
Low-cost instant CO generation at room temperature using formic acid, mesyl chloride and triethylamine†
Cedrick
Veryser‡
 ,
Seger
Van Mileghem‡
,
Brecht
Egle
,
Philippe
Gilles
and
Wim M.
De Borggraeve
*
,
Seger
Van Mileghem‡
,
Brecht
Egle
,
Philippe
Gilles
and
Wim M.
De Borggraeve
*
Department of Chemistry, Molecular Design and Synthesis, KU Leuven, Celestijnenlaan 200F, box 2404, 3001 Leuven, Belgium. E-mail: Wim.DeBorggraeve@chem.kuleuven.be
First published on 15th February 2016
Abstract
Three low-cost standard lab chemicals, formic acid, mesyl chloride and triethylamine, were used for instant carbon monoxide (CO) generation at room temperature. Subsequently this gas was implemented in palladium-catalysed aminocarbonylation chemistry. Moreover, 13C-enriched formic acid was used as one of the most economical CO precursors for 13C-carbonyl labelling.
Carbon monoxide is beyond doubt one of the most important C1 building blocks for organic synthesis.1 In addition, CO can also act as an excellent ligand in transition metal chemistry.2–4 Although carbonylation chemistry is very mild and versatile, safety issues constrain the direct utility of this highly toxic gas. Especially on lab scale in a typical research lab, storage and use of pure, pressurised carbon monoxide gas raise serious safety concerns. In order to improve the applicability of this gas, an alternative carbon monoxide source is highly desired.
Different CO sources have successfully been used: examples are metal carbonyl derivatives,5 aldehydes,6,7 formamides,8,9N-formyl saccharin10,11 and aryl formates.12,13 Unfortunately, these precursors are either very toxic themselves, require high temperatures for carbon monoxide liberation or generate a nucleophilic by-product that can participate in the carbonylation reaction.
The Skrydstrup group developed three efficient carbon monoxide generating systems: methyldiphenylsilacarboxylic acid,14 9-methyl-9H-fluorene-9-carbonyl chloride15 and tetraphenyldimethyldisilane in combination with carbon dioxide.16 These CO-precursors, however, are non-trivial specialty chemicals with an associated higher cost. It would be advantageous if inexpensive commodities could be used to release carbon monoxide at ambient temperature in a robust manner. Different groups are working towards optimising such precursor systems.
In 2015, rapid hydrolysis of chloroform to carbon monoxide by caesium hydroxide hydrate was described.17 As chloroform is an inexpensive bulk chemical, it is an interesting CO precursor with the possibility of 13C labelling since 13C-CHCl3 is commercially available. On the other hand, the heterogeneous hydroxide (CsOH·H2O) base is an expensive chemical and must be used in excess to ensure rapid hydrolysis of chloroform. Replacement of this caesium salt by a more cost efficient one leads to a drastic decrease in carbon monoxide production.
Generation of carbon monoxide by dehydration of formic acid18 in the presence of sulfuric acid is known as the Morgan reaction.19 The utility of this reaction was improved by adapting this CO-generating system to a continuous microflow system by using a tube-in-tube reactor or by making use of a two-chamber system.20 However, this system still requires corrosive concentrated sulfuric acid. Zeolites are known to be safe alternatives to strong and corrosive liquid acids.21 Although replacement of sulfuric acid by zeolites seems promising, only a few specific zeolites can adequately decompose formic acid at an elevated temperature (150 °C), hampering the robustness of the method.22
Recently, the Wu group discovered that formic acid releases carbon monoxide in the presence of acetic anhydride under basic conditions.23–25 Moderate to excellent yields were obtained when using this CO precursor in Pd-catalysed alkoxycarbonylation, carbonylative Suzuki and Sonogashira coupling of aryl halides. Unfortunately, the decomposition of the mixed anhydride is slow, while instant carbon monoxide generation is desired as it directly stabilises the catalytic system and shortens the reaction time.26 Furthermore, this method is not suitable for Pd-catalysed aminocarbonylation chemistry as the in situ formed acetic formic anhydride is known to be a formylating agent for amines.27
In this paper we report a state-of-the-art low-cost CO generating system based on the instant decomposition of formic acid by the addition of mesyl chloride (MsCl) and triethylamine (Et3N) at room temperature (Scheme 1). This system is 30 to 140 times more cost efficient than non formic acid precursors (see the ESI†), since all the required starting products are inexpensive commodity chemicals. The replacement of acetic anhydride by mesyl chloride was necessary to ensure instant CO generation at room temperature (see the ESI†). This replacement also contributes to the general safety aspects of the procedure, since mesyl chloride is a non-flammable chemical compared to acetic anhydride. As 13C-formic acid is also commercially available, this procedure provides an obvious source of 13CO. Comparing the costs of commercially available 13CO precursors, this system is probably one of the least expensive 13C-carbonylation methods (see the ESI†).
