
Towards scalable and controlled synthesis of metal–organic framework materials using continuous flow reactors
Peter W.
Dunne
*a,
Edward
Lester
*b and
Richard I.
Walton
*c
aSchool of Chemistry, Trinity College Dublin, College Green, Dublin 2, Ireland. E-mail: p.w.dunne@tcd.ie
bDepartment of Chemical and Environmental Engineering, University of Nottingham, University Park, Nottingham, UK. E-mail: edward.lester@nottingham.ac.uk
cDepartment of Chemistry, University of Warwick, Coventry, UK. E-mail: r.i.walton@warwick.ac.uk
First published on 23rd June 2016
Abstract
Metal–organic frameworks have emerged as one of the most diverse new families of materials in the past few years. Their hybrid structures, combinations of inorganic and organic moieties, give a wide range of complex architectures with resultant properties that are suitable for numerous important fields, including porosity for molecular sieving and sensing, heterogeneous catalysis, drug delivery, and energy storage. If applications of these materials are to be realised then scalable synthesis is required, taking laboratory batch reactions towards industrial production. Continuous flow reactors offer the most versatile method for scaling their solvothermal synthesis, with the largest range of materials accessible, in high yield, and with control over crystal form.
Peter Dunne received his BSc and PhD from the National University of Ireland, Galway in 2005 and 2010, respectively. He then joined the group of Prof. Richard Walton at the University of Warwick, before moving to the Chemical Engineering Department at the University of Nottingham as a postdoctoral fellow under Prof. Ed Lester. In 2016 he took up a post as Assistant Professor in Inorganic Energy Materials at Trinity College Dublin. His research is focussed on the development of synthetic methods for the production of inorganic nanomaterials. |
Edward Lester is Professor of Chemical Technologies at the University of Nottingham with a research focus around continuous solvothermal and hydrothermal reactors for nanomaterial synthesis. He is also Technical Director of Promethean Particles which is a materials discovery company that uses his continuous reactor to make and scale up nanomaterials for clients around the world. Until April 2016 he was also coordinator for the 4 year FP7 project called SHYMAN (Sustainable Hydrothermal Manufacture of Nanomaterials) which scaled this continuous technology to create a 1000 ton per annum plant. He is currently Secretary to the International Solvothermal and Hydrothermal Association. |
Richard Walton is Professor of Inorganic Chemistry at the University of Warwick and his research focuses on developing novel synthesis methods for solid-state materials and structural characterisation to develop structure–property relationships. He is also presently Royal Society Industry Fellow, working with Johnson Matthey plc to examine the applications of new materials in various technologies. His research regularly makes use of synchrotron X-ray and central neutron facilities for materials characterisation, with particular emphasis on in situ methods for following crystallisation. He is an editor of the Inorganic Materials book series, with Bruce and O'Hare. |
Background: developments in the materials chemistry of porous solids
Metal–organic frameworks (MOFs) are one of the fastest developing families of solid-state materials at the present time. The systematic study of the chemistry of these hybrid materials, combining metal ions or clusters with polydentate ligands, has developed intensively from the 1990s onwards,1,2 although much earlier reports of three-dimensional ‘coordination polymers’ date back to the 1930s if one considers three-dimensional cyanide networks.3 MOFs have now been defined by IUPAC as a subset of coordination networks, themselves a subset of coordination polymers, that possess ‘a coordination network with organic ligands containing potential voids’.4 This void space, and thus potential for porosity, is where interest in MOFs is largely focussed for practical applications, since it may span the microporous (pore sizes of less than 2 nm, exemplified traditionally by the zeolites and ‘zeotype’ analogues) to the mesoporous (pore sizes between 2 nm and 50 nm, such as seen in liquid-crystal templated silicas). Importantly, by judicious choice of metal and connecting ligand(s) one can envisage the ‘design’ of open frameworks with pore size, pore shape, pore connectivity and internal chemical reactivity that may be tuned for selectively binding (or releasing) molecules or ions. This chemical reactivity may be as a result of the application of functional-group chemistry on the organic ligands, or the chemistry associated with particular metal centres, such as those with a binding preference for certain substrate molecules. A further, unique characteristic of MOFs, among all classes of solid-state materials, is the possibility of colossal structural flexibility, where atom displacements of several Ångströms are possible whilst maintaining the connectivity, and often crystallinity, of the structure.2,5–7 This can lead to enzyme-like conformational changes,8 or porosity that is adjusted by external stimuli, such as temperature or pressure. One could thus envisage pre-planned synthesis of a MOF material with a solid structure with the desired characteristics to match the needs of a particular application that requires the controlled transport of small molecules through an internal structure, furthermore with adaptable functionality depending on application of an external stimulus. Fig. 1 shows structures of some prototypical MOFs, whose production we consider in the later sections of this article.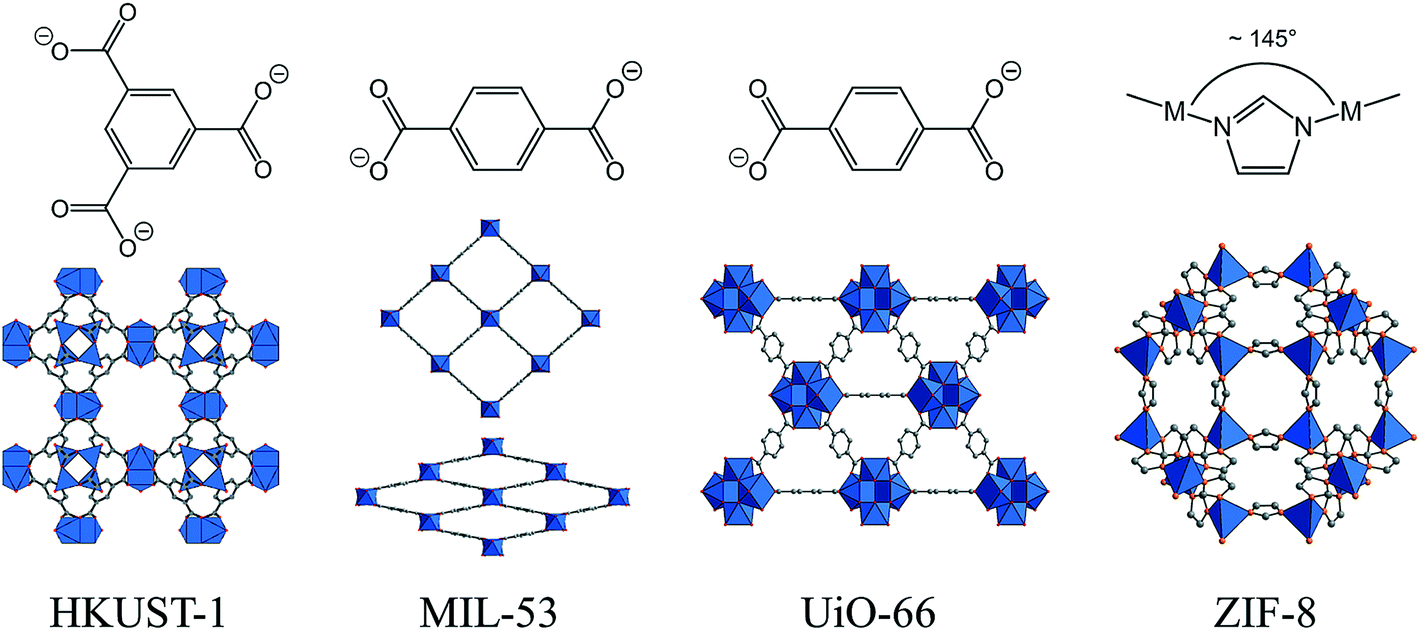 | ||
