinc–air battery could apply alkaline KOH as electrolyte and are possible to be developed as a safe and low cost battery system.5–8 Meanwhile, the oxygen reduction reaction (ORR) is the governing electrochemical reaction in overall performance including discharge rate and capacity of metal–air battery.9–11 ORR occurred at the cathode allows the reduction of oxygen to hydroxide ions in the alkaline electrolyte with the electrons obtained from the electrochemical oxidation of the metal anode.12,13 At the current state of art, ORR catalysts for alkaline system applied nickel, iron and cobalt based metal catalysts exhibited better performance than noble metal ORR catalyst in KOH.14,15 Other than metal-based catalyst, the nitrogen-doped (N-doped) carbon, of which the catalyst itself is embedded within the carbon matrix, has been attracting lots of attentions as next generation electrocatalyst since it provides additional advantages in terms of long term stability and durability.15–18 Theoretically, nitrogen doped in carbon matrix can induce defects due to the difference of atomic radius, bond length and electrical negativity. The conjugated hetero-atom and the surrounding sites become uneven distribution of electric charge state.19–22 As a result, carbon atoms are positively charged by the charge delocalization and promote reduction of oxygen molecules to water.23,24 For superior ORR activity of nitrogen-containing carbon material, in situ doping of nitrogen and the chemical states of nitrogen bonding are important.25
Four major bonding states of nitrogen could be observed: pyridinic, pyrrolic, quaternary and oxide-type nitrogen.26 Previous researches often report higher ORR activity was provided by pyridinic27–29 and/or quaternary-type30–32 of nitrogen. Quaternary-N normally constitutes a broad peak centered on 401.3 eV and can be further developed into double assignments:33 a center-type quaternary-N which located within the carbon matrix (also referred as graphitic-N) and valley-type quaternary-N which settled at the edge of the matrix. The center quaternary-N is less positively charged than valley-N, which the binding energy is shifted from 401 eV to 402.3 eV. Previous study has investigated the difference of ORR activity between two types of quandary-N, and suggested that valley-type quaternary-N with pyridinic-N showed superior ORR activity, where center-type quaternary-N has only limited effect on ORR mechanism.32 Recently, amino-N has also been reported to act as an electron donating group in N-doped carbon and attributed to advanced ORR activity.34–36 Since the effect on ORR activity induced by each bonding state is still debatable, many studies has focused on selective synthesis of specific C–N bonding configurations in order to further probe the relationship between each C–N bonding state and electrochemical performance. In general, selective synthesis of C–N bonding states are mostly conducted by bottom up synthesis using chemical vapor deposition (CVD),37 post-annealing with temperature varied from 550 to 1000 °C.32,38,39 However, these methods are complex process, high temperature and pressure, which make the approaches not economically favourable.40,41 Recently, a simple and effective synthesis method specific for in situ heteroatom-doped carbon has been reported. Solution plasma process (SPP), a cold plasma occurred in liquid under room temperature and atmospheric pressure, has been successfully applied to synthesize N-containing from pyridine,42,43 pyrazine,44 and N-methyl-2-pyrrolidone.45 Up to date, the precursors of N-containing carbon mostly focused on aromatic structure due to easier formation of sp2 graphitic carbon structure for higher conductivity. Panomsuwan et al. has firstly demonstrated the synthesis nitrogen-doped carbon by linear acrylonitrile.46 Although these results showed the possibility to apply room temperature solution plasma for ORR electrocatalysis synthesis, further discussion regarding selective C–N bonding via the process has yet been investigated. Herein, the comparison of chemical bonding states of nitrogen and its effect on ORR catalytic activity by aromatic and linear precursor was firstly reported. Detail of analyses for structure, chemical bonding states and electrochemical measurements were conducted in order to give significant insight to the corresponding chemical bonding states of nitrogen in N-containing carbon. Also, the impact of selective C–N bonding on ORR catalytic activity will be discussed in detail by electrochemical analysis. We expect that our work provides a simple process to synthesis highly selective nitrogen-doped carbon for advanced ORR activity.