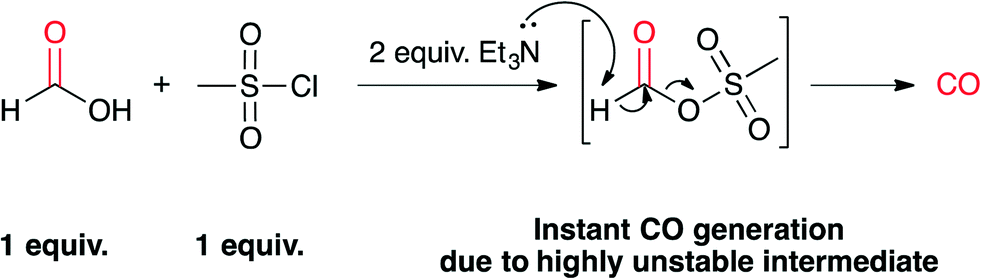 | ||
| Scheme 1 Proposed decomposition mechanism of formic acid in the presence of 1 equiv. mesyl chloride and 2 equiv. triethylamine leading to instant CO generation. | ||
To gain more insight into the steps involved in the decomposition of formic acid to carbon monoxide in the presence of mesyl chloride and triethylamine, a 13C-NMR study was conducted at room temperature (Scheme 2). First, 0.5 mmol of HCOOH was added to 600 μL of CD3CN (Scheme 2[A]). Then, 0.5 mmol of MsCl was added to this solution (Scheme 2[B]). At this point no changes seemed to occur in the NMR spectrum of the mixture, indicating that a base is needed to start the reaction. Subsequent dropwise addition of 1.0 mmol of Et3N resulted in vigorous gas development for several seconds. Afterwards the 13C-NMR spectrum was recorded (Scheme 2[C]) and complete decomposition of HCOOH was observed as the corresponding peak disappeared. MsCl also disappeared in the spectrum and a new peak at a lower ppm-value appeared (39.6 ppm), indicating the formation of the methanesulfonate anion and the triethylammonium cation (see the ESI†).
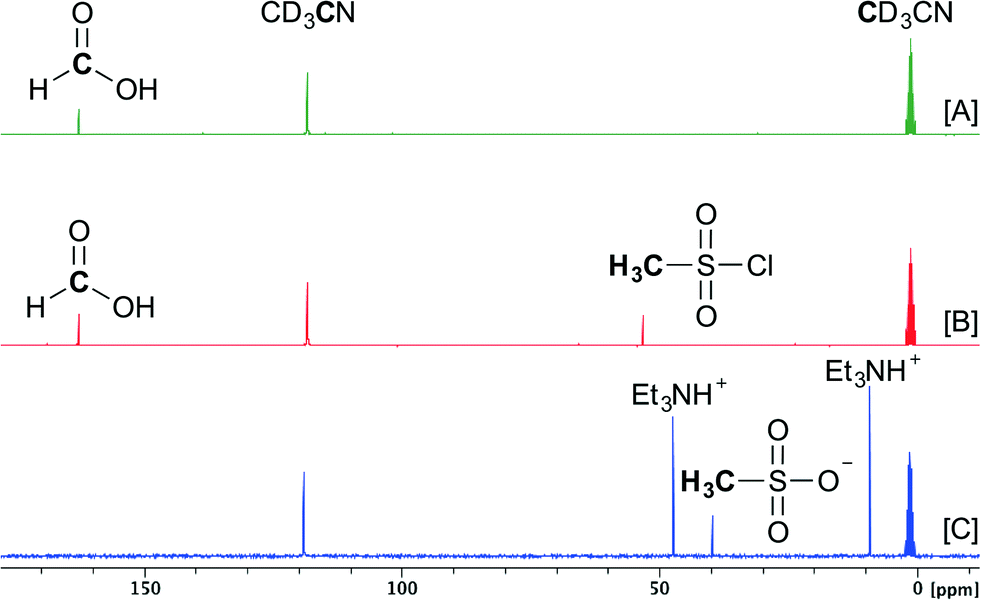 | ||
| Scheme 2 13C-NMR study of the CO precursor at room temperature. [A] 0.5 mmol of HCOOH in 600 μL of CD3CN. [B] 0.5 mmol of HCOOH and 0.5 mmol of MsCl in 600 μL of CD3CN. [C] 0.5 mmol of HCOOH, 0.5 mmol of MsCl and 1.0 mmol of Et3N in 600 μL of CD3CN. | ||
These results indicate that once the formic acid is deprotonated, it most probably reacts with mesyl chloride and forms a highly unstable mixed anhydride intermediate. A second deprotonation leads to instant CO formation, as the methanesulfonate is an excellent leaving group (Scheme 1). At this point it is not known whether the elimination of the sulfonate happens concerted with the deprotonation or not.
