
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of microfluidics to control product selectivity during non-catalytic oxidation of naphthenic-aromatic hydrocarbons†
M. N.
Siddiquee
,
A.
de Klerk
and
N.
Nazemifard
*
Department of Chemical and Materials Engineering, University of Alberta, Edmonton, Alberta T6G 2V4, Canada. E-mail: nn1@ualberta.ca
First published on 23rd May 2016
Abstract
Liquid phase oxidation of a simple naphthenic-aromatic hydrocarbon was studied in a microreactor over low temperatures. The goal was to investigate how local oxygen availability could be controlled during oxidation and how it affected both oxidation rate and product selectivity. A microfluidic reactor provides a unique opportunity to exactly manipulate local oxygen availability by changing the hydrodynamics of the reactor in a Taylor flow region. Ketone-to-alcohol selectivity in the microreactor was increased by an order of magnitude from less than 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 14:1 by increasing oxygen availability in the liquid phase at a near constant conversion, oxygen partial pressure, and temperature. Similar oxidation experiments were conducted in semi-batch and batch reactors and the results were compared to the microreactor experiment. The higher ketone selectivity in the microreactor could be explained in terms of the oxidation of the alcohol and/or alkoxy radicals formed. It was observed that the selectivity to alcohols and addition products increased at lower oxygen availabilities. In addition to selectivity, the oxygen mass transfer coefficient to the liquid phase was also calculated based on the microreactor geometry and hydrodynamics.
:1 to 14:1 by increasing oxygen availability in the liquid phase at a near constant conversion, oxygen partial pressure, and temperature. Similar oxidation experiments were conducted in semi-batch and batch reactors and the results were compared to the microreactor experiment. The higher ketone selectivity in the microreactor could be explained in terms of the oxidation of the alcohol and/or alkoxy radicals formed. It was observed that the selectivity to alcohols and addition products increased at lower oxygen availabilities. In addition to selectivity, the oxygen mass transfer coefficient to the liquid phase was also calculated based on the microreactor geometry and hydrodynamics.
Introduction
Liquid phase oxidation of hydrocarbons is widely applied for the production of petrochemicals.1–3 Many of these processes employ oxidation catalysts, but not all feed materials are amenable to catalytic conversion. Homogeneous oxidation catalysts used for the production of fine chemicals generate wastes; catalyst deactivation and recycling are also problematic.4 When dealing with complex mixtures that contain potential fouling agents and catalyst poisons, it may be necessary to resort to non-catalytic oxidation. The challenge in free radical oxidation is to achieve good selectivity.At low temperatures some reaction pathways are not favored, because they require higher activation energies, but in free radical processes selectivity cannot always be controlled by just the kinetics. In practice, oxidation selectivity is governed by temperature in combination with oxygen availability.5–7 It is the local concentration of oxygen in proximity to the free radical intermediates that influences selectivity and hence the reference to oxygen availability rather than oxygen partial pressure.3,8 Although it is convenient to think in terms of bulk concentrations, the reactions that determine selectivity are local events. Yet, despite the industrial importance of selective liquid phase oxidation of hydrocarbons, only a few studies dealt with reaction engineering.3,5,8–12
Oxygen availability is influenced by mass transport. The transport of oxygen from the gas phase to the liquid phase, as well as the transport of oxidation intermediates between the bulk liquid and the gas–liquid interface, are likely important. The oxidation intermediates are oxygenates that may be surface active in an apolar hydrocarbon medium.5 A proper description of oxygen availability and how it is influenced by mass transport must therefore take oxygen and oxygenate transport into account.
During liquid-phase oxidation of hydrocarbons with molecular oxygen, various oxygenates like hydroperoxides, alcohols and ketones are produced as primary oxidation products.5,13,14 Secondary oxidation products (oxidized multiple times), and in some cases addition products (dimerized/oligomerized), are also generated.15 Oxidation proceeds via a free radical mechanism, which is well studied and reviewed in literature.5,13,14 Alkyl free radicals (R˙) are formed (initiation) during the induction period, but initiation has no appreciable effect on product selectivity.5 The reaction between alkyl radicals (R˙) and molecular oxygen (O2) is very fast and follows zero order kinetics at an oxygen partial pressure above 13 kPa.3,8 Once the propagation starts, selectivity to form oxidation products is changed depending on the oxidation conditions and structure of the hydrocarbons. Alcohols and ketones are generally formed at a 1:1 ratio for primary and secondary hydrocarbons:5,13 however, depending on the oxygen availability, this ratio changes. During industrial applications, low conversion (less than 5%) is preferred to control the selectivity of hydrocarbon autoxidation; otherwise, different kinds of by-products are produced.16 It is desirable to control selectivity and preferable to control selectivity independently from conversion.
Continuous-flow liquid phase oxidation in microreactors has received a lot of attention as the miniaturized reactor provides high interfacial areas resulting in enhanced heat and mass transfer rates, safe use of hazardous oxidants, and scale-up potential. The goal of the present study was to investigate how oxygen availability could be manipulated during the liquid-phase oxidation of naphthenic-aromatic hydrocarbons with molecular oxygen and how it was related to both oxidation rate and product selectivity. In order to have a clearly defined and measurable gas–liquid interface area, the experimental work in this investigation was conducted using a microfluidic reactor.
One of the advantages of using microfluidic reactors is the exact control of liquid and gas flow rates, as well as channel geometry and dimensions. This made it possible to calculate the exact gas–liquid interfacial area and film thickness by performing experiments in the Taylor flow region. The Taylor flow region (also known as slug flow) is characterized by a liquid–gas two-phase flow in which the gas phase exists as bubbles separated by liquid “slugs”. The internal circulation within the liquid slug ensures good mixing,17–21 and, more importantly, could transport the oxygenates from the interface to the bulk liquid. Moreover, the short radial diffusion length of microfluidic reactors ensures better mixing and it leads to a narrow residence time distribution (RTD).22 These are potentially advantageous for preventing the over-oxidation of oxygenates in consecutive processes and for controlling the product selectivity. Another important advantage of microfluidic reactor is that oxidation can be conducted using pure oxygen with a reduced risk of explosion.9,17,23
In this manuscript, the microfluidic reactor was used as a tool to prove fundamentally how selectivity can be manipulated at near constant conversion. It is not suggested that the microfluidic reactor be used for the large-scale production required by industry. The insights from this study can be applied to select appropriate reactor technology to achieve the same results at a larger scale, for example, the use of loop reactors24–26 for improved mass transfer.
Experimental
Materials
The list of chemicals used in this study is given in Table 1. Tetralin, known as a hydrogen-donor solvent, was selected as the model hydrocarbon due to its relevance to the energy industry and the richness of kinetic data. Di-tert-butyl peroxide (DTBP) was used as the initiator for a few experiments to overcome the initiation limitation. The oxidized model compounds of tetralin-1,2,3,4-tetrahydro-1-naphthol and α-tetralone were used for identification by GC-MS (gas chromatography-mass spectrometry). Hexachlorobenzene (99%, analytical standard, Supleco) was used as an internal standard in GC-FID (gas chromatography with a flame ionization detector) analysis for the conversion calculation and chloroform (98%, HPLC grade, Fischer Scientific) was used as a solvent for GC analysis. Extra-dry oxygen (99.6% molar purity, Praxair Inc., Canada) was used as an oxidizing agent and nitrogen (99.999% molar purity, Praxair Inc., Canada) was used to control backpressure. Properties of oxygen and tetralin for all the experimental conditions are reported in the ESI† (Table S3).| Name | CASRNa | Formula | Purity (wt%) | Supplier | |
|---|---|---|---|---|---|
| Supplierb | Analysisc | ||||
| a CASRN = Chemical Abstracts Services Registry Number. b This is the purity of the material guaranteed by the supplier. c This is the purity based on peak area obtained by GC-FID analysis. d Compounds were decomposed while GC-FID analysis and purity was not determined. | |||||
| Tetralin | 119-64-2 | C10H12 | 99 | 98.9 | Sigma-Aldrich |
| Di-tert-butyl-peroxide | 110-05-4 | C8H18O2 | 99 | —d | Acros Organics |
| 1,2,3,4-Tetrahydro-1-naphthol | 529-33-9 | C10H12O | 97 | —d | Aldrich |
| α-Tetralone | 529-34-0 | C10H10O | 97 | —d | Aldrich |
| Hexachlorobenzene | 118-74-1 | C6Cl6 | 99 | 99.1 | Supleco |
| Chloroform | 67-66-3 | CHCl3 | 99.1 | 98.1 | Fisher Scientific |
Equipment and procedure
The experimental setup employed for this investigation is shown in Fig. 1. Oxidations were performed at low temperatures (120–160 °C) and pressure to control product selectivity. The barometric pressure at the test location was on average 90 kPa. Taylor flow conditions were maintained in which tetralin slugs were separated by elongated oxygen bubbles (Fig. 2). The slug flow was maintained due to the higher boiling point of tetralin (207 °C) and minor changes in density, surface tension and viscosity (Table S3†). In a typical experiment, tetralin was loaded into the 5 ml syringe. After the syringe was loaded and before tetralin was fed through the system, the system was pressurized to 90 kPa gauge by flowing oxygen through the system. Tetralin was then fed into the system using a syringe pump. Gas–liquid slugs were formed due to the co-feed of tetralin and oxygen and an applied backpressure using a backpressure regulator and nitrogen gas. The flow rate of the gas–liquid slug was controlled by observing the flow of the gas–liquid slug and by changing the setting of the backpressure regulator. The flow patterns of the gas and liquid were monitored during the experiment using a digital camera mounted above the microfluidic reactor. The tetralin and oxygen co-feed in Taylor flow conditions was continued for twenty minutes, and the system was then depressurized. After the reaction, oxidized tetralin was collected from the pressure vessel using a needle valve, and stored for the instrumental analyses. After each experiment the reactor was flushed with acetone and dried by flowing nitrogen. Oxygen availability was manipulated by performing experiments at different injection rates (1 to 15 μL min−1) of tetralin. Liquid slugs and gas bubbles of different sizes were obtained. All of the experiments were performed in triplicate. Ten different slugs and gas bubbles from each experiment were used to calculate hydrodynamic parameters, mass transfer characteristics and oxygen availability. Oxidative conversion and product selectivity were calculated from the triplicate gas chromatographic analyses of the liquid phase products from each experiment.