| Fig. 1 Structures of four prototypical MOFs whose flow synthesis has been studied: HKUST-1 (Cu3(BTC)2); MIL-53 (Al(BDC)(OH)); UiO-66 (Zr6O4(OH)4(BDC)6); and ZIF-8 (Zn(Im)2). The linkers present in the structures are drawn (BTC = 1,3,5-benzenetricarboxylate; BDC = 1,4-benzenedicarboxylate and Im = imidazolate) and the structures of the solids are represented with blue polyhedral units showing the local coordination of the metals. MIL-53 is shown in both its contracted and expanded forms, while the angle shown for the Im linker illustrates the analogy with zeolite structures (see text). | ||
The possible applications of MOFs stem from some of the areas in which microporous zeolites are used: separation of mixtures of small molecules (molecular sieving) and shape-selective heterogeneous catalysis. The larger range of pore sizes provided by MOFs, coupled with the potential for unique functionality and structural flexibility leads to new possibilities. It is beyond the scope here to review the wide ranges of structures and properties of MOFs, and indeed there are already collections of review articles9,10 and books available,11–14 so we will provide some of the key emerging uses of the materials, highlighted by recent published work. In the field of energy, the group of Yaghi has pioneered the development of ultra-high surface area MOFs that have exceptional capacity for gas uptake:15 they have recently proposed such materials as the basis of a carbon-neutral energy-cycle where gas storage and release is the key, with hydrogen as the fuel of choice in the long term, with methane an intermediate replacement and carbon dioxide capture used to counter climate change.16 In terms of catalysis, MOFs will never outperform zeolites in applications such as cracking of crude-oil derived hydrocarbons, because of their lower thermal stability, but some striking results have been obtained in other areas. The electrocatalytic reduction of CO2 to CO, for example, has been achieved using a MOF incorporating a cobalt porphyrin, thus taking a known molecular catalyst to a high surface area heterogeneous catalyst,17 while photocatalysis using the array of metal centres presented in a MOF structure has promise for oxidation of organics and synthesis of solar fuels.18 In separation science, MOFs can offer fine-tuned selectivity towards separation of hydrocarbon isomers by careful matching of pore size and connectivity to the shapes of substrate molecules,19 and certain flexible MOFs have been shown to offer counterintuitive pressure dependence, such as spontaneous desorption of guests at high pressure.20 The use of MOFs in biomedical applications is also emerging,21 while coating MOF particles with bioactive agents for controlled drug delivery has proved possible.22
Although it is anticipated that the applications of MOFs could be far reaching, their practical uses are really only beginning to be explored. Issues such as long-term stability (chemical and thermal) must be investigated under realistic conditions, but another crucial step in the availability of MOFs for real-life industrial use is finding efficient ways for their large-scale production. As with all scaled production of materials, these methods must yield reproducible batches of material in the most energy-efficient manner with minimal waste. In the case of polycrystalline powders it must be borne in mind that the crystal size and shape must often be tuned to match the needs of any application (high surface areas, or presentation of some particular crystal face(s) with specific reactivity, for example) or for any further processing steps (shaping and binding in pellets, for example), and this must be achieved with a tight dispersion of crystal size and shape. Furthermore, as with any materials with extended three dimensional structures, once synthesised there is rarely any possible scope for purification by recrystallisation: the material is produced essentially in the form it will be used in, although, for porous materials, washing or degassing may be used to remove excess solvent from the porous structure. This mini-review is concerned with the development of continuous processes for the scalable production of MOFs, to illustrate how the laboratory scale synthesis can be taken forward to industrial-scale manufacture.