We also found that other sulfonyl chlorides are equally able to decompose formic acid to carbon monoxide in the presence of triethylamine: tosyl chloride, triflyl chloride, nosyl chloride, etc. However, for reasons of atom economy, mesyl chloride was our reagent of choice.
Palladium-catalysed aminocarbonylation of aryl bromides was chosen as the test case to prove the utility of this CO precursor. As already mentioned, in situ formation of CO by e.g. acetic formic anhydride will lead to formylation of the amine. More generally, decomposition of any precursor will lead to the formation of by-products which could potentially cause side reactions. In our case, the amine moiety (needed to form an amide) could react with mesyl chloride and a mixture of compounds might be expected. Inspired by the elegant two-chamber setup of the Skrydstrup group, ex situ CO generation was implemented to address these difficulties.28
We began our optimisation by using the conditions reported by Buchwald et al. as a benchmark for the Pd-catalysed aminocarbonylation chemistry.29 Initially, chamber A was filled with 5 mol% Pd(OAc)2/Xantphos, Na2CO3 (3 equiv.), 3 mL of dry degassed toluene (0.167 M), bromobenzene (1 equiv.) and n-hexylamine (1.5 equiv.). Chamber B was loaded with 3 equiv. formic acid and mesyl chloride. Finally, Et3N (6 equiv.) was added by injection through the septum in chamber B at room temperature (Table 1). Performing the reaction at 60 °C resulted in 41% of the desired amide 3a after 18 hours (entry 1). To our delight an increased yield was observed at higher temperatures (entries 2 and 3). In contrast to our results at 100 °C, Buchwald reported an incomplete conversion of the starting material at this temperature. This unusual result was ascribed to decreased catalyst stability at higher temperature.29 However, in our system, CO pressure builds up in the closed system. We assume that the generated pressure stabilises the catalytic system and therefore higher temperatures and lower catalyst loadings can be applied.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | T (°C) | t (h) | CO eq. | Catalyst/ligand | Conc. (M) | Yieldb (%) |