 | ||
| Fig. 1 Experimental setup for tetralin oxidation by molecular oxygen. | ||
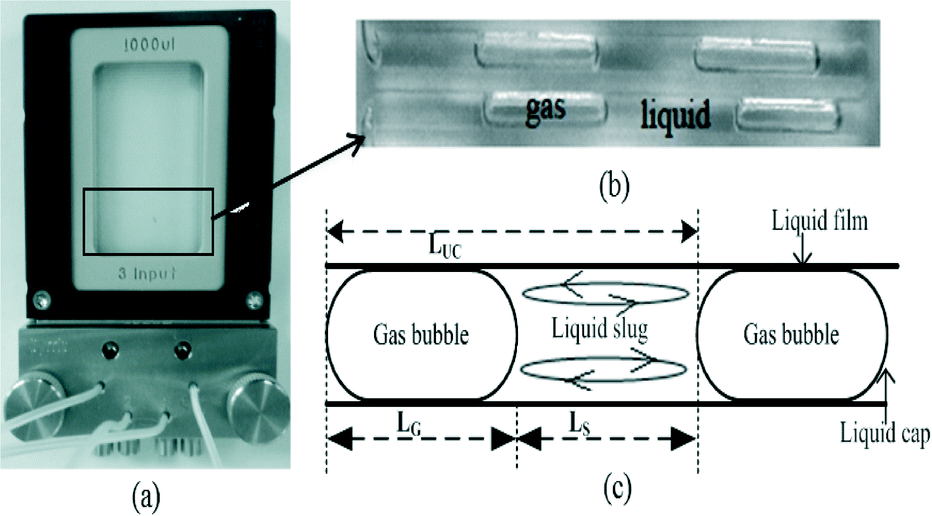 | ||
| Fig. 2 (a) Microfluidic reactor; (b) typical gas–liquid (tetralin–oxygen) slugs in a microfluidic reactor; (c) sketch of gas bubble–liquid slug to represent length of gas (oxygen) bubble (LG), length of liquid (tetralin) slug (LS), unit cell length (LUC), liquid film and liquid cap. Liquid can circulate within the liquid slug. (Reactor was placed horizontally during experiments). | ||
Pressure drop has an influence on the oxygen partial pressure and on the Taylor flow hydrodynamics that would change the bubble sizes during the experiments. Considering all the variations, many researchers have used the slug flow geometry to investigate different reaction kinetics.9–11
A 25 ml batch reactor manufactured from 316 standard stainless steel Swagelok fittings and tubing was used to compare the tetralin conversion and product selectivity with those of a microfluidic reactor. The reactor was equipped with a thermocouple and a pressure gauge to monitor the operating temperature and pressure, respectively. The batch reactor setup is shown in the ESI† (Fig. S2). Oxidation was performed at 130 °C for 2 min and 5 min (oxidation/residence time) after reaching the internal temperature within 1 °C of the heater temperature. Heating and cooling time are excluded from the oxidation time reported. The heat-up time required to reach an internal temperature within 1 °C of the heater temperature was six minutes. Tetralin was first charged into the reactor, and then oxygen was introduced into the reactor by maintaining the liquid-to-gas volume and the operating pressure at the same value as used for the microfluidic reactor. The reactor was then submerged into a temperature controlled preheated sand-bath heater (model: FSB-3, Omega Engineering, Inc., USA) to control the oxidation temperature. The reactor was removed from the sand bath heater at the end of oxidation and allowed to cool for 10 minutes. Sands from the surface of the reactor were cleaned. The reactor was depressurized and liquid oxidation products were collected for chromatographic analyses to calculate the oxidative conversion and product selectivity. A balance (Mettler-Toledo Model XP1203S, 1.2 kg weighing range, with 1 mg readability) was used to record the mass of the complete microreactor system after each step of the experiment.
Analyses
Gas chromatography with a flame ionization detector (GC-FID) was used for quantitative analysis. An Agilent GC-FID (Agilent 7890 A GC system) equipped with a DB-5 MS column 30 m × 0.25 mm × 0.25 μm column was employed for this purpose. The GC injector temperature was 250 °C, the split ratio was 10:1. The column flow of helium as the carrier gas was 2 mL min−1 and was kept constant. The oven temperature was 75 °C for 0.5 minutes and then the temperature was increased from 75 °C to 325 °C at a rate of 20 °C min−1, and, finally, the temperature was kept constant at 325 °C for 5 minutes. The samples were prepared using chloroform as solvent and hexachlorobenzene was added as an internal standard.
A GC-MS using an Agilent 7820A GC system (Agilent Technologies Canada Inc., Mississauga, ON) and an Agilent 5977E MSD (Agilent Technologies Canada Inc., Mississauga, ON) mass spectrometer was used to identify the oxidation products of tetralin. The products were separated on an HP-5 30 m × 0.25 mm × 0.25 μm column. The same temperature program as that in GC-FID analyses was used; the HP-5 and DB-5 MS columns also have similar separation characteristics. Oxygenates were classified as primary, secondary and addition products. Primary oxidation products are alcohol and ketones of tetralin, secondary oxidation products contain more than one ketone and/or alcohol functional groups, and addition products contain at least a dimer having different functional groups. Primary oxidation products were identified using GC-MS spectra of the commercially available alcohol and ketone of tetralin; secondary and addition products were identified using GC-MS spectra and the NIST library. Details of product identification are available in our previous study.15
A high-pressure differential scanning calorimeter (HP DSC) was used to evaluate the thermal behavior of the initiator (di-tert-butyl-peroxide). The instrument, Mettler-Toledo HP DSC 1, was equipped with an FRS-5 sensor employing 56 thermocouples, a Chiller (JULABO GmbHE, Germany) and a 400 W power amplifier. In a typical analysis, approximately 10 mg of sample was placed in an aluminum crucible with a pin (40 μL). The lid was pierced with a needle on a clean rubber surface and the crucible was sealed with a Mettler crucible sealing press. Measurements were conducted under a nitrogen atmosphere and at 1 MPa pressure. The temperature program was to heat the sample from 25 to 250 °C at different heating rates: 5 °C min−1, 10 °C min−1, 15 °C min−1 and 20 °C min−1.
Calculations
Different hydrodynamic parameters and mass transfer coefficients were calculated from the captured images of flow in the microfluidic reactor as follows:(a) Gas liquid interfacial area per unit liquid slug volume, a: this was calculated from the dimension of the rectangular channel reactor (h × w) and image analysis of gas bubbles and liquid slugs (Fig. 2(c)).
Surface area of gas bubble:
| SG = 2(wLG,actual + hLG,actual) + 4π((w + h)/4)2 | (i) |
| LG,actual = LG − (w + h)/2 | (ii) |
Volume of liquid slug:
| VL = whLS + wh[(w + h)/2] − (4/3)π[(w + h)/4]3 | (iii) |
Gas liquid interfacial area per unit liquid slug volume:
| a = SG/VL | (iv) |
Approximated radius of the cap (Fig. 2c) of liquid slug:
| rcap = dH/2 | (v) |
The approximation was made since geometry formed by the two liquid caps is not a complete sphere. dH is defined by eqn (xii).
(b) Superficial liquid slug velocity, UL, and gas bubble velocity, UG: these were calculated from the distance travelled by the slug and bubble in a particular time. Two phase superficial velocity (UTP) was calculated as follows:
| UTP = εGUG + (1 − εG)UL | (vi) |
Here, the volume fraction of gas bubble is:
 | (vii) |
V G is the volume of gas bubble:
| VG = whLG,actual + (4/3)π((w + h)/4)2 | (viii) |
(c) Average residence time: this was calculated from the two-phase superficial velocity (UTP) and the reactor length.
(d) Liquid film thickness surrounding a gas bubble, δ: this was calculated from the captured images and also using the correlations provided by Yun et al.27 for a rectangular microchannel as follows:
 | (ix) |
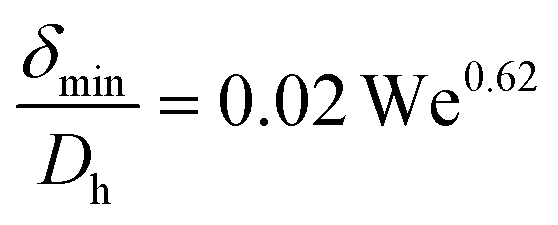 | (x) |
Here, the Weber number,
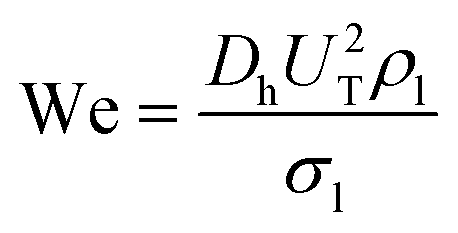 | (xi) |
Hydraulic diameter of the channel,
| Dh = 2[wh/(w + h)] | (xii) |
δ max and δmin are the maximum and minimum thicknesses of the liquid film (m), respectively.
U TP (m s−1) is the two phase superficial gas velocity, ρl is the density of liquid and σl is the surface tension of liquid (N m−1).
w and h are the width and depth of the reactor channel, respectively.
(e) Volumetric mass transfer coefficient, kLa (s−1): this was calculated using film theory:19,21
 | (xiii) |
Calculations performed using the results obtained by analysis of the products using gas chromatography were:
(f) Product selectivity: this was calculated from the relative peak area of the products as follows:
 | (xiv) |
Ketone-to-alcohol selectivity in primary oxidation products was calculated from the relative peak areas of ketones and alcohols in primary oxidation products.
(g) Calculation of the GC-FID response factor and tetralin conversion is included in the ESI.† Flame ionization detector response factors for the feed materials were used to calculate the conversion. Response factors for the products were not used in the selectivity calculations, due to the diversity of oxidation products.
Results
Role of oxygen availability on conversion and selectivity
The experimental parameter that influenced oxygen availability most, without changing the oxygen partial pressure, is the gas–liquid interfacial area. A series of experiments were conducted at different flow conditions to evaluate the role of oxygen availability on conversion and selectivity. Conditions of low conversion were selected so that the initial product selectivity values could be obtained.| Series | Experimentala | ||||||
|---|---|---|---|---|---|---|---|
| Length of liquid slug, LS × 103 (m) | Length of bubble, LG × 102 (m) | Superficial liquid velocity, US × 102 (m s−1) | Superficial bubble velocity, UG × 102 (m s−1) | Two-phase superficial velocity, UTP × 102 (m s−1) | Residence time (min) | Gas–liquid interfacial area, a × 10−4 (m2 m−3) | |
| A | 1.6 ± 0.5 | 2.1 × 101 ± 5.3 | 2.6 ± 1.0 | 2.6 ± 1.0 | 2.6 ± 1.0 | 1.5 ± 0.4 | 3.0 × 101 ± 5.2 |
| B | 2.7 ± 0.3 | 9.8 ± 4.1 | 2.1 ± 0.4 | 2.1 ± 0.4 | 2.1 ± 0.4 | 1.8 ± 0.4 | 1.5 × 101 ± 4.7 |
| C | 4.9 ± 1.9 | 5.1 ± 1.5 | 2.0 ± 0.7 | 2.0 ± 0.7 | 2.0 ± 0.7 | 2.0 ± 0.7 | 6.2 ± 1.8 |
| D | 4.3 ± 0.6 | 1.1 ± 4.1 × 10−1 | 1.6 ± 0.5 | 1.6 ± 0.5 | 1.6 ± 0.5 | 2.4 ± 0.8 | 1.6 ± 5.8 × 10−1 |
| E | 4.9 ± 0.6 | 0.4 ± 6.0 × 10−2 | 1.1 ± 0.2 | 1.1 ± 0.2 | 1.1 ± 0.2 | 3.5 ± 0.7 | 5.4 × 10−1 ± 1.3 × 10−3 |
| Series | Calculated based on eqn (x)a | Calculated based on eqn (ix)a | |||||
|---|---|---|---|---|---|---|---|
| Film thickness,cδmin × 106 (m) | Mass transfer coefficient,bkL × 103 (m s−1) | Liquid side volumetric mass transfer coefficient, kLa × 10−2 (s−1) | Film thickness,cδmax × 104 (m) | Mass transfer coefficient,bkL × 104 (m s−1) | Liquid side volumetric mass transfer coefficient, kLa × 10−1 (s−1) | ||
| a Based on 30 different slugs of each series of experiments. b Based on film theory and kL = DA/δ. c Maximum and minimum film thickness were calculated based on the correlation provided for Taylor flow in a rectangular channel by Yun et al. (2010).27 | |||||||
| A | 5.4 ± 3 × 10−2 | 6.3 ± 3.1 × 10−2 | 1.9 × 101 ± 3.3 | 1.6 ± 9.0 × 10−2 | 2.2 ± 1.1 × 10−1 | 6.4 ± 1.2 | |
| B | 5.4 ± 2 × 10−2 | 6.3 ± 2.4 × 10−2 | 9.6 ± 2.9 | 1.5 ± 4.8 × 10−2 | 2.2 ± 7.4 × 10−2 | 3.4 ± 1.1 | |
| C | 5.4 ± 3 × 10−2 | 6.3 ± 3.8 × 10−2 | 3.9 ± 1.1 | 1.5 ± 1.0 × 10−1 | 2.3 ± 1.5 × 10−1 | 1.4 ± 4.1 × 10−1 | |
| D | 5.4 ± 3 × 10−2 | 6.3 ± 4.0 × 10−2 | 1.0 ± 3.0 × 10−1 | 1.4 ± 9.8 × 10−2 | 2.4 ± 1.7 × 10−1 | 3.7 × 10−1 ± 1.3 × 10−1 | |
| E | 5.3 ± 2 × 10−2 | 6.4 ± 2.3 × 10−2 | 3.4 × 10−1 ± 8.0 × 10−2 | 1.3 ± 5.4 × 10−2 | 2.5 ± 1.0 × 10−1 | 1.4 × 10−1 ± 3.0 × 10−2 | |
The two-phase superficial velocity (UTP) varied with different sizes of the slugs and gas bubbles and was a consequence of the manipulation of gas–liquid flow ratio. The highest two-phase superficial velocity was 2.6 × 10−2 m s−1, which was obtained in the case of smaller liquid slugs and larger gas bubbles (series A). Residence time decreased with increasing two-phase superficial velocity (UTP).