Synthesis of MOFs: from laboratory to commercial production
MOFs are typically produced under solvothermal conditions, where a solvent heated above its boiling point is used as a reaction medium to bring about dissolution (partial or complete) of otherwise insoluble reagents, and then crystallisation of an extended network structure. Polar solvents are usually used to solubilise the bulky organic ligands used. In the laboratory synthesis is performed on a scale suitable for preparing specimens sufficient for analysis (powder X-ray diffraction, gas adsorption measurements, IR spectroscopy, thermogravimetric analysis, and ideally single crystal X-ray diffraction), so only a few hundred milligrams of solid are required and thus 20–50 mL batch reactors are used. Scale-up to larger batch reactors is of course feasible, and industrial manufacture has thus proved possible; for example, BASF hold patents for various methods of MOF manufacture and provide a set of MOFs that are commercially available with the tradename Basolite™. It may be noted that despite the thousands of MOF structures now known, only a relatively few are currently prepared on commercial scale. Although this situation may be compared with zeolite chemistry, with its long history and around 200 unique frameworks catalogued yet only a handful of materials produced industrially;23 given the sheer number of MOFs one might expect more will be applied eventually in practical applications. Although there have been reports on the mechanochemical production of certain MOFs using milling of solid precursors,24 continuous flow reactors are much more suited for MOF production: given the common use of solvents in MOF crystallisation, a far wider range of different MOF structures is likely to be accessed by continuous flow than in the absence of solvent. Furthermore, milling techniques may severely limit any control over crystal morphology and particle size of product.The continuous-flow systems which have been used in the production of metal organic frameworks can be broadly classed as three categories: (i) continuous-flow (hydro)solvothermal synthesis (CFHS/CFSS); (ii) continuous-stirred tank reactors (CSTR) and (iii) micro- or millifluidic systems. The basic geometries of these reactors are shown in Fig. 2. The CFHS system was initially developed for the supercritical hydrothermal production of fine metal oxide nanopowders by Adschiri et al.25 These high temperature, high pressure reactors comprise of at least two streams containing solvents/precursors. One stream (generally pure solvent) is passed through a pre-heating zone where it is heated to the desired reaction temperature and this superheated stream is then brought into contact with the precursor stream(s) at a mixing point inducing very rapid reactions. The geometry of this mixing point has been shown to have important implications for product quality and operational logistics,26,27 and a number of systems may also employ a post-mixing isothermal reaction zone to promote crystallisation and growth. After reaction the product stream passes through a heat exchanger before exiting the system via the back pressure regulator which maintains the system under the required pressures. These systems are readily scalable, as has been demonstrated by several groups,28,29 and until recently a full-scale (300 ton per year) CFHS plant was operated by Hanwha Chemicals, Ltd for the production of lithium iron phosphate.30 Continuous-stirred tank reactors (CSTRs) are among the classical reactor archetypes. They comprise of a heated large volume tank with mechanical stirring to which reagent solutions are continuously added while the products or reactor effluent is recovered. While they are rarely reported for the direct synthesis of inorganic materials, CSTRs have been used for the continuous co-precipitation production of carbonate and hydroxide-based precursors for the generation of battery materials.31,32 Microfluidic and millifluidic reactors, as the name suggests, involve extremely small reactor volumes and are often prepared by lithographic patterning or other microfabrication techniques to produce intricate nano/microchannels through which reagent solutions are flowed and mixed in various conformations.33 Increasingly micro- and millifluidic reactors are being constructed from readily available parts, such as small diameter PTFE tubing and micromixers. Frequently these systems employ an inert, immiscible phase, typically silicone oil, to segment the flow, creating “slugs” of reactant solution and further promoting internal mixing. The clear advantage of these microfluidic systems lies in the high degree of control which can be achieved thanks to the efficient heat and mass transport dynamics operating on such small scales.
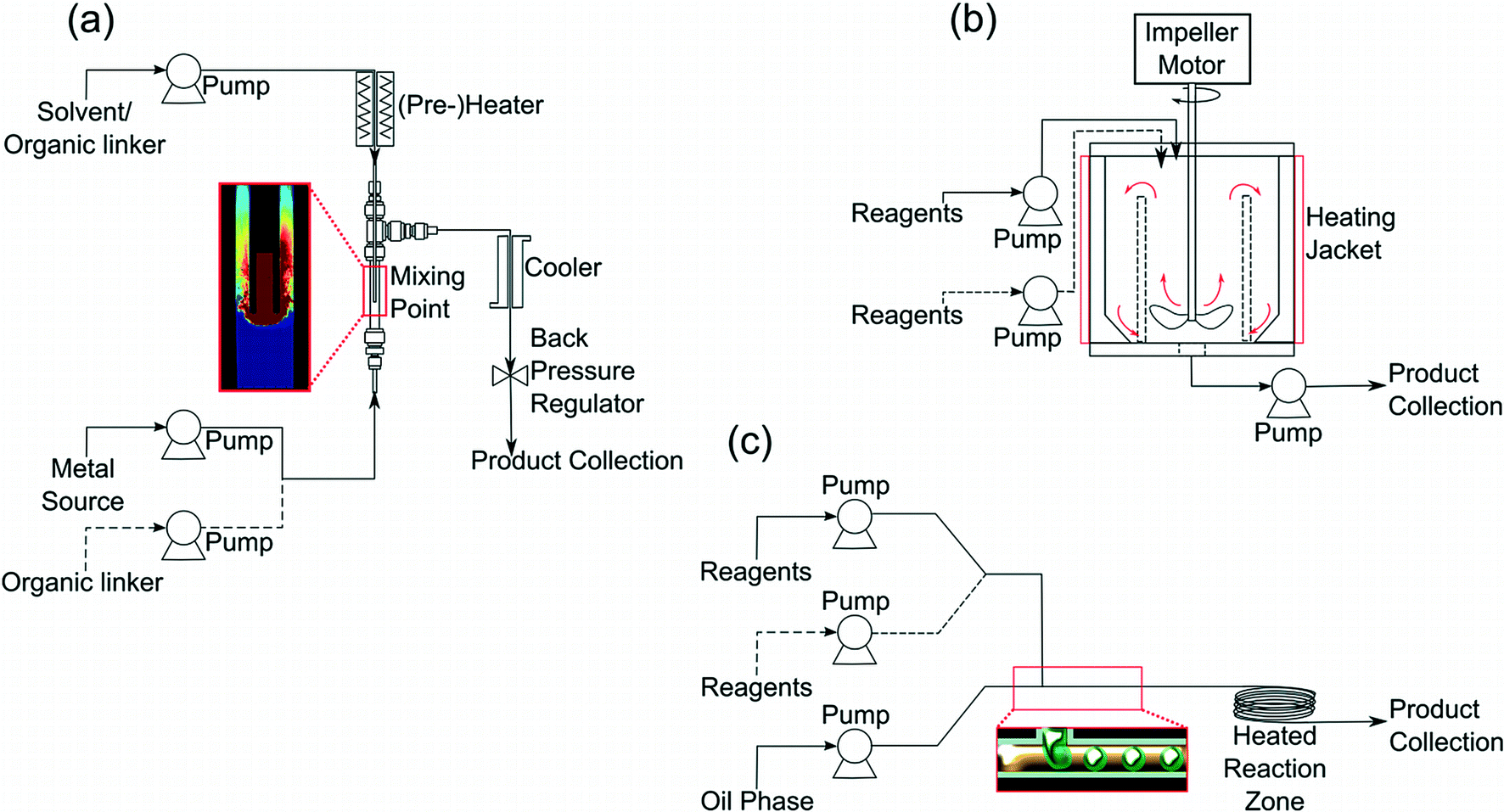 | ||
| Fig. 2 Schematics of various flow reactor geometries used for MOF synthesis: (a) continuous-flow (hydro)solvothermal synthesis (CFHS/CFSS); (b) continuous-stirred tank reactors (CSTR) and (c) micro- or millifluidic systems. | ||
MOFs prepared by continuous flow methods: towards controlled synthesis
Table 1 surveys the various reports of MOF synthesis by CFHS and CFSS methods, where we have provided comparative data concerning experimental details and, product yields and product characteristics. Four common MOF structures have been studied independently by a number of groups, and so these four structures are those shown on Fig. 1: these materials have been heavily investigated for their gas adsorption properties and many other applications, so are provided as representative MOF structures. The first continuous flow synthesis was reported by Gimeno-Fabra et al. in 2012,34 who described the production of two prototypical MOFs, the Cu(II) framework, HKUST-1 (Cu3(BTC)2, where BTC = 1,3,5-benzene dicarboxylate),35 which is also available commercially as Basolite C330, and the Ni(II) version of the CPO-27 structure (also known as MOF-74, with chemical formula ([M2(DHTP)(H2O)2]·8H2O where, DHTP = 2,5-dihydroxyterephthalate and M is a divalent metal cation).36 They used the CFSS “counter current mixing reactor” developed by the Nottingham group, in which preheated water is introduced through an inner pipe (downflow), meeting a precursor stream (upflow, injected at room temperature) at a mixing point. Thus metal salts and ligands were dissolved in the upstream (N,N-dimethylformamide and ethanol solvent mixture in this case) then nucleation and crystal growth did not occur until the mixing point; furthermore by controlling the reagent concentration in the upflow and temperature of downflow water, the size of the crystals formed could be tuned from a few microns to a few tens of microns for HKUST-1, and more dramatically for CPO-27(Ni) from the nanoscale to the micron-scale, Fig. 3.| Method | MOF | Solvent | Time | Temperature/°C (microwave power) | Production rate/g h−1 | Space–time yield/kg m−3 day−1 | Surface area/m2 g−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Pilot scale CFHS reactor. b Calculated based on information available. c No information available on yields or production rate. | ||||||||
| CFHS | MIL-53 (Al) | Water | 20 min | 250 | 1.5 | 1021b | 919 | 37 |
| MIL-53 (Al)a | Water | 20 min | 250 | 125 | 1300 | 1010 | 37 | |
| STA-12 (Cd)c | Water | 5–20 min | 70 | — | — | 134 | 55 | |
| ZIF-8 | Water | <5 s | 100 | 27 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 625 625 |
1806 | 43 | |
| ZIF-8a | Water | <5 s | 100 | 810 | — | 1780 | 43 | |
| CFSS | CAU-13 | DMF/AcOH | 20 min | 130 | 1.85 | 3049 | 401 | 55 |
| CPO-27 | DMF/H2O | <5 s | 300 | 10 | 1501 | 1030 | 34 | |
| HKUST-1 | EtOH | 20 | 250 | 2.1 | 730 | 1554 | 37 | |
| HKUST-1 | DMF/EtOH | <5 s | 300 | 30 | 4399 | 1950 | 34 | |
| HKUST-1 | EtOH | 1 min | 80 | 61.2 | 4533 | 1805 | 38 | |
| HKUST-1 | EtOH | 10 min | 80 | 1.48 | 592 | 1852 | 38 | |
| NOTT-400 | DMF/THF/H2O | 15 min | 85 | 2.78 | 741 | 1078 | 38 | |
| UiO-66 | DMF | 10 min | 130 | 1.68 | 672 | 1186 | 38 | |
| UiO-66 | DMF | 45 min | 120 | 0.26 | 428 | 1263 | 55 | |
| CFMW | HKUST-1 | DMF | 1 min | (360 W) | 79.4 | 64800 |
1550 | 39 |
| MIL-53 (Al) | DMF | 4 min | (200 W) | 7.1 | 3618 | 1376 | 39 | |
| UiO-66 | DMF | 7 min | (200 W) | 14.4 | 7204 | 1052 | 39 | |
 | ||