2. Experimental section
2.1 Experimental apparatus
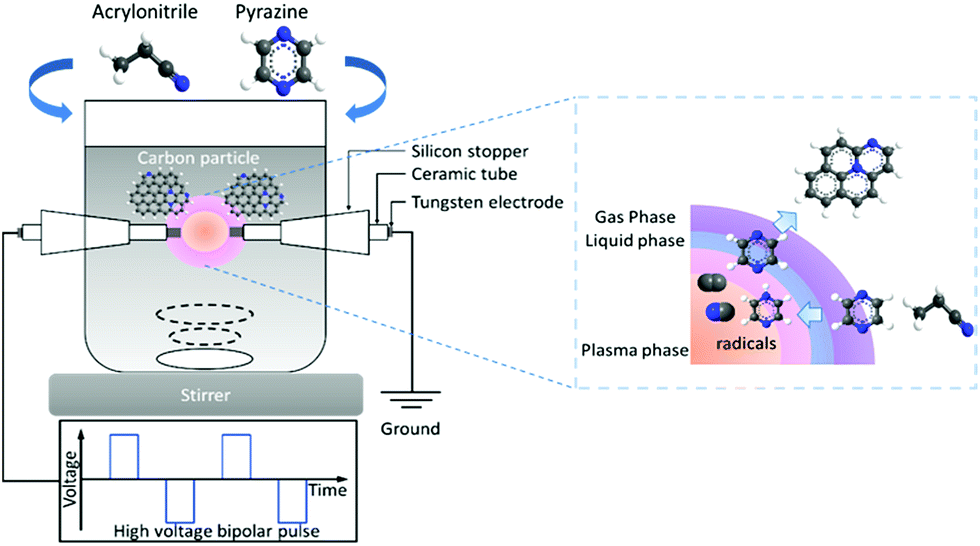
Fig. 1 shows the schematic image of SPP setup used in this study. Pure pyrazine (C4H4N2) (Wako Pure Chemical Industries, Ltd.) and pure acrylonitrile (CH2CHCN) (Kanto Chemical Co., Inc.) were applied as the precursors of N-doped carbon. The samples synthesized from pyrazine and acrylonitrile were hereafter denoted as PZ and AN, respectively. The theoretical ratios of carbon to nitrogen (C/N) of pyrazine and acrylonitrile are correspondingly 0.33 and 0.5. Solution plasma was generated in 100 mL of precursors under stirring and treatment time of 20 min. Diameter of 1 mm tungsten wire (Nilaco Corporation) was used as the electrodes, which were shielded with an insulating ceramic tube. A pair of tungsten electrodes was placed at the center of a glass reactor with a gap distance of 0.5 mm. A bipolar high voltage pulse of 2.0 kV was applied to the tungsten electrodes through a high-voltage bipolar pulse generator (Pekuris MPP-HV04). The pulse duration and repetition frequency were fixed at 1 μs and 20 kHz, respectively. Previous studies reported the precursors dissociated into active species (i.e., C2, CH, H, and CN) during plasma generation, then interacted and recombined with each other, resulting in forming carbon nanoparticles with a homogeneous incorporation of nitrogen atoms.43,46,47
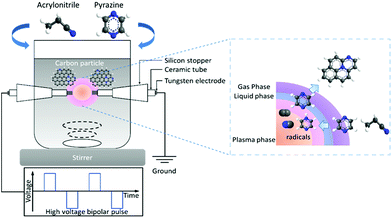 |
| | Fig. 1 Schematic image of solution plasma process for N-containing carbon synthesis using acrylonitrile and pyrazine. | |
After SP synthesis, all samples were separated by filtration using membrane filter, followed by repeated washing with ethanol (Kanto Chemical Co., Inc.), acetone (Kanto Chemical Co., Inc.), and ultrapure water in order to remove the remaining soluble organic compounds. The carbons were dried at 100 °C for 12 hours. Dried samples were ground with an agate mortar for further analyses.
2.2 Characterizations
Field emission scanning electron microscopy (FE-SEM) images were taken using a JEOL JSM-7610F scanning electron microscope at an accelerating voltage of 15 kV. The specific surface area was measured by Micromeritics Tristar II, Shimadzu for surface area and porosity analyses. Before the measurement, the carbon samples were annealed under vacuum at 300 °C for 3 hours. The phase structure was identified using a Rigaku Ultima IV X-ray diffractometer with monochromatic Cu Kα radiation (λ = 0.15418 nm) operating at 40 kV and 40 mA (1.6 kW). Raman spectra were recorded on a JASCO NRS-5100 Raman spectrometer equipped with a laser-excitation wavelength of 532.1 nm. The atomic ratios of carbon, hydrogen and nitrogen were calculated from data acquired by a Perkin Elmer 2400 Series II CHNS elemental analyser. X-ray photoelectron spectroscopy (XPS) measurements were carried out on a JEOL JPS-9010MC equipped with monochromatic Mg Kα X-ray radiation (1253.6 eV). The Mg X-ray source was operated at 25 mA and 10 kV. The binding energy was calibrated using the C 1s peak (284.5 eV) by the same method as our previous work.48 The fitting curves were analysed using a Gaussian line shape and Shirley background subtraction.
2.3 Electrochemical measurements
All electrochemical measurements were performed on a computer-controlled ALSCH model 704ES electrochemical analyser (CH instrument Inc.) with a typical three-electrode system in a 0.1 M KOH solution at room temperature. A platinum coil and Ag/AgCl (saturated KCl) were used as the counter and reference electrodes, respectively. The catalyst ink for the electrochemical measurements was prepared by dispersing 5 mg of finely ground catalyst in a mixture containing 900 μL of ethanol and 100 μL of Nafion®117 solution and ultrasonicating for 30 min. The catalyst loading was calculated to be 198 μg cm−2. Cyclic voltammetry (CV) measurements were performed in a 0.1 M KOH solution saturated with N2 and O2 at a scan rate of 50 mV s−1. The electrolyte was purged with high purity nitrogen or oxygen at a constant flow rate of 50 mL min−1 for 30 minutes prior to each measurement.