The maximum and minimum thicknesses of liquid film surrounding the gas bubble were calculated, respectively, based on eqn (ix) and (x). The minimum film thickness was almost identical for all test conditions at 5.4 × 10−6 m and maximum film thickness was ∼1.4 × 10−4 m for all experimental conditions.
Liquid side volumetric mass transfer coefficient was calculated using eqn (xiii). The mass transfer coefficient increased with increasing gas–liquid interfacial area, which was calculated using eqn (iv). The maximum liquid side volumetric mass transfer coefficient was 1.9 × 103 s−1 and was obtained for the minimum film thickness at the largest gas–liquid interfacial area, 3 × 105 m2 m−3 (series A).
| Series | Oxidative conversiona ×101 (wt%) | Selectivity of oxidation productsa (wt%) | Ketone-to-alcohol in primary oxidation productsa | ||
|---|---|---|---|---|---|
| Primary | Secondary | Addition | |||
| a Calculated based on the GC-FID relative peak area of triplicate runs of each experiment. | |||||
| A | 7.4 ± 1.5 | 100 | 0 | 0 | 1.4 × 101 ± 2.2 |
| B | 2.1 ± 1.0 × 10−1 | 100 | 0 | 0 | 7.0 ± 2.0 × 10−1 |
| C | 1.7 ± 1.0 × 10−1 | 100 | 0 | 0 | 1.6 ± 2.0 × 10−1 |
| D | 1.2 ± 1.0 × 10−1 | 100 | 0 | 0 | 1.3 ± 1.0 × 10−1 |
| E | 1.6 ± 1.0 × 10−1 | 100 | 0 | 0 | 1.3 ± 1.0 × 10−1 |
Conversion rates were very close for a low gas–liquid interfacial area (Fig. 3). When the gas–liquid interfacial area was increased by a factor ∼5 it resulted in an increase in tetralin conversion by a factor ∼8. Despite the magnitude of change in the conversion rate, the overall conversion remained low.
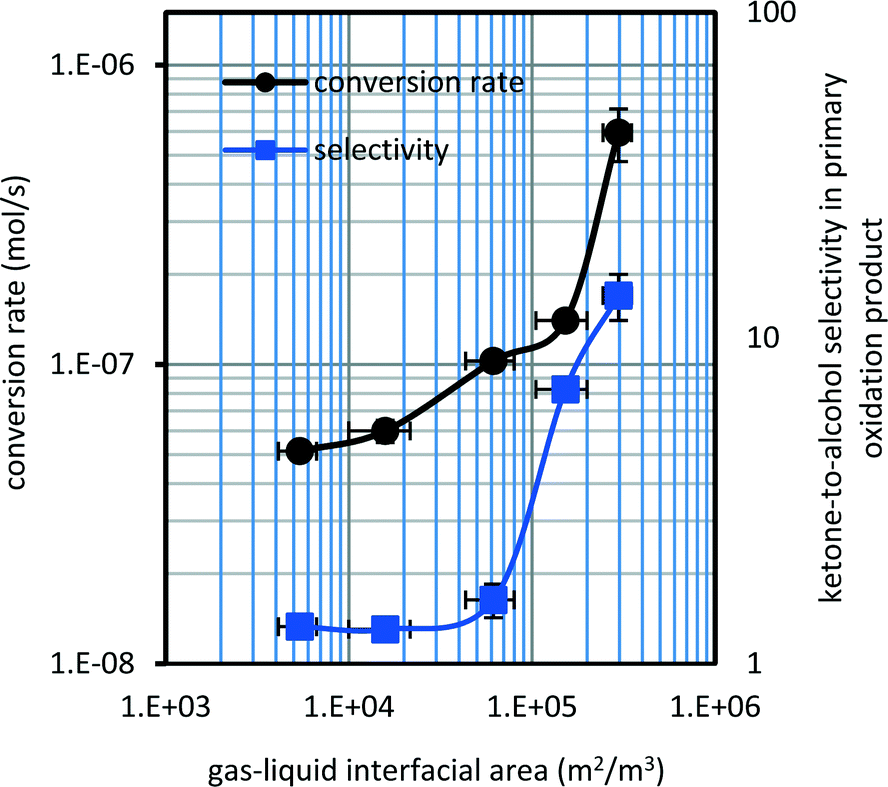 | ||
| Fig. 3 Effect of oxygen availability during tetralin oxidation with oxygen at 150 °C and a 90 kPa gauge pressure [triplicate results for the experiment series A–E: (●) conversion rate and (■) product selectivity]. | ||
The selectivity of the products varied with gas–liquid interfacial area (Fig. 3). Addition products and secondary oxidation products were not identified at the low conversion levels reported (Table 3). The ketone-to-alcohol selectivity in primary oxidation products increased by an order of magnitude as the gas–liquid interfacial area increased. The chromatograms of tetralin oxidized at 150 °C (series A and series E) are reported in the ESI† (Fig. S3) to illustrate the ketone-to-alcohol selectivity in the primary oxidation product.
Effect of temperature and initiator on conversion and selectivity
The conversion levels that could be achieved when manipulating the gas–liquid interfacial area in the microfluidic reactor was limited to less than 1 wt%. Although it enabled the initial reaction selectivities to be determined, it did not provide any information on the influence on secondary oxidation. Secondary oxidation required somewhat higher conversion. The conversion range could be extended in two ways. First, by adding an oxidation initiator to reduce the initiation period. Second, by increasing the temperature of oxidation. Both strategies were explored.Di-tert-butyl peroxide was used as an initiator and five different temperatures (120–160 °C) were maintained. The liquid flow rate was constant at 10 μL min−1. Like the oxygen availability investigation, experiments were performed at a low pressure (90 kPa gauge) and oxygen was used as the oxidizing agent.
| Series | Reactants | Temperature (°C) | Experimentala | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Length of liquid slug, LS × 103 (m) | Length of gas bubble, LG × 103 (m) | Liquid slug velocity, US × 102 (m s−1) | Bubble velocity, UG × 102 (m s−1) | Two-phase superficial velocity, UTP × 102 (m s−1) | Residence time (min) | Gas–liquid interfacial area, a × 10−4 (m2 m−3) | |||
| F | Tetralin | 120 | 3.5 ± 0.4 | 8.0 ± 1.7 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 | 3.4 ± 0.7 | 1.4 ± 3.1 × 10−1 |
| G | 130 | 3.8 ± 0.5 | 8.4 ± 2.8 | 1.8 ± 0.1 | 1.8 ± 0.1 | 1.8 ± 0.1 | 2.0 ± 0.7 | 1.4 ± 4.5 × 10−1 | |
| H | 140 | 3.7 ± 0.3 | 8.7 ± 2.4 | 1.3 ± 0.1 | 1.3 ± 0.1 | 1.3 ± 0.1 | 2.9 ± 1.1 | 1.4 ± 3.8 × 10−1 | |
| I | 150 | 4.3 ± 0.6 | 1.1 × 101 ± 4.1 | 1.6 ± 0.5 | 1.6 ± 0.5 | 1.6 ± 0.5 | 2.4 ± 0.8 | 1.6 ± 5.8 × 10−1 | |
| J | 160 | 3.7 ± 0.4 | 9.4 ± 2.3 | 1.4 ± 0.1 | 1.4 ± 0.1 | 1.4 ± 0.1 | 2.7 ± 1.3 | 1.5 ± 3.7 × 10−1 | |
| Kb | Tetralin with 0.5 wt% DTBPc | 120 | 3.4 ± 0.4 | 8.2 ± 2.2 | 1.5 ± 0.1 | 1.5 ± 0.1 | 1.5 ± 0.1 | 2.4 ± 0.6 | 1.4 ± 4.5 × 10−1 |
| Lb | 130 | 3.6 ± 0.4 | 7.9 ± 2.3 | 2.0 ± 0.8 | 2.0 ± 0.8 | 2.0 ± 0.8 | 2.0 ± 0.8 | 1.3 ± 4.2 × 10−1 | |
| Mb | 140 | 3.7 ± 0.5 | 7.8 ± 3.0 | 1.9 ± 0.1 | 1.9 ± 0.1 | 1.9 ± 0.1 | 2.0 ± 0.7 | 1.3 ± 5.1 × 10−1 | |
| Nb | 150 | 3.7 ± 0.3 | 6.9 ± 1.6 | 1.4 ± 0.4 | 1.4 ± 0.4 | 1.4 ± 0.4 | 2.6 ± 0.7 | 1.2 ± 2.7 × 10−1 | |
| Ob | 160 | 4.1 ± 0.3 | 9.8 ± 2.8 | 1.5 ± 0.0 | 1.5 ± 0.0 | 1.5 ± 0.0 | 2.5 ± 0.7 | 1.4 ± 3.4 × 10−1 | |
| Series | Reactants | Temperature (°C) | Calculated based on eqn (x)a | Calculated based on eqn (ix)a | |||||
|---|---|---|---|---|---|---|---|---|---|
| Film thickness,dδmin × 106 (m) | Mass transfer coefficient,ekL × 103 (m s−1) | Liquid side volumetric mass transfer coefficient, kLa × 10−1 (s−1) | Film thickness,dδmax × 104 (m) | Mass transfer coefficient,ekL × 104 (m s−1) | Liquid side volumetric mass transfer coefficient, kLa (s−1) | ||||
| a Based on 30 different slugs of each series of experiments. b The experiments performed in the presence of 0.5 wt% initiator (di-tert-butyl peroxide). c DTBP (di-tert-butyl peroxide) was used as the initiator. d Maximum and minimum film thicknesses were calculated based on the correlation provided for Taylor flow in a rectangular channel by Yun et al. (2010).27 e Based on film theory and kL = DA/δ. | |||||||||
| F | Tetralin | 120 | 5.3 ± 1.8 × 10−2 | 4.0 ± 1.4 × 10−2 | 5.6 ± 1.3 | 1.3 ± 5.1 × 10−2 | 1.6 ± 6.0 × 10−2 | 2.2 ± 5.0 × 10−1 | |
| G | 130 | 5.4 ± 2.6 × 10−2 | 4.7 ± 2.2 × 10−2 | 6.4 ± 2.1 | 1.5 ± 7.8 × 10−2 | 1.7 ± 9.2 × 10−2 | 2.4 ± 8.5 × 10−1 | ||
| H | 140 | 5.4 ± 3.3 × 10−2 | 5.5 ± 3.3 × 10−2 | 7.7 ± 2.0 | 1.4 ± 9.4 × 10−2 | 2.1 ± 1.4 × 10−1 | 3.0 ± 7.9 × 10−1 | ||
| I | 150 | 5.4 ± 3.3 × 10−2 | 6.3 ± 4.0 × 10−2 | 9.9 ± 3.6 | 1.5 ± 9.8 × 10−2 | 2.4 ± 1.7 × 10−1 | 3.7 ± 1.3 | ||
| J | 160 | 5.4 ± 3.3 × 10−2 | 7.3 ± 4.4 × 10−2 | 1.1 × 101 ± 2.7 | 1.4 ± 9.3 × 10−2 | 2.8 ± 1.9 × 10−1 | 4.3 ± 1.1 | ||
| Kb | Tetralin with 0.5 wt% DTBPc | 120 | 5.4 ± 1.8 × 10−2 | 4.0 ± 1.4 × 10−2 | 5.9 ± 1.8 | 1.4 ± 5.4 × 10−2 | 1.5 ± 5.9 × 10−2 | 2.2 ± 6.7 × 10−1 | |
| Lb | 130 | 5.4 ± 3.5 × 10−2 | 6.3 ± 4.1 × 10−2 | 8.4 ± 2.6 | 1.5 ± 1.1 × 10−1 | 2.3 ± 1.6 × 10−1 | 3.0 ± 9.0 × 10−1 | ||
| Mb | 140 | 5.4 ± 2.7 × 10−2 | 5.5 ± 2.7 × 10−2 | 7.1 ± 2.8 | 1.5 ± 8.0 × 10−2 | 2.0 ± 1.1 × 10−1 | 2.6 ± 1.0 | ||
| Nb | 150 | 5.4 ± 2.5 × 10−2 | 6.3 ± 3.0 × 10−2 | 7.6 ± 1.7 | 1.4 ± 7.5 × 10−2 | 2.4 ± 1.2 × 10−1 | 2.9 ± 6.1 × 10−1 | ||
| Ob | 160 | 5.4 ± 2.4 × 10−2 | 7.0 ± 4.3 × 10−2 | 1.0 × 101 ± 2.7 | 1.4 ± 7.3 × 10−2 | 2.6 ± 2.2 × 10−1 | 3.8 ± 9.2 × 10−1 | ||
Reported parameters were varied due to the variation of temperatures and the presence of the initiator. The size of liquid slugs varied between 3.4 × 10−3 m (series K) and 4.3 × 10−3 m (series I). Gas bubbles changed within 6.9 × 10−3 m (series N) and 1.1 × 10−3 m (series I). Slugs and gas bubbles of different sizes resulted in variation in the superficial liquid velocity (US), superficial bubble velocity (UG), two-phase superficial velocities (UTP), residence times and gas–liquid interfacial area (a). Gas–liquid interfacial area varied between 1.2 × 104 m2 m−3 (series N) and 1.6 × 104 m2 m−3 (series I).