| Fig. 3 Electron micrographs of MOF samples prepared by continuous flow. (a) and (b) show examples of CPO-27 ((Ni2(DHTP)(H2O)2)·8H2O where, DHTP = 2,5-dihydroxyterephthalate) with crystallite size controlled from the nanoscale to the micron-scale by reagent dilution (reproduced from ref. 34 with permission from The Royal Society of Chemistry); (c) and (d) are examples of ZIF-8 with crystallite shape controlled by inclusion of solution additives (reproduced from ref. 43 with permission from The Royal Society of Chemistry) and (e) and (f) are crystallites of HKUST-1 where size was tuned by downstream solvent temperature (reproduced from ref. 34 with permission from The Royal Society of Chemistry). | ||
It is informative to compare samples of the prototype MOF HKUST-1 prepared subsequently by various groups using different CFSS reactor geometries. Bayliss et al. used pure ethanol as the solvent with separate flows of solutions of the metal salt and ligand mixed and heated to 200 °C.37 Rubio-Martinez et al. also used pure ethanol as solvent with separate solutions pumped with HPLC and mixed in a T-micro mixer and investigated the effect of residence time over the form of the product:38 they proposed control of particle size, although this was over a very small and overlapping range of distributions, from 181 ± 32 nm to 233 ± 82 nm average particle diameter. Taddei et al. used pure DMF as solvent with solutions of metal salts and of ligand mixed in a microwave heated at 4 bar in a plug-flow reactor;39 this gave exceptional space-time yield of ∼64800 kg d−1 m−3, the highest reported yet for any MOF material produced by continuous flow methods (see Table 1). Each of these samples of HKUST-1 has been shown to possess surface areas (once rendered free of adsorbed solvent) comparable to materials prepared by conventional synthesis methods, between 1500 and 1900 m2 g−1, and so would be perfectly suitable for applications that make use of their porosity.
The case of the Zr MOF UiO-66 (Zr6O4(OH)4(BDC)6), where BDC = 1,4-benzenedicarboxylate) is also useful to consider since this MOF is one of unusual thermal (up to around 400 °C) and chemical stability (under hydrothermal conditions at extremes of pH, for example) and has been intensively studied for its properties, particularly in catalysis.40 The groups of Rubio-Martinez et al.38 and Taddei et al.39 used the same CFSS reactor cells as for HKUST-1 mentioned above, with the microwave reactor of the second group giving again high yields, but with the use of water (necessary to promote formation of zirconium oxoclusters as secondary building units) and acetic acid as a ‘modulator’ (i.e. a crystal growth modifier). The material MIL-53 (M(BDC)(OH), where M is a trivalent metal cation) is another interesting case, since this is an example of a MOF with a flexible framework, showing massive expansion or contraction once water, or other guest molecule is removed from the pores, after the material has been prepared.41 Bayliss et al. produced MIL-53(Al) using the sodium salt of terephthalic acid and water as the solvent,37 while Taddei et al. used DMF as solvent in their continuous microwave reactor, giving much superior space-time yields.39 Surface area data proved the high quality of the samples produced, being consistent with the expected structural flexibility of the material.
ZIF-8 (M(Im)2, where Im = imizadolate, and M is a divalent metal cation) is one of a family of ‘zeolite imizadolate frameworks’, commonly prepared using Zn2+ as the metal ion, that have structures that resemble zeolite frameworks due to the similarity in the values of intra-tetrahedral Zn–imizadole–Zn angles and the Si–O–Si in corner-shared silicate units.42 ZIF-8 has a structure with the SOD zeolite topology (Fig. 1). Its production by CFHS has been reported by Munn et al. from water using very short residence time by virtue of the rapid mixing afforded by the counter current mixing reactor.43 The crystallite size could be varied from submicron to a few microns in dimension, while crystal shape could be adjusted from cubic to rhombic dodecahedral by the inclusion of ammonium hydroxide or triethylamine in the reaction mixture, respectively, or by changing temperature (between 100 and 400 °C) in the case of the ammonium hydroxide mediated reaction. Significantly, the process could be scaled to a pilot plant operating with a flow-rate of 900 mL min−1 and furthermore allowed continuous activation of the MOF sample, by pumping the as-made suspension of ZIF-8 crystallites through a heated coil downstream from the reaction point.
There are also two reports of the use of continuous-stirred tank reactor for MOF syntheses, which could be optimised to give similar yields to the flow reactors: Schoenecker et al. used a baffled reactor to produce UiO-66 from DMF solvent,44 whereas McKinstry et al. reported the synthesis of MOF-5 crystals in a continuous-stirred tank reactor and found that increased space-time-yields required 4 hour residence time.45
Microfluidic synthesis of MOFs has also been the focus of some attention, which although not immediately scalable to produce materials at industrially viable levels, allows the fundamental variables defining continuous production to be investigated easily. The reports so far in the literature are summarised in Table 2, and this includes a range of unusual materials, including MOFs containing precious metals, such as Ru, and, in the case of Ce5(BDC)7.5(DMF)4 (DMF = N,N-dimethylformamide), a novel material isolated from the reactor, which the authors subsequently prepared in a batch reactor as large crystals to determine its crystal structure.46 Beyond the efficient heat and mass transport that permits the rapid synthesis in these systems, a particular advantage of micro- and milli-fluidic synthesis lies in the ease with which different reactor configurations may be fabricated and assessed. This flexibility has facilitated the generation of core–shell MOF@MOFs (Co3BTC2@Ni3BTC2, MOF-5@dimethyl-MOF-5), as well as composites of ZIF-8 with magnetite nanoparticles, allowing the immobilisation or separation of active MOF catalysts through the use of multiple microfluidic reactors in series.47
| Method | MOF | Solvent | Time | Temperature (°C) | Production rate (g h−1) | Space-time yield (kg m−3 day−1) | Surface area (m2 g−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Multi-stage reactions. b Calculated based on information available. | ||||||||
| μ-CFSS | Ce5(BDC)7.5(DMF)4 | DMF | 30 s | 230 | 46 | |||
| HKUST-1 | EtOH | 5 min | 60 | 2.04 | 26800b |
1673 | 56 | |
| ZIF-8 | MeOH | 15 s | RT | 26.64 | 21000 |
1770 | 57 | |
| Segmented microfluidics | Fe-MIL-88B | H2O/DMF | 240 s | 95 | 58 | |||
| Fe-MIL-88B-Br | H2O/DMF | 360 s | 95 | 58 | ||||
| Fe-MIL-88B-NH2 | H2O/DMF | 20 s | 95 | 58 | ||||
| HKUST-1 | DMF/H2O/EtOH | 3 min | 90 | 0.0042 | 5.8 | 1911 | 47 | |
| IRMOF-3 | DMF/H2O/EtOH | 3 min | 120 | 2428 | 47 | |||
| MOF-5 | DMF/H2O/EtOH | 3 min | 120 | 3185 | 47 | |||
| Ru3BTC2 | H2O/EtOH | 160 | 550 | 47 | ||||
| UiO-66 | DMF/H2O/EtOH | 15 | 140 | 1059 | 47 | |||
| Co3BTC2@Ni3BTC2a | H2O | 5, 5 min | 140 | 47 | ||||
| MOF-5@DiMeMOF-5a | DMF/H2O/EtOH | 5, 15 min | 120 | 47 | ||||
| Fe3O4@ZIF-8a | H2O/MeOH | 2, 5 min | 80, 50 | 47 | ||||
The use of continuous microfluidic technology has been taken further by other groups to form MOF membranes. Brown et al. produced ZIF-8 films in hollow poly(amide–imide) fibres by using a flow of Zn2+ in octanol introduced into an aqueous solution of the ligand 2-methylimidazole held in a reactor.48 This allow formation of an 8.8 ± 1.4 μm membrane on the inner surface of the fibres that showed high H2/C3H8 and C3H6/C3H8 separation factors. Cacho-Bailo et al. used a similar approach with ZIF-8, ZIF-7 and ZIF-98 to form internally coated polymer fibres and proved molecular sieving properties,49,50 whereas Biswal et al. extended the work to include HKUST-1 coatings.51 Since in separation applications many porous materials are in reality used as films and membranes (rather than as polycrystalline powders), the use of continuous microfluidics points towards a future fabrication technology of devices.
Conclusions and outlook
The first continuous flow synthesis of MOFs was reported in 2012 and in the past 4 years there have been increasing numbers of reports of how such methods can be easily tailored to form high quality samples of some of the prototypical MOFs, which would be suitable for applications that require high surface area adsorbents. Established synthesis recipes have been taken from laboratory-scale batch reactors and adapted to large scale production in geometries such as continuous stirred tank reactors and continuous-flow hydrothermal and solvothermal synthesis reactors. These flow techniques have proven themselves as viable methods for the productions of increasingly important MOF materials on industrially relevant scales. Microfluidic reactors offer the opportunity to assess quickly reaction conditions for the identification of viable routes for the continuous production of MOFs, as well as screening conditions for the generation of novel frameworks and composites.Continuous-flow synthetic methods offer fast mixing, high heat transfer, and are inherently safer, with proportionally smaller volumes under reactive conditions at any given time. Together these factors conspire to make continuous methods highly scalable, and industrially preferred in many cases. In addition to the scalability and “batch-to-batch” reproducibility of continuous-flow systems, as solution based methods they offer a greater degree of control over crystal morphology and particle size of product, relative to mechanochemical techniques.
Certain MOFs lend themselves well to the step from batch to continuous since they readily form in relatively mild conditions, e.g., HKUST-1 and ZIF-8. Some MOFs also show lower dependence on the choice of solvent carrier which also makes them good initial targets for scale-up of proof-of-concept work. The key variable that will remain a challenge when considering continuous processing is time. The kinetics of formation is a key (possibly the most important) variable and this can be impacted by manipulation of temperature, pH, pressure, choice of solvent, precursor concentration and precursor type. However, if the growth kinetics remain relatively slow then mixing in the reactor will probably be less of an issue than developing a system that allows long residence times (10s of minutes to several hours), possibly under elevated pressures and temperatures.
Although a number of continuous flow synthesis methods have been successfully applied for MOF production, independently by a number of groups, and a variety of analysis data have been presented, there is still much further work required to characterise fully the MOF samples prepared in this work. For example, most literature reports have included scanning electron microscopy images to assess particle size and shape, but these are typically single images with no size analysis with statistical consideration, and no analysis using complementary techniques, such as light scattering, which would provide particle size distributions more representative of the bulk. Likewise the possibility of defects in the MOF samples prepared by continuous flow has not been addressed. This is important to consider, since there is a growing body of literature that shows how linker defects (i.e. partially occupied ligand sites within the MOF structure) can dramatically adjust the porosity and gas adsorption properties of MOFs.52 This may be an important consideration when samples have been prepared by rapid mixing of solutions, as in the continuous methods that we have reviewed, where there is no possibility of defect annealing following nucleation. Another issue in continuous synthesis of MOFs that has not yet been addressed, to our knowledge, is batch-to-batch variability. This requires careful analysis considering factors such as the length of operation of the reactor and sensitivity of crystallisation to purity of reagents and solvents, as well as independent verification of sample analysis by different laboratories. With the emergence of applications of MOFs, these aspects of manufacture will undoubtedly become the focus of attention.
The continuous flow synthesis of MOFs is clearly only just being started to be explored, and while extensive and systematic optimisation is needed, there are glimpses of some exciting possibilities in fine-tuning materials for practical application. For example, the fine control of particle size offered by dilution and/or flow rate through a heated zone, gives the possibility to control of the balance of nucleation and growth so to access crystals from the nanoscale up to the micron-scale. The use of activation immediately post-synthesis in a coupled reactor, means that a material can be prepared fit for use in one process. Not yet explored for MOFs, but investigated for the formation of oxide and phosphate materials in continuous flow, is the injection of capping agents downstream from the crystallisation point to supress further crystal growth and simultaneously surface-modify crystallites. This could conceivably be adapted to form core–shell particles, MOF@MOF, with the addition of further ligand(s) and metal salts so that crystal growth of a second MOF occurs on already formed MOF crystals. The MOF@MOF concept is proving to be an interesting strategy in tuning the properties of materials,53,54 so such systems could well be the future target of scaled synthesis. Another interesting possibility is the chance of crystallisation of new MOFs by continuous flow reactions, not yet seen in batch reactors; given the vast number of MOF materials being reported in the literature at the present time whose successful synthesis can be a fine balance of reaction conditions and reagents, exploratory synthesis using, for example, microfluidic reactors, could be an interesting strategy for automated materials discovery. Since the industrial-scale solvothermal synthesis using continuous reactors has proved possible for other inorganic materials (such as for lithium iron phosphate at 300 ton per year, as mentioned earlier), there is no reason to assume that similar processes will not be viable for MOF production.
Acknowledgements
Our work in this field work has been funded by the European Union's Seventh Framework Programme (FP7/2007–2013), grant agreement no. FP7-NMP4-LA-2012-280983, SHYMAN.References
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- J. F. Keggin and F. D. Miles, Nature, 1936, 137, 577–578 CrossRef CAS.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Ohrstrom, M. O'Keeffe, M. P. Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109–119 RSC.
- S. Bureekaew, S. Shimomura and S. Kitagawa, Sci. Technol. Adv. Mater., 2008, 9, 14108 CrossRef.
- G. Férey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC.
- J. Rabone, Y. F. Yue, S. Y. Chong, K. C. Stylianou, J. Bacsa, D. Bradshaw, G. R. Darling, N. G. Berry, Y. Z. Khimyak, A. Y. Ganin, P. Wiper, J. B. Claridge and M. J. Rosseinsky, Science, 2010, 329, 1053–1057 CrossRef CAS PubMed.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Metal-Organic Frameworks, Chem. Rev., 2012, 112(2), 673–1268 CrossRef CAS PubMed.
- H.-C. Zhou and S. Kitagawa, Metal-Organic Frameworks (MOFs) Themed Issue, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- Functional Metal-Organic Frameworks: Gas Storage, Separation and Catalysis, ed. M. Schröder, Springer-Verlag Berlin Heidelberg, 2010 Search PubMed.
- Metal-Organic Frameworks: Applications from Catalysis to Gas Storage, ed. D. Farrusseng, Wiley-VCH Verlag, Weinheim, 2011 Search PubMed.
- Metal Organic Frameworks, ed. L. R. MacGillivray and C. M. Lukehart, John Wiley & Sons Ltd, Chichester, 2014 Search PubMed.
- The Chemistry of Metal-Organic Frameworks: Synthesis, Characterization, and Applications, ed. S. Kaskel, Wiley-VCH Verlag, Weinheim, 2016 Search PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- A. Schoedel, Z. Ji and O. M. Yaghi, Nature Energy, 2016, 1, 16034 CrossRef.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- M. A. Nasalevich, C. H. Hendon, J. G. Santaclara, K. Svane, B. van der Linden, S. L. Veber, M. V. Fedin, A. J. Houtepen, M. A. van der Veen, F. Kapteijn, A. Walsh and J. Gascon, Sci. Rep., 2016, 6, 23676 CrossRef CAS PubMed.
- Z. R. Herm, B. M. Wiers, J. A. Mason, J. M. van Baten, M. R. Hudson, P. Zajdel, C. M. Brown, N. Masciocchi, R. Krishna and J. R. Long, Science, 2013, 340, 960–964 CrossRef CAS PubMed.
- S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Többens, S. Zander, R. S. Pillai, G. Maurin, F. O.-X. Coudert and S. Kaskel, Nature, 2016, 532, 348–352 CrossRef CAS PubMed.
- M. Gimenez-Marques, T. Hidalgo, C. Serre and P. Horcajada, Coord. Chem. Rev., 2016, 307, 342–360 CrossRef CAS.
- V. Agostoni, P. Horcajada, M. Noiray, M. Malanga, A. Aykac, L. Jicsinszky, A. Vargas-Berenguel, N. Semiramoth, S. Daoud-Mahammed, V. Nicolas, C. Martineau, F. Taulelle, J. Vigneron, A. Etcheberry, C. Serre and R. Gref, Sci. Rep., 2015, 5, 7 Search PubMed.
- R. Xu, W. Pang, J. Yu, Q. Huo and J. Chen, Chemistry of Zeolites and Related Porous Materials, John Wiley & Sons (Asia) Pte Ltd, Singapore, 2007 Search PubMed.
- D. Crawford, J. Casaban, R. Haydon, N. Giri, T. McNally and S. L. James, Chem. Sci., 2015, 6, 1645–1649 RSC.
- T. Adschiri, K. Kanazawa and K. Arai, J. Am. Ceram. Soc., 1992, 75, 1019–1022 CrossRef CAS.
- P. J. Blood, J. P. Denyer, B. J. Azzopardi, M. Poliakoff and E. Lester, Chem. Eng. Sci., 2004, 59, 2853–2861 CrossRef CAS.