| a Reaction conditions: chamber A – bromobenzene (0.5 mmol), n-hexylamine (0.75 mmol, 1.5 equiv.), Na2CO3 (1.5 mmol, 3.0 equiv.), Pd(OAc)2 (0.005 mmol), Xantphos (0.005 mmol) and 1 mL of dry degassed toluene; chamber B – formic acid (0.65 mmol, 1.3 equiv.), methanesulfonyl chloride (0.65 mmol, 1.3 equiv.) in 2 mL of dry degassed toluene. Finally, Et3N (1.3 mmol, 2.6 equiv.) was added by injection through the septum in chamber B at room temperature. After 2 minutes the reactor was placed in an oil-bath at 100 °C. b Isolated yield. | ||||||
| 1 | 60 | 18 | 3.0 | 5 mol% Pd(OAc)2/Xantphos | 0.167 | 41 |
| 2 | 80 | 18 | 3.0 | 5 mol% Pd(OAc)2/Xantphos | 0.167 | 80 |
| 3 | 100 | 18 | 3.0 | 5 mol% Pd(OAc)2/Xantphos | 0.167 | 96 |
| 4 | 100 | 18 | 2.0 | 5 mol% Pd(OAc)2/Xantphos | 0.167 | 95 |
| 5 | 100 | 18 | 1.3 | 5 mol% Pd(OAc)2/Xantphos | 0.167 | 93 |
| 6 | 100 | 18 | 1.3 | 2 mol% Pd(OAc)2/Xantphos | 0.167 | 96 |
| 7 | 100 | 18 | 1.3 | 1 mol% Pd(OAc)2/Xantphos | 0.167 | 94 |
| 8 | 100 | 18 | 1.3 | 1 mol% Pd(P(Ph3))4 | 0.167 | 19 |
| 9 | 100 | 2 | 1.3 | 1 mol% Pd(OAc)2/Xantphos | 0.167 | 95 |
| 10 | 100 | 2 | 1.3 | 1 mol% Pd(OAc)2/Xantphos | 0.250 | 96 |
| 11 | 100 | 2 | 1.3 | 1 mol% Pd(OAc)2/Xantphos | 0.500 | 98 |

In order to increase the cost efficiency of this system, a reduction of CO and catalyst loading was investigated (entries 4–7). Performing the reaction with only 1 mol% Pd(OAc)2/Xantphos in the presence of 1.3 equiv. CO (assuming full conversion of the precursor) affords the amide in 94%. Further improvements were made by reducing the reaction time and increasing the concentration to 2 hours (entry 9) and 0.5 M (entry 11), respectively.
The ex situ generated CO was successfully implemented in the system comprising bromobenzene and n-hexylamine yielding 3a in 98%. With the optimised conditions in hand, the scope of the Pd-catalysed aminocarbonylation chemistry was further explored. As is shown in Scheme 3, different benzamides and Weinreb amides were synthesised in good to excellent yields.
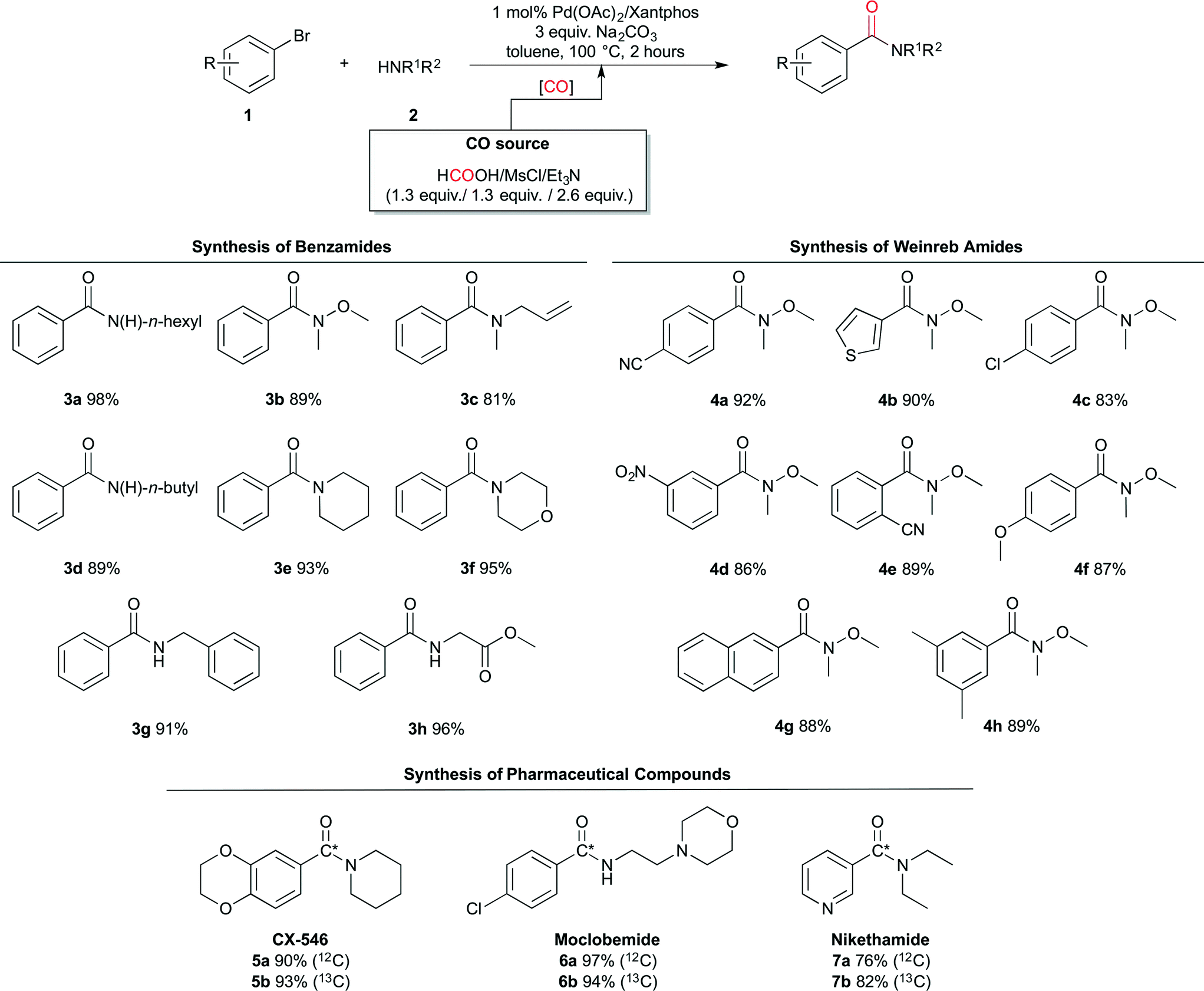 | ||
| Scheme 3 Palladium-catalysed aminocarbonylation by using the formic acid, mesyl chloride and triethylamine system as the CO source. a Reaction conditions: chamber A – bromobenzene (0.5 mmol), amine (0.75 mmol, 1.5 equiv.), Na2CO3 (1.5 mmol, 3.0 equiv.), Pd(OAc)2 (0.005 mmol), Xantphos (0.005 mmol) in 1 mL of dry degassed toluene; chamber B – HCOOH (0.65 mmol, 1.3 equiv.) and MsCl (0.65 mmol, 1.3 equiv.) in 2 mL of dry degassed toluene. Finally, Et3N (1.3 mmol, 2.6 equiv.) was added by injection through the septum in chamber B at room temperature. After 2 minutes the reactor was immersed in an oil-bath at 100 °C. | ||