Like manipulating the oxygen availability investigation, the calculated minimum thicknesses of a liquid film surrounding the gas bubble was almost identical for all experimental conditions, 5.4 × 10−6 m and the maximum film thickness was ∼1.4 × 10−4 m for all the experimental conditions.
The liquid side mass transfer coefficient (kL) was increased from 4 × 10−3 m s−1 to 7 × 10−3 m s−1 for the minimum film thickness and 2.2 m s−1 to 4.3 m s−1 for the maximum film thickness as temperature increased from 120 °C to 160 °C. The liquid side volumetric mass transfer coefficient (kLa) increased with increasing temperature and gas–liquid interfacial area, which was calculated using eqn (iv). The maximum liquid side volumetric mass transfer coefficient was 1.1 × 102 s−1 and was obtained at 160 °C and at 1.5 × 105 m2 m−3 (series J) gas–liquid interfacial area.
 | ||
| Fig. 4 Effect of temperature on conversion rate [triplicate results: tetralin (♦) and tetralin with initiator (▲)] and product selectivity [triplicate results: tetralin (■) and tetralin with initiator (●)] during tetralin oxidation with oxygen in the presence or absence of the initiator (0.5 wt% di-tert-butyl peroxide) at 90 kPa gauge pressure. | ||
Table 5 summarizes the oxidative conversion product selectivity of tetralin oxidized at different temperatures and oxygen availabilities with and without the initiator. Primary products (alcohol and ketone) were observed in all cases; but secondary and addition products were observed during tetralin oxidation performed with the 0.5 wt% initiator at temperatures of 150 °C and 160 °C. The ketone-to-alcohol formation was varied with temperature and initiator (di-tert-butyl peroxide). In the case of tetralin oxidation at temperatures of 120–140 °C without the initiator, alcohol formation dominated over ketone formation (Table 5); however, at temperatures of 150–160 °C, tetralin oxidations without the initiator formed ketones as the major products compared to alcohols in primary oxidation products. Oxidation performed in the presence of the initiator resulted in a dominant ketone formation compared to alcohol as the primary oxidation products. The maximum ketone-to-alcohol selectivity in the primary oxidation product was 2.1 (series N) which was obtained at a temperature of 150 °C, but the ratio decreased to 1.6 (series O) at a temperature of 160 °C.
| Series | T (°C) | Oxidative conversionsb,c ×102 (wt%) | Selectivity of oxidation productsb,c (wt%) | Ketone-to-alcohol in primary oxidation productsb (×101) | ||
|---|---|---|---|---|---|---|
| Primary | Secondary | Addition | ||||
| a The experiments conducted in the presence of 0.5 wt% initiator (di-tert-butyl peroxide). b Calculated based on the GC-FID relative peak area of triplicate runs of each experiment. c Selectivity and conversion of tetralin oxidized in the presence of the 0.5 wt% initiator (di-tert-butyl peroxide) was calculated by subtracting the product formed due to the initiator before oxidation in the microfluidic reactor. | ||||||
| F | 120 | 3.0 ± 1.0 | 100 | 0 | 0 | 5.4 ± 1.0 × 10−1 |
| G | 130 | 4.0 ± 1.0 | 100 | 0 | 0 | 5.7 ± 5.0 × 10−1 |
| H | 140 | 6.0 ± 1.0 | 100 | 0 | 0 | 6.4 ± 3.0 × 10−1 |
| I | 150 | 1.2 × 101 ± 4.0 | 100 | 0 | 0 | 1.3 × 101 ± 6.0 × 10−1 |
| J | 160 | 4.4 × 101 ± 6.0 | 100 | 0 | 0 | 1.3 × 101 ± 8.0 × 10−1 |
| Ka | 120 | 1.0 × 101 ± 1.0 | 100 | 0 | 0 | 1.2 × 101 ± 6.0 × 10−1 |
| La | 130 | 1.8 × 101 ± 4.0 | 100 | 0 | 0 | 1.4 × 101 ± 6.0 × 10−1 |
| Ma | 140 | 5.0 × 101 ± 9.0 | 100 | 0 | 0 | 1.5 × 101 ± 8.0 × 10−1 |
| Na | 150 | 1.6 × 102 ± 1.6 × 101 | 95.5 | 2.8 | 1.4 | 2.1 × 101 ± 9.0 × 10−1 |
| Oa | 160 | 3.3 × 102 ± 1.3 × 101 | 90.0 | 5.1 | 4.9 | 1.6 × 101 ± 2.0 × 10−1 |
Microfluidic reactor compared to batch and semi-batch reactors
Gas–liquid interfacial area and hence oxygen availability was also manipulated by using reactors of different geometries. Oxidation was performed in batch and semi-batch reactors to compare the oxidative conversion and product selectivity with oxidation performed in a microfluidic reactor. The oxidation induction period was also determined from the oxidation performed in a semi-batch reactor.Oxidation performed in all three reactors in our experiments showed very low conversion (about 0.1 wt%) after 2 to 6 minutes at the oxidation conditions (Table 6). The low oxidation conversion enabled a direct comparison of selectivities and provided information about the initial selectivity of oxidation in the different reactors.
| Reactor type | Series | Temperature | Residence/oxidation time (min) | Gas–liquid interfacial aread (a, m2 m−3) | Oxidative conversione (wt%) | Selectivity of oxidation productse (wt%) | Ketone to alcohol in primary oxidation productse | ||
|---|---|---|---|---|---|---|---|---|---|
| Primary | Secondary | Addition | |||||||
| a Oxidation performed with pure oxygen at 90 kPa pressure gauge at the tetralin input rate of 10 μL min−1. b Oxidation performed with pure oxygen at 90 kPa pressure gauge using tetralin-to-oxygen volume identical to that of the microfluidic reactor. c Oxidation performed with pure oxygen at near atmospheric pressure. d Calculated from the gas–liquid surface area (m2) and the volume of the liquid (m3) used in the reactor. e Calculated based on the GC-FID relative peak area of triplicate runs. | |||||||||
| Microfluidica | G | 130 | 2.0 | 1.4 × 104 | 0.1 | 100 | 0 | 0 | 0.6 |
| I | 150 | 2.4 | 1.6 × 104 | 0.1 | 100 | 0 | 0 | 1.3 | |
| Batchb | P | 130 | 2 | 2.4 × 101 | 0.1 | 100 | 0 | 0 | 0.6 |
| Q | 130 | 5 | 2.4 × 101 | 0.1 | 100 | 0 | 0 | 0.8 | |
| Semi-batchc | R1 | 150 | 2 | 9.8 × 102 | 0.1 | 100 | 0 | 0 | 1.3 |
| R2 | 150 | 4 | 9.8 × 102 | 0.1 | 100 | 0 | 0 | 1.3 | |
| R3 | 150 | 6 | 9.8 × 102 | 0.1 | 100 | 0 | 0 | 1.4 | |
| R4 | 150 | 10 | 9.8 × 102 | 0.2 | 100 | 0 | 0 | 1.8 | |
| R5 | 150 | 15 | 9.8 × 102 | 0.5 | 100 | 0 | 0 | 2.0 | |
| R6 | 150 | 30 | 9.8 × 102 | 2.0 | 96 | 1 | 3 | 2.0 | |
| R7 | 150 | 45 | 9.8 × 102 | 2.9 | 91 | 2 | 7 | 2.0 | |
| R8 | 150 | 60 | 9.8 × 102 | 4.5 | 88 | 1 | 11 | 1.1 | |
Ketone-to-alcohol selectivity in the primary oxidation products was identical (0.6:1.0) for the oxidation performed at 130 °C in microfluidic and batch reactors for two minutes. Identical ketone-to-alcohol selectivity (1.3:1.0) was also observed for the oxidation completed in the microfluidic reactor and semi-batch batch reactor (series R1–R3) at 150 °C.
Induction time was fourteen min (Fig. 5) for the oxidation of tetralin in a semi-batch reactor at 150 °C having an oxygen flow rate of 124–130 mL h−1 per g tetralin. During the induction period, oxygen consumption and oxidation rate was low; ketone-to-alcohol selectivity in primary oxidation products was 1.3:1.0 in first 6 minutes.