- L. Zhou, S. Wang, D. Xu and Y. Guo, Ind. Eng. Chem. Res., 2014, 53, 481–493 CrossRef CAS.
- P. W. Dunne, C. L. Starkey, A. S. Munn, S. V. Y. Tang, O. Luebben, I. Shvets, A. G. Ryder, Y. Casamayou-Boucau, L. Morrison and E. H. Lester, Chem. Eng. J., 2016, 289, 433–441 CrossRef CAS.
- R. I. Gruar, C. J. Tighe and J. A. Darr, Ind. Eng. Chem. Res., 2013, 52, 5270–5281 CrossRef CAS.
- T. Adschiri, Y.-W. Lee, M. Goto and S. Takami, Green Chem., 2011, 13, 1380–1390 RSC.
- J. Camardese, D. W. Abarbanel, E. McCalla and J. R. Dahn, J. Electrochem. Soc., 2014, 161, A890–A895 CrossRef CAS.
- D. Wang, I. Belharouak, G. M. Koenig, G. Zhou and K. Amine, J. Mater. Chem., 2011, 21, 9290–9295 RSC.
- Y. Song, J. Hormes and C. S. S. R. Kumar, Small, 2008, 4, 698–711 CrossRef CAS PubMed.
- M. Gimeno-Fabra, A. S. Munn, L. A. Stevens, T. C. Drage, D. M. Grant, R. J. Kashtiban, J. Sloan, E. Lester and R. I. Walton, Chem. Commun., 2012, 48, 10642–10644 RSC.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- P. D. C. Dietzel, P. A. Georgiev, J. Eckert, R. Blom, T. Strässle and T. Unruh, Chem. Commun., 2010, 4962–4964 RSC.
- P. A. Bayliss, I. A. Ibarra, E. Perez, S. Yang, C. C. Tang, M. Poliakoff and M. Schröder, Green Chem., 2014, 16, 3796–3802 RSC.
- M. Rubio-Martinez, M. P. Batten, A. Polyzos, K.-C. Carey, J. I. Mardel, K.-S. Lim and M. R. Hill, Sci. Rep., 2014, 4, 5443 CAS.
- M. Taddei, D. A. Steitz, J. A. van Bokhoven and M. Ranocchiari, Chem. – Eur. J., 2016, 22, 3245–3249 CrossRef CAS PubMed.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- F. Millange, C. Serre and G. Férey, Chem. Commun., 2002, 822–823 RSC.
- B. R. Pimentel, A. Parulkar, E.-K. Zhou, N. A. Brunelli and R. P. Lively, ChemSusChem, 2014, 7, 3202–3240 CrossRef CAS PubMed.
- A. S. Munn, P. W. Dunne, S. V. Y. Tang and E. H. Lester, Chem. Commun., 2015, 51, 12811–12814 RSC.
- P. M. Schoenecker, G. A. Belancik, B. E. Grabicka and K. S. Walton, AIChE J., 2013, 59, 1255–1262 CrossRef CAS.
- C. McKinstry, R. J. Cathcart, E. J. Cussen, A. J. Fletcher, S. V. Patwardhan and J. Sefcik, Chem. Eng. J., 2016, 285, 718–725 CrossRef CAS.
- L. D'Arras, C. Sassoye, L. Rozes, C. Sanchez, J. Marrot, S. Marre and C. Aymonier, New J. Chem., 2014, 38, 1477–1483 RSC.
- M. Faustini, J. Kim, G.-Y. Jeong, J. Y. Kim, H. R. Moon, W.-S. Ahn and D.-P. Kim, J. Am. Ceram. Soc., 2013, 135, 14619–14626 CAS.
- A. J. Brown, N. A. Brunelli, K. Eum, F. Rashidi, J. R. Johnson, W. J. Koros, C. W. Jones and S. Nair, Science, 2014, 345, 72–75 CrossRef CAS PubMed.
- F. Cacho-Bailo, G. Caro, M. Etxeberria-Benavides, O. Karvan, C. Tellez and J. Coronas, Chem. Commun., 2015, 51, 11283–11285 RSC.
- F. Cacho-Bailo, S. Catalan-Aguirre, M. Etxeberria-Benavides, O. Karvan, V. Sebastian, C. Tellez and J. Coronas, J. Membr. Sci., 2015, 476, 277–285 CrossRef CAS.
- B. P. Biswal, A. Bhaskar, R. Banerjee and U. K. Kharul, Nanoscale, 2015, 7, 7291–7298 RSC.
- D. S. Sholl and R. P. Lively, J. Phys. Chem. Lett., 2015, 6, 3437–3444 CrossRef CAS PubMed.
- S. Furukawa, K. Hirai, Y. Takashima, K. Nakagawa, M. Kondo, T. Tsuruoka, O. Sakata and S. Kitagawa, Chem. Commun., 2009, 5097–5099 RSC.
- K. Koh, A. G. Wong-Foy and A. J. Matzger, Chem. Commun., 2009, 6162–6164 RSC.
- S. Waitschat, M. T. Wharmby and N. Stock, Dalton Trans., 2015, 44, 11235–11240 RSC.
- J. Kim, D. Kim, B. Veriansyah, J. Won Kang and J.-D. Kim, Mater. Lett., 2009, 63, 1880–1882 CrossRef CAS.
- A. Polyzoidis, T. Altenburg, M. Schwarzer, S. Loebbecke and S. Kaskel, Chem. Eng. J., 2016, 283, 971–977 CrossRef CAS.
- L. Paseta, B. Seoane, D. Julve, V. Sebastián, C. Téllez and J. Coronas, ACS Appl. Mater. Interfaces, 2013, 5, 9405–9410 CAS.
| This journal is © The Royal Society of Chemistry 2016 |