3. Results and discussion
3.1 Characteristics of solution plasma
For the physical phenomena, the discharges in organic solution were observed in two different stages, the initiation of plasma followed closely by the formation of discharge. Due to extremely low conductivity, the discharge could only be succeeded when the applied voltage is larger than 1 kV. The maximum discharge current reached between 3 to 10 A. Also, previous studies discovered that CN and C2 radicals generated during discharge under pyridine. These two radicals were suggested to initiate the formation of nitrogen-containing carbon.43,48
3.2 Morphology of as-prepared N-doped carbon
The morphology of carbon samples was observed under SEM as shown in Fig. 2. The images demonstrated that nano-sized agglomerates were formed. The specific surface area and porosity characteristics of each sample were measured by Brunauer–Emmett–Teller (BET) absorption, as shown in Table 1. The particle size formed by AN and PZ were 24.5 and 35.4 nm, respectively. Also, the surface area and pore volume of AN was comparatively higher than that of PZ. In general, a higher surface area and pore volume might have positive impact on ORR catalytic activity since it provides a higher interaction interface between oxygen and the catalyst.
 |
| | Fig. 2 SEM morphology of carbon samples formed by the (a) acrylonitrile (AN) and (b) pyrazine (PZ). | |
Table 1 Surface area, pore volume, pore size and average particle size of AN and PZ obtained from BET adsorption
| |
Surface area (m2 g−1) |
Pore volume (m3 g−1) |
Pore size (nm) |
Average particle size (nm) |
| AN |
245.44 |
1.94 |
5.09 |
24.5 |
| PZ |
169.3 |
1.17 |
5.6 |
35.4 |
3.3 Structural properties of as-prepared N-doped carbon
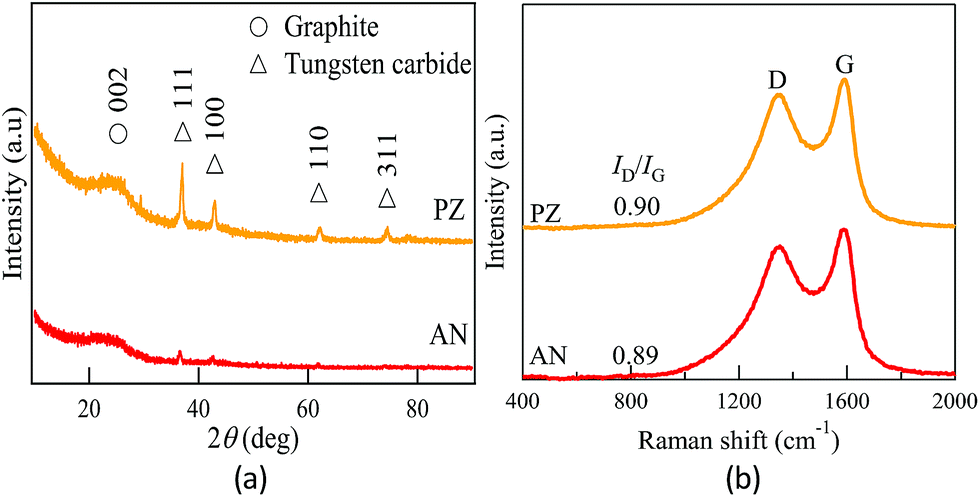
The XRD patterns of the carbon synthesized by SPP are shown in Fig. 3a. The broad peak at 2θ = 24.4° corresponded to graphite 002 reflection. The broad peak indicated low graphitic ordering in the carbon frameworks, regardless of the type of precursor. A slightly sharper peak indicated at 2θ = 24.4° in PZ indicated that the crystallinity was slightly higher compared to that of AN. The deference of crystallinity might be contributed by the original structure of precursor. During plasma process, the growth of carbon tends to remain its planar structure when pyrazine was applied as the main precursor, since the solvent originally appeared as ring/planar structure. Meanwhile, linear structure of acrylonitrile limited the carbon formation along the planar direction. Accompany with the results obtained from BET adsorption, it confirmed that the physical properties were dependent on the original structure of precursor.
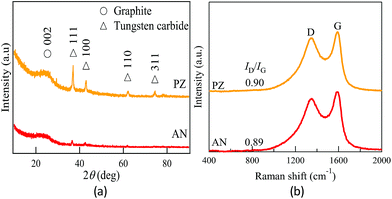 |
| | Fig. 3 (a) X-ray diffraction pattern and (b) Raman spectroscopy corresponding to AN and PZ. | |
Some sharp peaks observed at 2θ = 36.98°, 42.89°, 62.03° and 74.20° are identified as tungsten carbide (WC1−x). It can be observed that higher density of metal nanoparticles was presented in PZ. According to the qualitative study by energy dispersive X-ray (EDX) spectroscopy, the relative atomic concentration of tungsten were approximately 0.26 and 0.47 at%, respectively, in AN and PZ (Table S1†). Previous studies have reported tungsten electrodes were formed by occasional electrode sputtering and dispersed on carbon sample randomly.49 The size has been reported ranging from 5–10 nm.
Raman spectroscopy provides additional information about structural properties of carbon material. Raman spectra of carbon samples were shown in Fig. 3b. Two evident peaks appeared at approximately 1600 cm−1 and 1350 cm−1. The G peak at around 1600 cm−1 and D peak at around 1350 cm−1 are contributed, respectively, by the graphite-like structure corresponding to symmetry group of graphite crystal planes and the defect sites of the graphite-like structure. The increase of D peak implies higher defects within the carbon matrix, which also indicates successful doping of nitrogen atom into the graphitic framework. According to the research of Ferrari et al., the intensity ratio of D peak to G peak (ID/IG) can be used to estimate the degree of disorder for the carbon samples.50–52 The calculated ID/IG values were 0.89, and 0.90 for AN and PZ, correspondingly. The similar ID/IG ratio implied that the degree of defect in both catalysts were resemble.
3.4 Elemental composition and bonding configuration of N-doped carbon
The bulk elemental compositions were measured by elemental analysis and shown in Table 2. The nitrogen contents in AN and PZ were, respectively, 2.97 and 8.58 at%. Since nitrogen atoms were detected in all carbon samples, indicating doping of nitrogen into carbon was successfully via SPP. The original C/N ratio in precursors in AN and PZ were 0.33 and 0.5, respectively. The synthesized carbons have a much lower C/N ratio compared to that of original precursors. However, PZ still presented three times higher nitrogen in atomic percentage compared to that of AZ. The results illustrated that the nitrogen percentage was dependent on the original C/N ratio of precursor.
Table 2 Elemental compositions of C, H and N of each sample
| Elements |
Elemental composition (at%) |
| AN |
PZ |
| C |
77.25 |
63.45 |
| H |
10.2 |
17.87 |
| N |
2.97 |
8.58 |
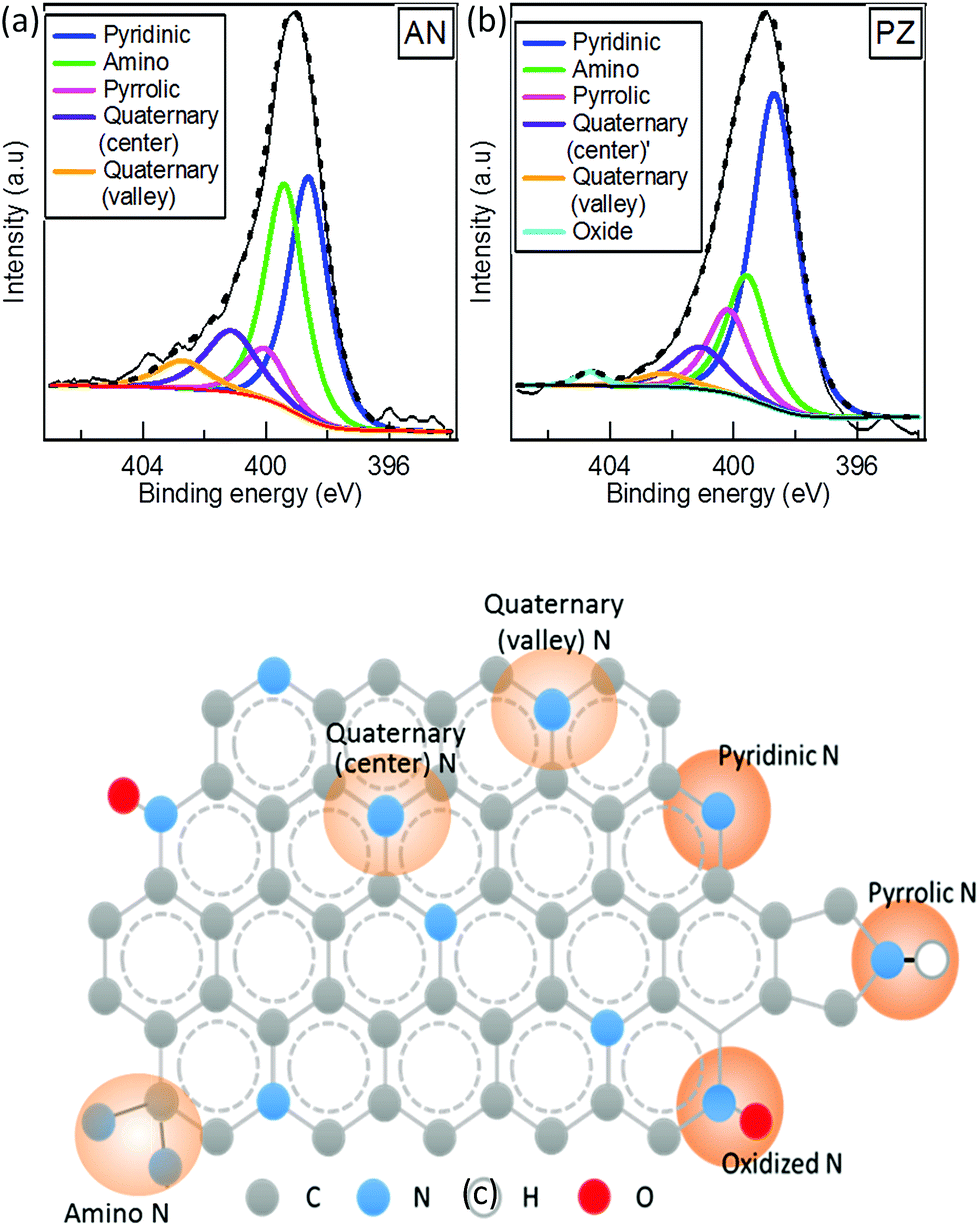
Compared to other SP-synthesized N-doped carbon, the hydrogen content was much higher (>10%) in our catalysts. It should be noted that all synthesized N-doped carbon in this study was dried at 100 °C, where N-doped carbons in other studies were mostly treated at post annealing treatment at 800 °C.42–44 The elimination of post-annealing process resulted at higher nitrogen and hydrogen contents. Further analysis of chemical bonding state was conducted by XPS. The wide survey spectra of carbon samples composed of C 1s, N 1s, and O 1s peaks (Fig. 4), which again confirmed successful incorporation of nitrogen atoms into carbon matrix. On the contrary, W 4f peak assigned for tungsten was hardly observed in both catalysts. The surface atomic concentrations of C, N, O and W were calculated from the corresponding peak area of the spectra (Table S2†). The total surface content of W in PZ and AN were, respectively, 0.5 and 0.8 at%. It is comparatively higher than the results obtained from EDS. The observation implies that tungsten carbide formed during the solution plasma process might prefer to adhere on the surface of the carbon. To investigate the bonding states of nitrogen atoms in the carbon catalysts, XPS N 1s narrow spectra were acquired and demonstrated in Fig. 5a and b, where the content of various nitrogen moieties was quantitatively reported in Table 3. XPS N 1s spectra were deconvoluted into six peaks including pyridine-N (398.5 ± 0.1 eV), amino-N (399.5 eV), pyrrolic-N (400.2 eV ± 0.2 eV), quaternary-N (center) (401.2 ± 0.2 eV), quaternary-N (valley) (402.3 ± 0.2 eV) and oxidized-N (403.9 ± 0.2 eV). The dominated peaks of AN were composed of both pyridinic-N and amino N, followed by quaternary-N (center). In PZ, the most pronounced peak was attributed by pyridinic N, which was almost twice the amount of that in AN. The relative concentration of amino-N was significantly different between two catalysts, where AN consisted of a significant higher quantity of amino N compared to PZ. The total concentration of quaternary-N was higher in AN as well. It is interesting to note that the ratio between center and valley types of quaternary-N were also different between two catalysts. The percentages of valley-N to total quaternary-N were 30.9% and 19.4% in AN and PZ, respectively. It can be generally accepted that pyridinic-N was highly favourable when the original precursor composed of pyridinic structure, where the functionalization occurred in higher opportunity at the edge when linear structure precursor was applied. The schematic of nitrogen bonding configurations of carbon sample is illustrated in Fig. 5c.
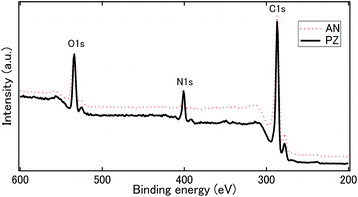 |
| | Fig. 4 XPS Wide spectra of AN and PZ. | |
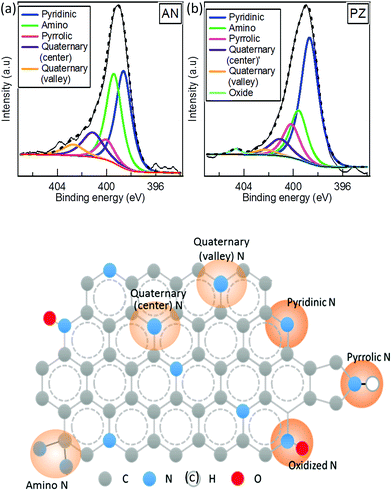 |
| | Fig. 5 XPS N 1s narrow spectra of (a) AN, (b) PZ and (c) the schematic of various types of nitrogen bonding states in N-doped carbon. | |
Table 3 Bonding states of nitrogen in AN and PZ from detailed deconvolution of XPS N 1s spectra
| Bonding |
Peak position (eV) |
Relative percentage (%) |
| AN |
PZ |
| Pyridinic |
398.5 ± 0.1 |
37.9 |
56.0 |
| Amino |
399.5 |
34.9 |
19.9 |
| Pyrrolic |
400.2 ± 0.2 |
8.5 |
13.5 |
| Quaternary (center) |
401.2 ± 0.2 |
12.8 |
8.1 |
| Quaternary (valley) |
402.3 ± 0.2 |
5.74 |
2.1 |
| Oxide |
403.9 ± 0.2 |
— |
1.39 |
3.5 Electrocatalytic activities towards ORR
Electrocatalytic activity of carbon samples was investigated by cyclic voltammetry (CV). Since the surface area of two catalysts was significantly different (Table 1), the current density in CV was normalized based the total surface area ratio of two samples, which denoted as normalized current density. In Fig. 7, the reduction current remained featureless in N2-saturated solution (dotted line), where a prominent cathodic peak was clearly observed under O2-saturated solution between −0.2 to −0.5 V (solid line). These results suggested that the synthesized catalysts possessed catalytic activity for ORR. The onset and peak ORR potentials of AN was, respectively, −0.23 and −0.31 V, which was more positive than that of PZ (−0.26 and −0.38 V). On the contrary, the peak current density of PZ was almost 40% higher than AZ at the peak potential. A slightly higher crystallinity of PZ was observed by XRD, which might have positive effect on current density. However, the effect is expected to be limited since the catalysts were still at its amorphous form. Although the existence of WC1−x was observed in both catalysts, the metal particle should not play an important role in current density as well. Previous study from our group demonstrated that annealing treatment at 800 °C was required to transform WC1−x to WC for decent ORR catalytic activity.43 The catalysts synthesized in this study was dried at 100 °C without further heat treatment. Also, the metal nanoparticles in the catalyst exhibited as purely WC1−x. Thus, it can be elucidated that the ORR activity was mainly provided by N-doped catalysts. Generally, a more positive ORR peak potential with higher current density indicates higher ORR catalytic activity. Herein, PZ exhibited a higher current density, where AN demonstrated a more positive ORR onset and peak potential. Since the two catalysts exhibited major differences in nitrogen bonding structure, therefore it is beneficial to elucidate the correlation between particular nitrogen functionalities and its specific role in ORR activity (Fig. 6).
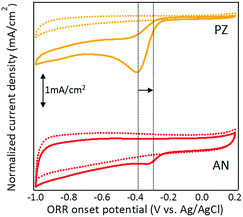 |
| | Fig. 6 CV curves of AN and PZ under N2 (dotted) and O2 (solid) saturated 0.1 M KOH solution. | |
3.6 Exploration of the roles of nitrogen states in ORR
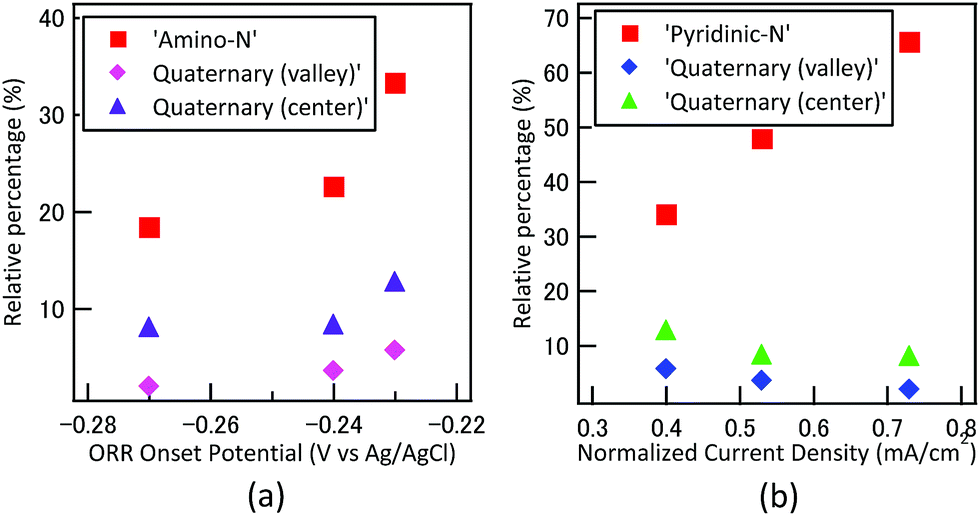
From elemental analysis, the atomic percentage of nitrogen of PZ was reported as 8.58%, which was much higher than that of AN (2.95%). On the contrary, the results from electrochemical measurement showed more positive ORR onset and peak potential in AN. Thus, it can be easily elucidated that the key factor for high ORR activity in nitrogen-containing carbon should not be attributed to the amount of nitrogen, but the type of chemical bonding states of nitrogen. By correlating the ORR measurements with the XPS surface analysis, the relationship between ORR onset potential, normalized current density and three major nitrogen states were summarized in Fig. 7. Catalyst synthesized by another heterocyclic C–N precursor pyridine (C5H5N), denoted as PD hereafter, was introduced as the third sample. The deconvoluted XPS N1S spectra, detailed bonding states of N 1s and CV of PD were demonstrated correspondingly in Fig. S4, Table S4 and Fig. S5.† Based on Fig. 7, we observed that both amino-N and quaternary-N exhibited positive effect on ORR onset potential, where the current density increased proportional to pyridinic-N concentration.
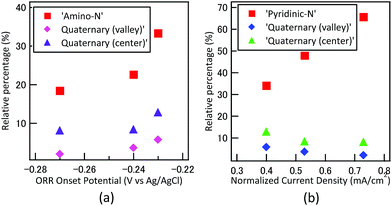 |
| | Fig. 7 Correlations between (a) ORR onset potential, amino-N type and quaternary-N bonding, (b) normalized current density, pyridinic-N and quaternary-N type bonding. | |
According to Zhang et al., the role of amino group has significant contribution to the onset potential and electron transfer number, where pyridinic-N was a key factor to enhance current density.35 From the electrochemical analysis, we observed similar trends as the Zhang et al. The normalized current of PZ (0.73 mA cm−2) was much higher than that of AN (0.4 mA cm−2). The ORR onset and peak potential, were both higher in the case of AN, as the carbon catalysts consisted of almost twice the quantity of amino N than PZ. The effect of quaternary-N with center and valley structure on ORR activity has been explored by Sharifi et al. The study precisely transformed the desired nitrogen functionality from already synthesized samples with varying heat treatment temperatures.32 The results suggested that valley-type quaternary-N with pyridinic structure showed superior ORR activity, especially on current density. On the other hand, center-type quaternary-N exhibited only limited effect on ORR mechanism. From our results, we observed that both types of quaternary-N appeared to have positive effect on ORR onset potential, but not significant to current density. It should be noted that the relative percentage of quaternary-N (center) and quaternary-N (valley) in N-doped carbon catalysts reported by Sharifi et al. were over 30 and 10%, respectively, which was much higher than our catalysts. The effect of quaternary-N might show more significant effect towards ORR with higher percentage.
Other researchers proposed the positive effect in ORR catalytic activities by pyridinic-N,53 quaternary-N,54 or the combination of both types of C–N bonding.55,56 Other types of nitrogen bonding including oxidized-N and pyrrolic-N presented at relatively low percentages in the SP-synthesized nitrogen-doped carbon. These two bonding states of nitrogen might not have significant effect on ORR activity according to previous study.57 Although the effect on ORR activity induced by each bonding state is still debatable, based on our current results, we would like to propose the synergic effect of enhanced ORR activity should be included when combining amino-N, quaternary-N and pyridinic-N within the carbon matrix.
4. Conclusions
In this work, we have reported a simple and effective way to selective doped particular nitrogen bonding states within carbon via solution plasma process. The original structure of nitrogen and carbon precursor has a significant effect on the types of nitrogen bonding. Amino-type N was more favorable when linear structure precursor, while the formation of pyridinic-N was preferred when heterocyclic structure was applied as precursor. Within all types of nitrogen bonding, amino-N and quaternary-N found to be effective on ORR onset potential, where the percentage of pyridinic-N has positive effect on current density. Also, we have proved that the amount of nitrogen content within the carbon matrix was not the key factor for high ORR activity. The above results suggested solution plasma synthesis method has the superiority in designing N-doped electrocatalyst for advanced ORR activity, and it can be highly application to the development of metal–air battery.
Notes and references
- Z. Wang, D. Xu, J. Xu and X. Zhang, Chem. Soc. Rev., 2014, 43, 7746 RSC.
- J. Xiao, D. H. Wang, W. Xu, D. Y. Wang, R. E. Williford, J. Liu and J. G. Zhang, J. Electrochem. Soc., 2010, 157, A487 CrossRef CAS.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172 RSC.
- M. Mirzaeian and P. J. Hall, J. Power Sources, 2010, 195, 6817 CrossRef CAS; F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172 RSC.
- X. Yang, P. He and Y. Y. Xia, Electrochem. Commun., 2009, 11, 1127 CrossRef CAS.
- J. S. Lee, S. Kim, R. Cao, N. S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34 CrossRef CAS.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257 RSC.
- H. Ma and B. Wang, RSC Adv., 2014, 4, 46084 RSC.
- T. Thippani, S. Mandal, G. Wang, V. K. Ramani and R. Kothandaraman, RSC Adv., 2016, 6, 71122 RSC.
- K. Kinoshita, Electrochemical Oxygen Technology, Wiley, New York, 1992 Search PubMed.
- K. Harting, U. Kunz and T. Turek, J. Phys. Chem., 2012, 226, 151 CAS.
- C. Tran, X. Q. Yang and D. Qu, J. Power Sources, 2010, 195, 2057 CrossRef CAS.
- C. R. Raj, A. Samanta, S. H. Noh, S. Mondal, T. Okajima and T. Ohsaka, J. Mater. Chem. A, 2016, 4, 11156 CAS.
- R. P. Hamlen and T. B. Atwater, in Metal/air batteries, Handbook of Batteries, ed. D. Linden and T. B. Reddy, McGraw-Hill, New York, 3rd edn, 2002, ch. 38 Search PubMed.
- J. Xiao, D. H. Mei, X. L. Li, W. Xu, D. Y. Wang, G. L. Graff, W. D. Bennett, Z. Nie, L. Saraf, I. Aksay, J. Liu and J. Zhang, Nano Lett., 2011, 11, 5071 CrossRef CAS PubMed.
- M. Prabu, P. Ramakrishnan, H. Nara, T. Momma, T. Osaka and S. Shanmugam, ACS Appl. Mater. Interfaces, 2014, 6, 1654C Search PubMed.
- K. Liu, Z. Zhou, H. Wang, X. Huang, J. Xu, Y. Tang, J. Li, H. Chu and J. Chen, RSC Adv., 2016, 6, 55552 RSC.
- D. U. Lee, P. Xu, Z. P. Cano, A. G. Kashkooli, M. G. Park and Z. Chen, J. Mater. Chem. A, 2016, 4, 7107 CAS.
- K. A. Kurak and A. B. Anderson, J. Phys. Chem. C, 2009, 113, 67 Search PubMed.
- G. L. Tian, Q. Zhang, B. Zhang, Y. G. Jin, J. Q. Huang, D. S. Su and F. Wei, Adv. Funct. Mater., 2014, 24, 5956 CrossRef CAS.
- Y. Tang, B. L. Allen, D. R. Kauffman and A. Star, J. Am. Chem. Soc., 2009, 131, 13200 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed.
- K. Parvez, S. B. Yang, Y. Hernandez, A. Winter, A. Turchanin, X. L. Feng and K. Müllen, ACS Nano, 2012, 6, 9541 CrossRef CAS PubMed.
- Y. Feng, F. Li, Z. Hu, X. Luo, L. Zhang, X. Zhou, H. Wang, J. J. Xu and E. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155454 CrossRef.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193 CrossRef CAS.
- V. Caramia and B. Bozzini, Mater. Renew. Sustain. Energy, 2014, 3, 28–40 CrossRef.
- C. V. Rao, C. R. Cabrera and Y. Ishikawa, J. Phys. Chem. Lett., 2010, 1, 2622 CrossRef CAS.
- S. Kundu, T. Nagaiah, W. Xia, Y. Wang, S. Dommele, J. Bitter, M. Santa, G. Grundmeier, M. Bron, W. Schuhmann and M. Muhler, J. Phys. Chem. C, 2009, 113, 14302 CAS.
- S. Maldonado and K. J. Stevenson, J. Phys. Chem. B, 2005, 109, 4707 CrossRef CAS PubMed.
- Y. Su, H. Jiang, Y. Zhu, X. Yang, J. Shen, W. Zou, J. Chen and C. Li, J. Mater. Chem. A, 2014, 2, 7281 CAS.
- P. Wang, Z. K. Wang, L. X. Jia and Z. L. Xiao, Phys. Chem. Chem. Phys., 2009, 11, 2730 RSC.
- T. Sharifi, G. Hu, X. Jia and T. Wagberg, ACS Nano, 2012, 6, 8904 CrossRef CAS PubMed.
- J. R. Pels, F. Kapteijin, J. A. Moulijin, Q. Zhu and K. M. Tomas, Carbon, 1995, 33, 1641 CrossRef CAS.
- H. Kim, K. Lee, S. I. Woo and Y. Jung, Phys. Chem. Chem. Phys., 2011, 13, 17505 RSC.
- C. Zhang, R. Hao, H. Liao and Y. Hou, Nano Energy, 2013, 2, 88 CrossRef CAS.
- Z. Jiang, Z. J. Jiang, X. Tian and W. Chen, J. Mater. Chem. A, 2014, 2, 441 CAS.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7935 Search PubMed.
- C. K. Chang, S. Kataria, C. Kuo, A. Ganguly, B. Wang, J. Hwang, K. Huang, W. Yang, S. Wang, C. Chuang, M. Chen, C. Huang, W. Pong, K. Song, S. Chang, J. Guo, Y. Ta, M. Tsujimoto, S. Isoda, C. Chen, L. Chen and K. H. Chen, ACS Nano, 2013, 7, 1333 CrossRef CAS PubMed.
- Z. Luo, S. Lim, Z. Tian, J. Shang, L. Lei, B. MacDonald, C. Fu, Z. Shen, T. Yu and J. Lin, J. Mat. Chem., 2011, 21, 8038 RSC.
- Z. Wang, P. Li, Y. Chen, J. Liu, H. Tian, J. Zhou, W. Zhang and Y. Li, J. Mater. Chem. C, 2014, 2, 7396 RSC.
- L. Ci, L. Song, C. Jin, D. Jariwala, D. Wu, Y. Li, A. Srivastava, Z. Wang, K. Storr, L. Balicas, F. Liu and P. M. Ajayan, Nat. Mater., 2010, 9, 430 CrossRef CAS PubMed.
- D. Kim, O. L. Li and N. Saito, Phys. Chem. Chem. Phys., 2015, 17, 407 RSC.
- D. Kim, O. Li, P. Pootawang and N. Saito, RCS Adv., 2014, 4, 16813 CAS.
- G. Panomsuwa, S. Chiba, Y. Kaneko, N. Saito and T. Ishizaki, J. Mater. Chem. A, 2015, 2, 18677 Search PubMed.
- K. Hyun, T. Ueno, O. L. Li and N. Saito, RSC Adv., 2016, 6, 6990 RSC.
- G. Panomsuwan, N. Saito and T. Ishizaki, Phys. Chem. Chem. Phys., 2015, 17, 6227 RSC.
- J. Kang, O. L. Li and N. Saito, Carbon, 2013, 60, 292 CrossRef CAS.
- G. Panomsuwan, T. Ishizaki and N. Saito, Carbon, 2016, 98, 411 CrossRef CAS.
- O. L. Li, H. Hayashi, T. Ishizaki and N. Saito, RSC Adv., 2016, 6, 51864 RSC.
- A. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS.
- A. Ferrari and J. Robertson, Philos. Trans. R. Soc., A, 2004, 362, 2477 CrossRef CAS PubMed.
- J. Robertson, Relat. Mater., 1994, 3, 361 CrossRef CAS.
- J. Wu, L. Ma, R. Yadav, Y. Yang, X. Zhang, R. Vajtai, J. Lou and P. Ajayan, ACS Appl. Mater. Interfaces, 2015, 7, 14763 CAS.
- W. He, C. Jiang, J. Wang and L. Lu, Angew. Chem., Int. Ed., 2014, 53, 9503 CrossRef CAS PubMed.
- G. Liu, X. Li, P. Ganesan and B. N. Popov, Electrochim. Acta, 2010, 55, 2853 CrossRef CAS.
- B. Merzougui, S. Bukola and R. Zaffou, Materials Today: Proceedings, 2016, 3, 691 CrossRef.
- G. Liu, X. Li, J. Lee and B. N. Popov, Catal.: Sci. Technol., 2011, 1, 207 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24546c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.