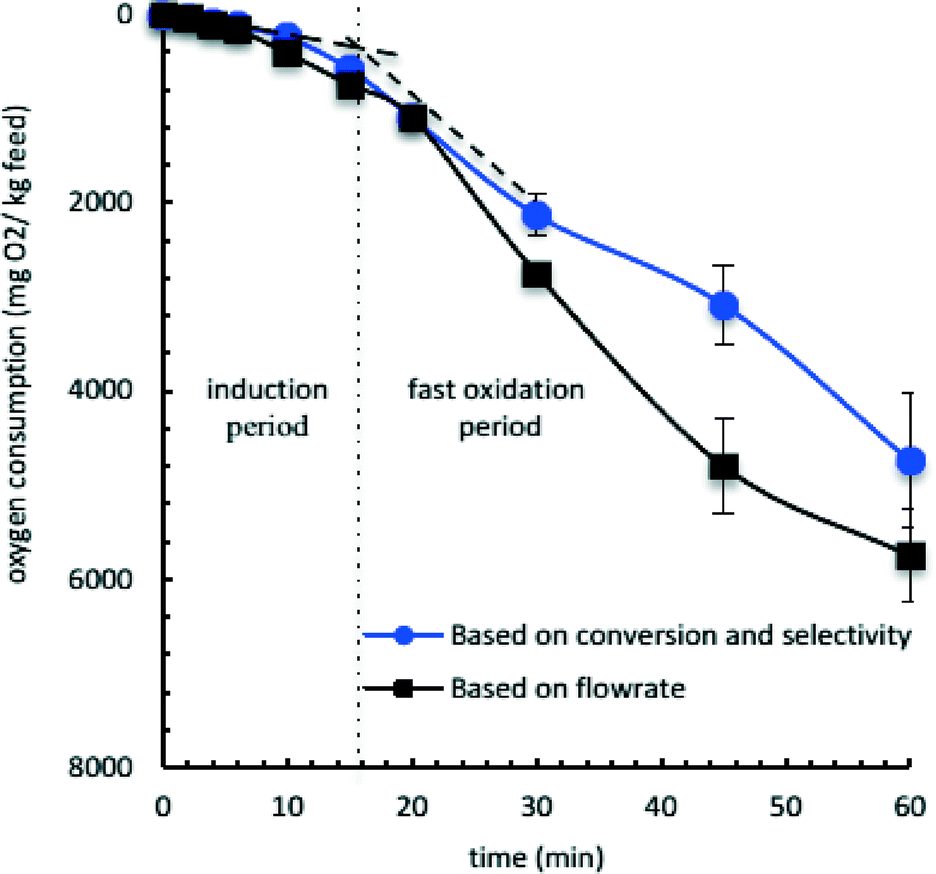 | ||
| Fig. 5 Oxygen consumption over time during the oxidation of tetralin at 150 °C using 124–130 mL oxygen per h per g feed [triplicate results: (●) based on conversion and product selectivity (water generation was not considered) and (■) based on oxygen flowrates]. | ||
Changes in selectivity with conversion
Tetralin was oxidized in a semi-batch reactor at 150 °C at near atmospheric pressure for longer oxidation times (Table 6) to understand the role of conversion on selectivity beyond the induction time.Oxidation rate was increased after the induction time (14 minutes). A maximum conversion of 4.5 wt% was obtained in one hour of oxidation (series R). Multifunctional groups containing oxidation products (secondary products) and addition products (dimerized products containing functional groups) were observed after 30 minutes of oxidation in semi-batch reactor. Those products were not observed in the case of the microfluidic and batch reactors at the studied experimental conditions. The maximum addition product selectivity was 11 wt%, which was observed after one hour of oxidation at 150 °C (series R).
After the induction period (15–45 minutes), ketone-to-alcohol selectivity in the primary oxidation products was constant at 2.0:1.0, but the ratio decreased to 1.1:1.0 after 45 minutes of oxidation, indicating increasing alcohol production compared to that of ketone. A similar observation was noticed for the oxidation of tetralin in the microfluidic reactor in the presence of di-tert-butyl peroxide initiator (Fig. 4 and Table 5).
Approximately 1000 mg H2O/kg tetralin was produced in the course of 1 hour of oxidation of tetralin in a semi-batch reactor at 150 °C having an oxygen flow rate of 124–130 mL h−1 per g tetralin. The change in CO2 was insignificant (0.4 mg CO2/kg tetralin) and no acid product was identified during one hour of oxidation. The online mass spectrometer also did not detect any other gases except the feed gases.
Discussion
Oxidation selectivity
It is clear from all the selectivity data that alcohol and ketones are the two main primary oxidation products regardless of experimental conditions and of the reactor types. Although alcohols and ketones are generally formed at a 1:1 ratio according to reaction (6) of Table 7 for compounds having secondary carbons,5,13,28 different alcohol-to-ketone selectivities are observed that describe the nature of the propagation and termination. Alcohol and ketone selectivities are discussed separately:
| Reaction | Activation energy, Ea × 10−1 (kJ mol−1) | Pre-exponential factorb (A) | Reaction rate constantb (kr) | |
|---|---|---|---|---|
| a Not reported. b Calculated for the reaction temperature, 150 °C. | ||||

|
(0) | 8.71 | 3.5 × 103 L2 mol−2 s | 7.1 × 10−8 L2 mol−2 s |

|
(1) | 0.0 | 6.8 × 107 L mol−1 s | 6.8 × 107 L mol−1 s |
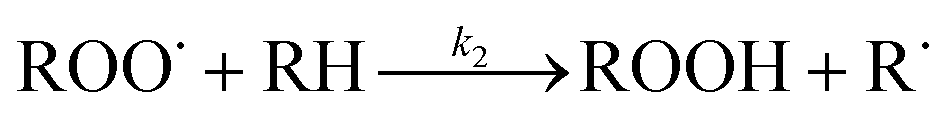
|
(2) | 1.9 | 2.5 × 104 L mol−1 s | 120 L mol−1 s |

|
(3) | 1.0 × 101 | 9.3 × 108 s−1 | 2.29 × 10−4 s−1 |
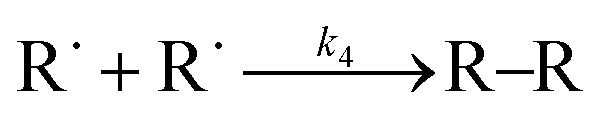
|
(4) | 1.1 | 5.5 × 108 L mol−1 s | 2.5 × 107 L mol−1 s |

|
(5) | —a | —a | —a |

|
(6) | 1.7 × 10−1 | 4.2 × 107 L mol−1 s | 2.6 × 107 L mol−1 s |
| 2ROOH → RO˙ + ROO˙ + H2O | (7) |
| ROOH + RO˙ → ROH + RO˙ + O2 | (8) |
Alkoxy radicals (RO˙) form in all cases and play an important role in changing the product selectivity. They can form alcohol via hydrogen abstraction from the α-position of the aromatic ring of tetralin as illustrated by the generic reactions (9) and (10).5
| RO˙ + RH → ROH + R˙ | (9) |
| ROOH + RH → [RO˙ + ˙OH] + RH → ROH + R˙ + HO˙ | (10) |
Alkoxy radicals can also form alcohol through hydrogen disproportionation, reaction (11), and alkyl peroxide via free radical addition, reaction (12).29
RO˙ + RO˙ → R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + ROH O + ROH | (11) |
| RO˙ + RO˙ → ROOR | (12) |
Alcohols can readily form by reaction (8). Reactions (9) and (10) result in alcohol only if the alkoxy radical (RO˙) exists without an in situ reaction within the molecular cage.5 Alcohol production through reaction (11) also produces ketone in a 1:1 ratio and cannot be the cause of higher alcohol than ketone selectivity. Hence, reaction (8) would explain the higher alcohol than ketone selectivity during the oxidation of tetralin without an initiator at low temperatures (series F–H in Table 5 and series G, P, Q in Table 6).
| ROOH → [RO˙ + ˙OH] → RO + H2O | (13) |
 | (14A) |
 | (14B) |
At very high oxygen availability reaction (15) takes place31 and produces ketone and the hydroperoxy radical. The hydroperoxy radical can react with other free radicals by reaction (16) or tetralin by reaction (17).
| RO˙ + O2 → RO + HOO˙ | (15) |
| ROO˙ + HOO˙ → ROOH + O2 | (16) |
| RH + HOO˙ → R˙ + H2O2 | (17) |
Tolman and co-workers, however, reported that the oxidation of alcohol formed during oxidation (Fig. 6) would result in a higher ketone-to-alcohol ratio rather than eqn (15).29 They heated a solution of dicyclohexyl peroxide (ROOR) at 160 °C under N2 and under 3.45 MPa (500 psi) O2, and obtained dominantly alcohol (ketone-to-alcohol (K/A) = 1/4) and dominant ketone (K/A = 15/1) selectivities, respectively. In order to support their claim, they added cyclopentyl alcohol in their experiments and found no cyclopentanone under N2; but 80% cyclopentyl alcohol was converted into cyclopentanone under O2. This explained that the autoxidation of alcohol to ketone gave a higher ketone-to-alcohol ratio.
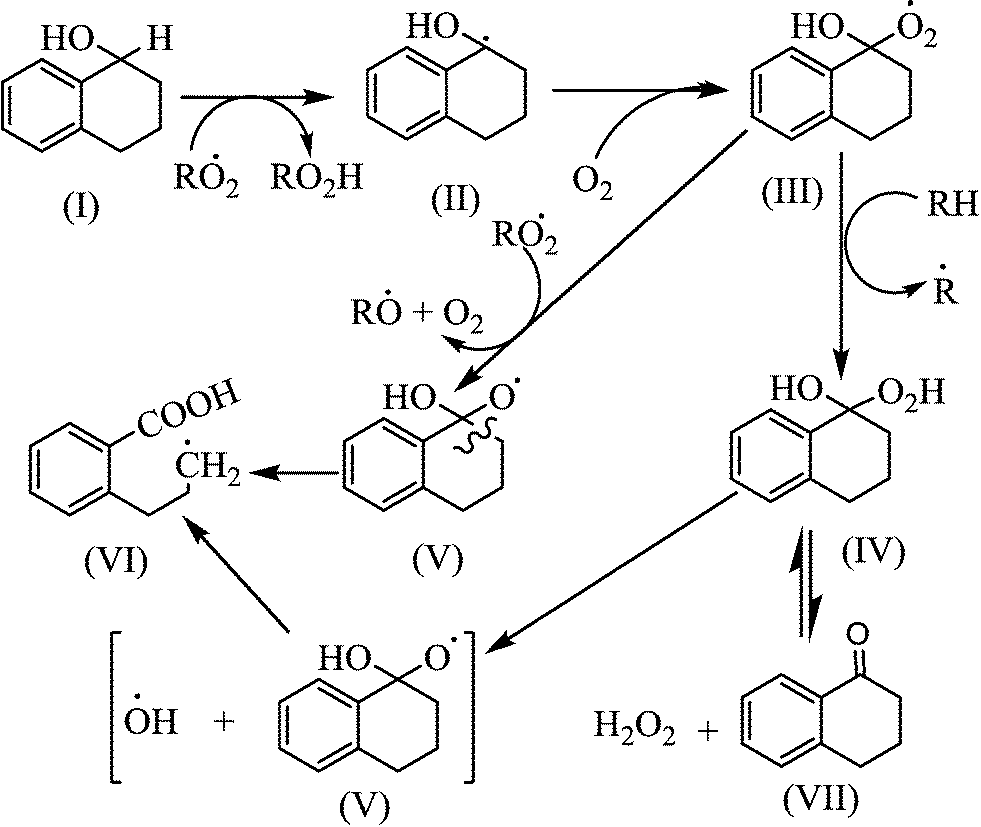 | ||
| Fig. 6 Illustration of the oxidation of 1-tetranol (alcohol of tetralin). | ||
Oxidation of alcohol not only produces ketone, but also leads to the formation of ring opening products (Fig. 6). Alcohols can readily lose hydrogen from the C–H bond of the α-position that is also the α-position from the benzene ring.29 Ring opening products (structure VI) can also be formed; but these were not observed at the studied experimental conditions.
A higher ketone level can also be formed by the oxidation of alcohol to produce ketone and water (reaction (18)).
| 2ROH + 0.5 O2 → 2RO + H2O | (18) |
Parameters that affected oxidation selectivity
In the introduction it was postulated that temperature and the reactor hydrodynamics would affect oxidation selectivity. Based on all of the results presented, the following parameters appear to affect oxidation selectivity at a near constant conversion:Although hydrocarbon oxidation with oxygen is exothermic, a few of the initial steps, especially the free radical formation steps via bond dissociations, are endothermic, slow, and require activation energy (Table 7).3,13,29 During the induction period, hydrogen abstraction via C–H bond dissociation and decomposition of peroxides (O–O) are two main free radical formation steps that are influenced by temperature. Free radicals are formed from the hydrocarbon via C–H bond dissociation. C–H bond strength varies (typically 300–420 kJ mol−1) depending on the carbon type, for example, primary, secondary, tertiary, aromatic, or benzylic.13,29,32 Tetralin has four aromatic C–H bonds which are very strong, four aliphatic C–H bonds of intermediate strength and four benzylic C–H bonds which are weak by comparison. Hydrogen can be readily abstracted from benzylic C–H by increasing the temperature. The bond strength of peroxides (O–O) (typically 150–180 kJ mol−1) varies depending on the group attached to the oxygen atom and decomposes typically at a temperature of about 150 °C.5,13,29,32 Decomposition of peroxides increases the formation of free radical intermediates. Hence, the formation of free radical intermediates would increase with temperature which would change the product selectivity.
After the induction period, sufficient free radical intermediates are available for autocatalytic oxidation to proceed and the temperature has a comparatively lower effect on free radical formation. For instance, hydroxyl radicals (HO˙) formed from the decomposition of hydroperoxides is strong enough to abstract hydrogen exothermically from a paraffinic hydrocarbon, and hydrogen abstraction by alkoxy radicals is mildly endothermic.29 Ketone formation also facilitates peroxide decompositions.13,29 However, our focus was on the initial product selectivity during the induction period.
The strength of O–H bond that is formed via hydrogen abstraction would determine the degree of product selectivity.29 Regardless of the reactor types, alcohol selectivity (initial product selectivity) dominated at temperatures below 140 °C; but ketone selectivity was higher at temperatures of 150 °C or higher (Tables 5 and 6). As discussed earlier, reaction (8) would explain the higher alcohol selectivity during the oxidation of tetralin without an initiator at low temperatures (series F–H in Table 5 and series G, P, Q in Table 6), and oxidation of the alkoxy radical (reaction (15)) or oxidation of the alcohol (Fig. 6 and reaction (18)) would explain the dominant ketone selectivity compared to alcohol.
Free radicals formed via H–abstraction during the initiation would react depending on the oxygen availability by reactions (1) and (4) of Table 7. If oxygen is present, reaction (1) will take place, because it requires zero activation energy (Table 7). In the absence of oxygen or oxygen partial pressure below 13 kPa, reaction (4) would take place.3,8 Although this indicates the product immediately after the initiation, it could happen for the reaction between any free radicals and/or oxygen.
Both aspects that describe the higher ketone formation (oxidation of alkoxy radical and oxidation of alcohol) require oxygen. But ketone formation viareaction (15) would require oxygen in the molecular cage where the alkoxy radical (RO˙) is formed. The most significant ketone-to-alcohol ratio in the primary oxidation product (14:1, series A, Table 3) was obtained at a gas–liquid interfacial area of 3.0 × 105 m2 m−3 (Table 2) where oxygen availability was the highest. Due to the longer gas bubbles (Table 2) and the smaller film thickness a higher amount of oxygen was available for the oxidation to take place in the cage conditions. Hence, reaction (15) could be dominant pathway to form more ketone. As noted earlier, the longevity of alkoxy radical (RO˙) might play an important role for such high ketone selectivity in tetralin (14:1).
In the cage condition, if oxygen is available reactions (9), (10), (11) and (12) would not take place because the reaction between the alkoxy radical and oxygen would be very fast. This would also support a higher ketone formation viareaction (15). If alcohol is formed, however, as described earlier by eqn (8), (9), (10) and (11), the oxidation of alcohol would be the dominant reaction for the higher ketone formation.
In the case of a constant oxygen supply, oxygen availability would change with conversion and oxidation time, which could lead to different product selectivities. This aspect is evaluated in following sections.
It is mentioned earlier that sufficient free radical intermediates are available for autocatalytic oxidation after the induction period. The concentration of free radical intermediates would increase with oxidative conversion and oxidation time. Immediately after the induction period (14 minutes), dissolved oxygen and supplied oxygen might be sufficient for the available free radical intermediates to produce more ketones compared to alcohol (2:1). However, a further increase in free radical intermediates and decrease in dissolved oxygen would influence the product selectivity. This situation is found 15 minutes after the induction time (series R6–R8 of Table 6) when secondary oxidation products and addition products are observed.
A near constant amount of oxygen was supplied as bubbles. Yet, the oxygen distribution might not be homogeneous due to transport effects and the local consumption of oxygen in the presence of a higher concentration of free radicals. Hence, some parts would oxidize more to produce secondary oxidation products; but some parts would form addition products due to oxygen starvation. This would explain the secondary and addition product formation after 30 minutes of oxidation (series R6–R8). Addition products could form via olefin formation as reported in our previous study.15
Another notable observation was a decrease in ketone-to-alcohol selectivity (2.0:1.0 to 1.1:1.0) in primary oxidation products (series R8 of Table 6). This indicates ketone-to-alcohol selectivity changed and had a local maximum (series R of Table 6) during oxidation. A similar observation was also made in our previous study of autoxidation (oxidation with air) of tetralin in a semi-batch reactor at 130 °C for 6 hours (Table S4†).15 The decrease in ketone-to-alcohol selectivity can be explained by the oxygen starvation compared to the free radical intermediates available at higher conversions and longer oxidation times. This observation also supports addition product formation at low oxygen availability.
It is worthwhile to mention here that tetralin was partly oxidized immediately after adding the initiator (Table 8) without performing oxidation in the microfluidic reactor and the main identified products were addition products of tetralin. Ketone was not observed at 0.03 wt% and 0.1 wt% di-tert-butyl peroxide. A lower amount of ketone compared to alcohol was formed at 0.5 wt% di-tert-butyl peroxide. Secondary oxidation products were not identified in any cases. As described earlier, oxygen unavailability could play a role for such product selectivities. However, 0.5 wt% initiator was selected to perform the experiments as it was the amount which could provide higher conversion without interfering too much with product identification.
| Di-tert-butyl peroxide in tetralin (wt%) | Tetralin conversion ×102a (wt%) | Selectivity of oxidation productsa (wt%) | Ketone-to-alcohol in primary oxidation products | ||
|---|---|---|---|---|---|
| Primary | Secondary | Addition | |||
| a Calculated based on the GC-FID relative peak area of triplicate runs. b Ketone to alcohol ratios were not reported as no ketone was identified. | |||||
| 0.03 | 7.0 ± 2.0 | 22.5 | 0 | 77.5 | —b |
| 0.1 | 1.0 × 101 ± 1.0 | 19.1 | 0 | 80.9 | —b |
| 0.5 | 2.2 × 101 ± 1.0 | 18.7 | 0 | 81.3 | 0.8 |
The conversion rate greatly increased in the presence of the initiator (di-tert-butyl peroxide) at 150 °C or higher temperatures that affected the product selectivity (series N and O). Two distinct observations in product selectivity were noticed: secondary and addition product formation increased and the ketone-to-alcohol ratio passed through a maximum with increasing temperature (Fig. 4). These can be explained by the decomposition temperature of the initiator (di-tert-butyl peroxide) and peroxides formed during oxidation.
The decomposition temperature of di-tert-butyl peroxide was reported at 150 °C by thermal screening unit (TSu) and the thermokinetic parameters of DTBP were measured by an accelerating rate calorimeter (ARC) and reported as heat of reaction = 4.6 × 102 kJ kg−1, reaction order n = 1, activation energy (Ea) = 1.70 × 102 kJ mol−1 and pre-exponential factor (A) = 1.06 × 1020 s−1.33 Our DSC experiments performed in a N2 atmosphere at different heating rates showed that decomposition started at a temperature greater than 133 °C (Fig. S4†). At 150 °C or higher, the initiator would decompose. Hydroperoxide (ROOH) and/or (HOOH) formed via oxidation would also decompose at this temperature or higher. As mentioned earlier, the decomposition of peroxides leads to the formation of free radical intermediates that would help in subsequent oxidations and would increase the oxidative conversion and also affect product selectivity.
The most noticeable changes in product selectivities were observed for the oxidation performed at 150 °C and 160 °C with equal amounts of oxygen supply. But, supplied oxygen might not be sufficient for converting the free radicals produced at 160 °C into oxygenates and that might be the cause of the decrease in ketone-to-alcohol selectivity at 160 °C and increase in addition product formation. However, a decreasing ketone-to-alcohol selectivity at 160 °C does not mean that there was a decrease in the yields of alcohol and ketone, it is just the ratio that changed. The overall yields of alcohol and ketone at 160 °C were much higher than that obtained at 150 °C.
The changes in product selectivities at a higher conversion were analogous to the observation reported for tetralin oxidation after the induction time in a semi-batch reactor (series R5–R8 of Table 6) and in our previous study (Table S4†).15 These illustrate that oxidation performed in different reactors could proceed in a similar way regardless of oxidants (air or oxygen) and temperatures. However, manipulating oxygen availability could change the product selectivity.
The influences of temperature, oxygen availability, conversion and reaction time were anticipated from literature.5–7,9,13,29 Our interest was mainly in the influence of oxygen availability, which is related to reactor hydrodynamics.
Mass transfer coefficients
The first parameter that affected oxidation selectivity and that was affected by hydrodynamics is the mass transfer coefficient. The mass transfer coefficient is affected by film thickness and gas–liquid-interfacial area, which is related to the reactor design and operation.Improving the mass transfer coefficient would increase the oxygen transfer to tetralin and hence increase the oxygen availability in the liquid phase. Oxygen transfer to tetralin can be described by three different models: film model, penetration model, and surface renewal model.34–36 The main parameter that changes with different models is the liquid side mass transfer coefficient (kL).19,34 In most cases the results are very similar.35–38 Film theory is simple and was used in this study to explain the process. According to film theory, the liquid side mass transfer coefficient is kL = DA/δ and it is assumed that the film is stagnant and uniform.19,34,36 Hence, the oxygen transfer rate to tetralin can be presented as:
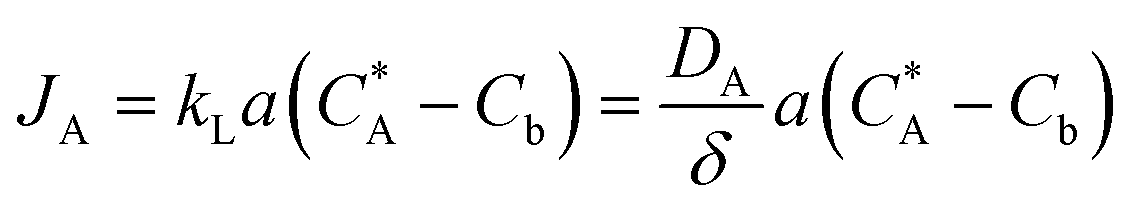 | (xv) |
Here, JA is the mass transfer rate across the gas–liquid interface (mol m−3 s), DA is the diffusivity of oxygen in tetralin (m2 s−1), kL is the liquid side mass transfer coefficient (m s−1), a is the gas–liquid interfacial area (m2 m−3), δ is the film thickness (m), C*A is the concentration of gas at the interface (mol m−3), and Cb is the concentration of gas in bulk liquid (mol m−3). As DA and C*A can be changed only to a limited extent, δ and a are the two most important parameters to enhance the gas–liquid mass transfer. These two aspects are discussed separately:
Liquid film thickness depends on reactor geometry, liquid agitation, interfacial properties of the channel, etc.20,34 For a rectangular microchannel, liquid film thickness (δ) surrounding a gas bubble is expected to be different at different positions: diagonally, laterally, and at the top and bottom of the bubble (Fig. 7) depending on the channel aspect ratio w/h.20,27,37 This can be explained by the Capillary number, eqn (xvi). Generally, film thickness increases with the Capillary number.39
| Capillary number , Ca = UGμ/σ | (xvi) |
 | ||
| Fig. 7 Sketch of bubble profiles in a rectangular channel under different flow conditions: (a) interface shape at Ca <10−3; (b) bubble shape at Ca >10−3 in a rectangular channel with the aspect ratio of width (w)/depth (h) >1.5.37,40,41 | ||
For values of Ca less than 10−3, the film surrounding the gas bubble would be very thin except in the corners (Fig. 7a). For values of Ca greater than 10−3, the liquid film occupies a significant portion of the microchannel (Fig. 7b).37,40 Since Ca is a function of bubble velocity, increasing velocity would increase the film thickness. Moreover, thicknesses would be different laterally and at the top and bottom of the channel due to the velocity differences. In this study, the calculated Ca value of 2.1 × 10−4 – 5.4 × 10−4 (Table 9) suggests that Fig. 7(a) would represent the most likely film thickness pattern.
| Dimensionless number | Value |
|---|---|
| a Based on minimum film thickness. b Based on maximum film thickness. c Calculated for reaction (1) in Table 7. | |
| ReL = ULdHρL/μL | 9–27 |
| ReG = UGdHρG/μG | 0.5–0.2 |
| ReTP = UTPdHρL/μL | 9–27 |
| We = DhU2TPρL/σL | 2.2 × 10−3–1.7 × 10−2 |
| Ca = UGμL/σL | 2.1 × 10−4–5.4 × 10−4 |
| Bo = (ρL − ρg)d2Hg/σ | 1.1 × 10−1–1.4 × 10−1 |
| PeL = δ2LUL/DA | 8.2 × 10−6 –2.2 × 10-5a |
| 5.2 × 10−3–2.0 × 10-2b | |

|
32–33a,c |
| 800–984b,c | |
| Da = (krC*Aδ2)/DA | 1.0 × 103–1.1 × 103a,c |
| 6.4 × 105–9.4 × 105b,c | |
Film thickness could also vary axially from the head side to the tail side of the bubble when inertial effects are not negligible.20,27 The bubble caps can be assumed to be spherical at a low Capillary number (Ca), but ripples start to form at the tail side with an increase in Ca.39 At Ca >10−3, the effect of inertia cannot be ignored and with an increase in the Reynolds number the amplitude of the ripples increases and the tail side becomes flattened, which would result in an increased film thickness.39 This would not be the case, however, in this study due to Ca < 10−3 (Table 9).
Yun et al.27 investigated the variation in film thickness for a rectangular microchannel experimentally and suggested correlations (eqn (ix) and (x)) to calculate the maximum and minimum film thickness. As the calculated maximum film thickness δmax (∼1.6 × 10−4 m) is ∼30 times higher than the minimum film thickness δmin, (∼5.4 × 10−6 m), both are considered during the analysis of the results.
Film thicknesses (lateral) calculated from the captured images were identical to those calculated using the correlation for the δmin, eqn (x). It was difficult to measure the exact film thickness, because proper film thickness determining methods, for instance, Laser Induced Fluorescence (LIF) or confocal Laser Scanning Microscopy (LSM),42 were not available to us. Also, our intention was to know only the order of magnitude of film thickness instead of investigating film thickness per se.
Since the thickness of the liquid film calculated using either eqn (ix) or (x) was almost identical (Table 2) and the diffusivity of oxygen in tetralin was expected to be constant at 150 °C, a change in the liquid side volumetric mass transfer coefficient (kLa) was mostly due to the increase in interfacial area. Hence, the gas–liquid interfacial area would be the most important parameter to assess the role of oxygen availability during the oxidation of tetralin with molecular oxygen.
Gas–liquid interfacial area proportionally increases with the mass transfer as shown in eqn (xv). It is clear from Table 2 that the gas–liquid interfacial area influences the mass transfer coefficient that increases the oxygen availability. The impact of the gas–liquid interfacial area on the conversion rate and product selectivity is clear from the Fig. 3. The conversion rate rather than conversion was calculated, because conversion is affected by the flow rate.
At the inflexion point, liquid film formation (not the film thickness, rather the length of the film) was increased (series B, Table 2) compared to series C, D and E (Table 2). Increased film formation ensured a higher gas–liquid interfacial area and hence oxygen availability for the oxidation to take place, and resulted in a higher conversion rate and changed the product selectivity. As explained earlier, due to the very high oxygen availability, a higher ketone compared to alcohol selectivity (14:1) was attributed to either the oxidation of the alkoxy radical or the alcohol with molecular oxygen.
Although gas–liquid interfacial areas were also manipulated by performing oxidation with different reactor geometries (Table 6), the initial ketone-to-alcohol selectivities did not change due to the longer induction period (14 minutes) at the studied oxygen availability. Thus, it was quite surprising to get an order of magnitude higher ketone level (series A, Table 3) at very high oxygen availability, but an even shorter residence time. Two possible things could happen: the induction time changed at a very high oxygen supply at 150 °C, or alcohol and/or alkoxy radicals reacted very fast at a very high oxygen availability and increased ketone formation.
The mass transfer coefficient is greatly influenced by the gas–liquid interfacial area compared to film thickness. Gas–liquid interfacial area and hence oxygen availability can be manipulated by changing the reactor geometry and operating conditions that would change the product selectivity.
Additional effects
Additional effects that can potentially influence the product selectivity and that are affected by reactor hydrodynamics are mixing by liquid circulation within the slug (Marangoni effects (Fig. 2c)), pressure drop, residence time distribution, etc. However, these aspects were not investigated.Characteristic dimensionless numbers
Although the hydrodynamics of microscale reactors can be explained by the principles used for macroscale reactors, many physical characteristics, for instance, surface tension forces, viscous forces, area-to-volume ratio, diffusion, cannot be scaled up linearly. Different characteristic dimensionless numbers may be useful to approximate the gas–liquid two-phase flow behavior in microchannel reactors.Characteristic dimensionless numbers (Table 9) were calculated from the studied gas–liquid systems.
A Reynolds number (Re) from 9 to 27 indicates laminar flow throughout the microchannel, typical for Taylor flow in microchannels.18,39 A smaller Re also indicated that viscous forces dominated over inertial forces.
The smaller Weber numbers (We) indicated that inertial forces were not high enough to overcome surface tension forces and to break down the slug between two phases.
The Capillary number (Ca) characterizes the relative importance of viscous forces relative to surface tension forces. A smaller Ca indicated that surface tension had a larger effect than viscous force. Both the Ca and We indicated that surface tension is dominant and that slug formation should be anticipated. As noted earlier, the film thickness could be estimated from these numbers.
The Peclet number (Pe) represents the relative importance of diffusive and convective transport. For both the minimum and maximum film thickness, Pe ≪ 1, which suggests diffusive transport far outweighs convective transport through the thin film.
The bond number (Bo) estimates the magnitude of gravitational forces over surface tension forces. Bo values of 1.1 × 10−1–1.4 × 10−1 (Table 9) suggest that the value of the surface tension has a larger influence on the system than gravitational forces.
The Hatta modulus (MH) and Damköhler number (Da) indicate whether a reaction is kinetically controlled or mass transfer limited. Since the calculated MH ≫ 3 and Da ≫ 1 for reaction (1) (Table 7), the reaction would be mass transfer limited, and based on these values the reaction would be completed in the film.35,43,44 This would be true for any reaction between a free radial and oxygen molecule. The experimental observations do not support such an interpretation, since it was possible to manipulate ketone-to-alcohol selectivity through the microfluidic reactor operation to control oxygen availability in the liquid phase at a near constant conversion, oxygen partial pressure and temperature.
Application of microfluidic devices to control selectivity
Microfluidic devices are not suitable for large-scale production, although microfluidic reactors may have some niche applications for the production of low volume high value materials. The real benefit of applying microfluidic devices is for the study and understanding of conversion processes. This knowledge can then be applied to the design of reactor types that are amenable to large-scale production. This is particularly important in the field of selective oxidation, where microfluidic devices also provide a safer means for exploring energetic reactions.Conclusions
The oxidation of a naphthenic-aromatic compound (tetralin) was investigated mainly in a microfluidic reactor to understand how product selectivity could be manipulated by ensuring oxygen availability and how mass transfer would affect the oxidation process. The main observations and conclusions are as follows:(i) Liquid phase mass transfer (kLa) in the microfluidic reactor could be manipulated over two orders of magnitude. This was possible mainly through changing the gas–liquid interfacial area (a). The mass transfer coefficient (kL) was fairly insensitive to changes in operation.
(ii) It was possible to increase the ketone-to-alcohol ratio in the product by an order of magnitude at near similar conversion, pressure and temperature. This was possible only through the manipulation of hydrodynamics in a microfluidic reactor.
(iii) The influence of operating conditions on oxidation selectivity could be explained in terms of the reaction network. The ketone-to-alcohol selectivity ratio was strongly affected by oxygen availability in the liquid phase, which promoted alcohol and/or alkoxy radical oxidation to produce ketones.
(iv) Addition products were not observed during tetralin oxidation at high oxygen availability. For example, when oxidation was performed with oxygen instead of air in a microfluidic reactor, no addition products were observed. When oxygen availability decreased, or the reaction rate was increased to decrease oxygen availability in relation to the reaction rate, addition products were observed. Hence, some addition products were noticed in the presence of the initiator (di-tert-butyl peroxide) at a higher temperature (150 °C and higher). Addition products were also observed after 30 minutes of tetralin oxidation performed in a semi-batch reactor. There was also a concomitant increase in alcohol selectivity with an increase in addition product selectivity. This was anticipated from the reaction network that favored ketone selectivity at a high oxygen availability, but favored alcohol and addition product selectivity at a lower oxygen availability.
(v) A higher Damköhler number (Da) and Hatta modulus (MH) suggested that gas–liquid oxidation might take place at the liquid film surrounding the gas bubbles rather than the liquid bulk. This was not consistent with the experimental observations, which recorded meaningful selectivity control based on the control of oxygen availability in the liquid phase through the microfluidic reactor operation at near constant conversion, pressure and temperature.
Nomenclatures
| a | Gas–liquid interfacial area, (m2/m3) |
| A | Pre-exponential factor |
| Bo | Bond number, Bo = (ρL − ρg)d2Hg/σ |
| Ca | Capillary number, Ca = UGμL/σL |
| C *A | Concentration of gas at the interface, (mol m−3) |
| C b | concentration of gas in bulk liquid (mol m−3) |
| Da | Damköhler number; Da = (krC*Aδ2)/DA |
| D A | diffusivity of oxygen in tetralin (m2 s−1) |
| d H | hydraulic diameter; dH = 2[wh/(w + h)], (m) |
| DTBP | Di-tert-butyl peroxide |
| E a | Activation energy, (kJ mol−1) |
| FID | Flame ionization detector |
| GC | Gas chromatography |
| h | Depth of the reactor, (m) |
| HP DSC | High-pressure differential scanning calorimeter |
| HPLC | High performance liquid chromatography |
| J A | Mass transfer rate across the gas–liquid interface (mol m−3 s) |
| k L | Mass transfer coefficient; kL = DA/δ, (m s−1) |
| k L a | Overall mass transfer coefficient, (s−1) |
| k r | Reaction rate constant |
| L G | Length of gas bubble, (m) |
| L S | Length of liquid slug, (m) |
| M H | Hatta modulus;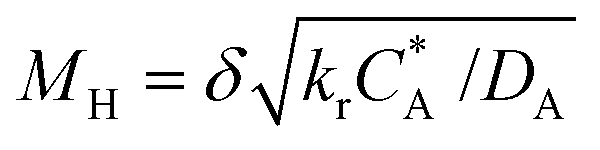 |
| MS | Mass spectrometry |
| NIST | National Institute of Standards and Technology |
| PeL | Peclet number; PeL = δ2LUL/DA |
| PTFE | Poly tetra fluoro ethylene |
| ReG | Gas-phase Reynolds number; ReG = UGdHρG/μG |
| ReL | Liquid-phase Reynolds number; ReL = ULdHρL/μL |
| ReTP | Two-phase Reynolds number; ReTP = UTPdHρL/μL |
| RTD | Residence time distribution |
| S G | Surface area of gas bubble, (m2) |
| U G | Superficial gas bubble velocity, (m s−1) |
| U L | Superficial liquid slug velocity, (m s−1) |
| U TP | Two phase superficial velocity, (m s−1) |
| V G | Volume of gas bubble, (m3) |
| V L | Volume of liquid slug, (m3) |
| w | Width of the reactor, (m) |
| We | Weber number; We = DhU2TPρL/σL |
Greek letters
| δ | Thickness of liquid film, (m) |
| ε G | Volume fraction of gas bubble |
| ρ G | Density of gas, (kg m−3) |
| ρ l | Density of liquid, (kg m−3) |
| μ G | Gas viscosity, (Pa s) |
| μ L | Liquid viscosity, (Pa s) |
| σ | Surface tension, (N m−1) |
Subscripts
| b | Bulk |
| G | Gas |
| L | Liquid |
| TP | Two-phase |
Acknowledgements
This work was financially supported by Cenovus FCCL Ltd as the Operator for the FCCL partnership in conjunction with the NSERC-CRD (Natural Sciences and Engineering Research Council of Canada through the Co-operative Research and Development) program.References
- C. D. Pina, E. Falletta and M. Rossi, Liquid phase oxidation of organic compounds by supported metal-based catalysts with a focus on gold, in Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications, ed. M. G. Clerici and O. A. Kholdeeva, Wiley, Hoboken, 1st edn, 2013, pp. 221–262 Search PubMed.
- N. Dimitratos, J. A. Lopez-Sanchez and G. J. Hutchings, Supported metal nanoparticles in liquid-phase oxidation reactions, in Handbook of Advanced Methods and Processes in Oxidation Catalysis: From Laboratory to Industry, ed. D. Duprez and F. Cavani, Imperial College Press, London, 2014, pp. 631–678 Search PubMed.
- A. K. Suresh, M. M. Sharma and T. Sridhar, Ind. Eng. Chem. Res., 2000, 39, 3958–3997 CrossRef CAS.
- C. L. Hill and O. A. Kholdeeva, Selective liquid phase oxidation in the presence of supported polyoxometalates, in Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications, ed. M. G. Clerici and O. A. Kholdeeva, Wiley, Hoboken, 1st edn, 2013, pp. 263–319 Search PubMed.
- A. De Klerk, Ind. Eng. Chem. Res., 2003, 42, 6545–6548 CrossRef CAS.
- A. Goosen and D. H. Morgan, J. Chem. Soc., Perkin Trans. 2, 1994, 557–562 RSC.
- R. Jevtic, P. A. Ramachandran and M. P. Dudukovic, Ind. Eng. Chem. Res., 2009, 48, 7986–7993 CrossRef CAS.
- C. C. Hobbs, E. H. Drew, H. A. Van't Hof, F. G. Mesich and M. J. Onore, Ind. Eng. Chem. Prod. Res. Dev., 1972, 11, 220–225 CAS.
- U. Neuenschwander and K. F. Jensen, Olefin Autoxidation in Flow, Ind. Eng. Chem. Res., 2014, 53, 601–608 CrossRef CAS.
- R. Jevtic, P. A. Ramachandran and M. P. Dudukovic, Chem. Eng. Res. Des., 2010, 88, 255–262 CrossRef CAS.
- A. Leclerc, M. Alame, D. Schweich, P. Pouteau, C. Delattreb and C. de Bellefon, Lab Chip, 2008, 8, 814–817 RSC.
- J. Fischer, T. Lange, R. Boehling, A. Rehfinger and E. Klemm, Chem. Eng. Sci., 2010, 65, 4866–4872 CrossRef CAS.
- N. M. Emanuel, E. T. Denisov and Z. K. Maizus, Liquid-phase oxidation of hydrocarbons, Plenum Press, New York, 1967 Search PubMed.
- G. H. Twigg, Chem. Eng. Sci., 1954, 8, 5–16 CrossRef.
- M. N. Siddiquee and A. De Klerk, Energy Fuels, 2014, 28, 6848–6859 CrossRef CAS.
- I. Hermans, J. Peeters and P. A. Jacobs, Top. Catal., 2008, 50, 124–132 CrossRef CAS.
- L. Vanoye, A. Aloui, M. Pablos, R. Philippe, A. Percheron, A. Favre-Réguillon and C. de Bellefon, Org. Lett., 2013, 15, 5978–5981 CrossRef CAS PubMed.
- A. Günther, S. A. Khan, M. Thalmann, F. Trachsel and K. F. Jensen, Lab Chip, 2004, 4, 278–286 RSC.
- L. K. Doraiswamy and D. Üner, Chemical Reaction Engineering, Beyond the Fundamentals, CRC Press, Boca Raton, 2014 Search PubMed.
- P. Sobieszuk, J. Aubin and R. Pohorecki, Chem. Eng. Technol., 2012, 35, 1346–1358 CrossRef CAS.
- H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noël, Chem. Soc. Rev., 2016, 45, 83–117 RSC.
- L. Kiwi-Minsker and A. Renken, Catal. Today, 2005, 110, 2–14 CrossRef CAS.
- M. Hamano, K. D. Nagy and K. F. Jensen, Chem. Commun., 2012, 48, 2086–2088 RSC.
- E. H. Stitt, Chem. Eng. J., 2002, 90, 47–60 CrossRef CAS.
- P. H. M. R. Cramers and A. A. C. M. Beenackers, Chem. Eng. J., 2001, 82, 131–141 CrossRef CAS.
- P. Havelka, V. Linek, J. Sinkule, J. Zahradník and M. Fialová, Chem. Eng. Sci., 2000, 55, 535–549 CrossRef CAS.
- J. Yun, Q. Lei, S. Zhang, S. Shen and K. Yao, Chem. Eng. Sci., 2010, 65, 5256–5263 CrossRef CAS.
- G. A. Russell, J. Am. Chem. Soc., 1957, 79, 3871–3877 CrossRef CAS.
- C. A. Tolman, J. D. Druliner, M. J. Nappa and N. Herron, Alkane oxidation studies in du Pont's central research department, in Activation and Functionalization of Alkanes, ed. C. L. Hill, Wiley, New York, 1989, pp. 303–360 Search PubMed.
- D. G. Hendry, C. W. Gould, D. Schuetzle, M. G. Syz and F. R. Mayo, J. Org. Chem., 1976, 41, 1–10 CrossRef CAS.
- J. E. Bennett, G. Brunton, J. R. L. Smith, T. M. F. Salmon and D. J. Waddington, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 2433–2447 RSC.
- W. A. Pryor, Free Radicals, McGraw−Hill, New York, 1966 Search PubMed.
- L. Jiayu, C. Wanghua, C. Liping, T. Yingtao and S. Xin, Procedia Eng., 2012, 43, 312–317 CrossRef.
- P. V. Danckwerts, Gas–Liquid Reactions, McGraw-Hill, New York, 1970 Search PubMed.
- F. Kaštánek, J. Zaharadnîk, J. Kratochvîl and J. Čermák, Chemical Reactors for Gas–Liquid Systems, Ellis Horwood, West Sussex, 1st edn, 1993 Search PubMed.
- E. L. Cussler, Diffusion: Mass Transfer in Fluid Systems, Cambridge University Press, Cambridge, 3rd edn, 2009 Search PubMed.
- N. Völkel, Design and Characterization of Gas–Liquid Microreactors, Ph. D. Thesis, Universite de Tolousé, December 2009 Search PubMed.
- A. K. Suresh, T. Sridhar and O. E. Potter, AIChE J., 1988, 34, 81–93 CrossRef CAS.
- V. Hessel, P. Angeli, A. Gavriilidis and H. Löwe, Ind. Eng. Chem. Res., 2005, 44, 9750–9769 CrossRef CAS.
- A. L. Hazel and M. Heil, J. Fluid Mech., 2002, 470, 91–114 Search PubMed.
- H. Wong, S. Morris and C. J. Radke, J. Colloid Interface Sci., 1992, 148, 317–336 CrossRef CAS.
- D. M. Fries, F. Trachsel and P. R. von Rohr, Int. J. Multiphase Flow, 2008, 34, 1108–1118 CrossRef CAS.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed.
- E. V. Rebrov, T. Duisters, P. Löb, J. Meuldijk and V. Hessel, Ind. Eng. Chem. Res., 2012, 51, 8719–8725 CrossRef CAS.
- H. Fadaei, B. Scarff and D. Sinton, Energy Fuels, 2011, 25, 4829–4835 CrossRef CAS.
- D. R. Snead and T. F. Jamison, Angew. Chem., Int. Ed., 2015, 54, 1521–3773 CrossRef PubMed.
- G. Rothenberg, Catalysis: Concepts and Green Applications, Wiley, Weinheim, 2008, pp. 1–38 Search PubMed.
- P. L. Alsters, J. M. Aubry, W. Bonrath, C. Daguenet, M. Hans, W. Jary, U. Letinois, V. Nardello-Rataj, T. Netscher, R. Rarton, J. Schütz, J. Van Soolingen, J. Tinge and B. Wüstenberg, Selective oxidation in DSM: Innovative catalysts and technology, in Handbook of Advanced Methods and Processes in Oxidation Catalysis: From Laboratory to Industry, ed. D. Duprez and F. Cavani, Imperial College Press, London, 2014, pp. 382–419 Search PubMed.
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6re00010j |
| This journal is © The Royal Society of Chemistry 2016 |