The reaction conditions tolerate a wide variety of functional groups on both the aryl bromide and the amines, including allyl (3c), nitrile (4a and 4e), nitro (4d), aryl chlorides (4c and 6a) and esters (3h). In addition, also heteroaryl bromides (thiophene 4b and pyridine 7a) gave good to excellent yields.
This work was finalised with the synthesis of three relevant 13C-labelled pharmaceuticals: CX-54630 (5a and 5b), Moclobemide31 (6a and 6b), and Nikethamide32 (7a and 7b). These compounds were labelled using 13C-formic acid. Notably, this enriched precursor is at least 9 times less expensive per mmol of 13CO generated than any other reported commercial 13CO precursor (see the ESI†).
In conclusion, we report a state-of-the-art low-cost system to instantly generate CO at room temperature, which is composed of formic acid, mesyl chloride and triethylamine. Since these reagents are inexpensive commodity chemicals, which are available in research labs, this system belongs to one of the most economical and readily available CO precursor systems. The ex situ generated CO is successfully applied in Pd-catalysed aminocarbonylation chemistry resulting in high yielding amide formation. Remarkably, this is the first report on 13C-carbonylation labelling in a Pd-catalysed reaction by decomposition of 13C-HCOOH into 13CO.
Acknowledgements
CV and BE thank the FWO (PhD Fellowship of Research Foundation – Flanders) for fellowships received. WDB thanks KU Leuven for financial support via project OT/14/067.Notes and references
- I. Cipres, J. Jenck and P. Kalck, J. Mol. Catal., 1990, 58, 387–392 CrossRef CAS.
- G. Kiss, Chem. Rev., 2001, 101, 3435–3456 CrossRef CAS PubMed.
- A. Brennfuhrer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed.
- C. J. Mallia and I. R. Baxendale, Org. Process Res. Dev., 2015 DOI:10.1021/acs.oprd.5b00222.
- A. Wieckowska, R. Fransson, L. R. Odell and M. Larhed, J. Org. Chem., 2011, 76, 978–981 CrossRef CAS PubMed.
- T. Morimoto, K. Fuji, K. Tsutsumi and K. Kakiuchi, J. Am. Chem. Soc., 2002, 124, 3806–3807 CrossRef CAS PubMed.
- T. Morimoto, K. Yamasaki, A. Hirano, K. Tsutsumi, N. Kagawa, K. Kakiuchi, Y. Harada, Y. Fukumoto, N. Chatani and T. Nishioka, Org. Lett., 2009, 11, 1777–1780 CrossRef CAS PubMed.
- K. Hosoi, K. Nozaki and T. Hiyama, Org. Lett., 2002, 4, 2849–2851 CrossRef CAS PubMed.
- T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2012, 14, 5370–5373 CrossRef CAS PubMed.
- T. Ueda, H. Konishi and K. Manabe, Angew. Chem., Int. Ed., 2013, 52, 8611–8615 CrossRef CAS PubMed.
- T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2013, 15, 5370–5373 CrossRef CAS PubMed.
- T. Fujihara, T. Hosoki, Y. Katafuchi, T. Iwai, J. Terao and Y. Tsuji, Chem. Commun., 2012, 48, 8012–8014 RSC.
- N. Alonso, J. D. Munoz, B. Egle, J. L. Vrijdag, W. M. De Borggraeve, A. de la Hoz, A. Diaz-Ortiz and J. Alcazar, J. Flow Chem., 2014, 4, 105–109 CrossRef.
- S. D. Friis, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 18114–18117 CrossRef CAS PubMed.
- P. Hermange, A. T. Lindhardt, R. H. Taaning, K. Bjerglund, D. Lupp and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 6061–6071 CrossRef CAS PubMed.
- C. Lescot, D. U. Nielsen, I. S. Makarov, A. T. Lindhardt, K. Daasbjerg and T. Skrydstrup, J. Am. Chem. Soc., 2014, 136, 6142–6147 CrossRef CAS PubMed.
- S. N. Gockel and K. L. Hull, Org. Lett., 2015, 17, 3236–3239 CrossRef CAS PubMed.
- R. E. DeRight, J. Am. Chem. Soc., 1933, 55, 4761–4764 CrossRef CAS.
- J. S. Morgan, J. Chem. Soc., Trans., 1916, 109, 274–283 RSC.
- C. Brancour, T. Fukuyama, Y. Mukai, T. Skrydstrup and I. Ryu, Org. Lett., 2013, 15, 2794–2797 CrossRef CAS PubMed.
- M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072–1133 CrossRef CAS PubMed.
- P. Losch, A. S. Felten and P. Pale, Adv. Synth. Catal., 2015, 357, 2931–2938 CrossRef CAS.
- X. X. Qi, L. B. Jiang, C. L. Li, R. Li and X. F. Wu, Chem. – Asian J., 2015, 10, 1870–1873 CrossRef CAS PubMed.
- L.-B. J. Xinxin Qi, Hao-Peng Li and Xiao-Feng Wu, Chem. – Eur. J., 2015, 21, 17650–17656 CrossRef PubMed.
- X. Qi, C.-L. Li, L.-B. Jiang, W.-Q. Zhang and X.-F. Wu, Catal. Sci. Technol., 2016 10.1039/C5CY01957E.
- F. Aubke and C. Wang, Coord. Chem. Rev., 1994, 137, 483–524 CrossRef CAS.
- W. Stevens and A. Vanes, Recl. Trav. Chim. Pays-Bas, 1964, 83, 863–872 CrossRef CAS.
- R. H. Taaning, P. Hermange, A. T. Lindhardt, S. D. Friis and T. Skrydstrup, Denmark Pat., PA201070546, 2010 Search PubMed.
- J. R. Martinelli, D. A. Watson, D. M. M. Freckmann, T. E. Barder and S. L. Buchwald, J. Organomet. Chem., 2008, 73, 7102–7107 CAS.
- T. Lipina, K. Weiss and J. Roder, Neuropsychopharmacology, 2007, 32, 745–756 CrossRef CAS PubMed.
- G. Laux, Psychiatr Prax., 1989, 16, 37–40 Search PubMed.
- O. Feinsilver, Curr. Ther. Res. Clin. Exp., 1962, 4, 165–177 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 13C-NMR study of the CO generating system, influence of MsCl and Ac2O on the decomposition rate of HCOOH, price comparison of recently published CO and 13CO generating systems, detailed experimental procedures and copies of 1H-NMR and 13C-NMR data. See DOI: 10.1039/c6re00006a |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |