
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N 6-Methyladenine hinders RNA- and DNA-directed DNA synthesis: application in human rRNA methylation analysis of clinical specimens†
Shaoru
Wang‡
a,
Jiaqi
Wang‡
a,
Xiaoe
Zhang‡
a,
Boshi
Fu
a,
Yanyan
Song
a,
Pei
Ma
b,
Kai
Gu
a,
Xin
Zhou
b,
Xiaolian
Zhang
c,
Tian
Tian
*a and
Xiang
Zhou
*a
aCollege of Chemistry and Molecular Sciences, Key Laboratory of Biomedical Polymers of Ministry of Education, The Institute for Advanced Studies, Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: xzhou@whu.edu.cn; ttian@whu.edu.cn; Fax: +86-27-68756663; Tel: +86-27-68756663
bZhongnan Hospital, Wuhan University, Wuhan 430071, Hubei Province, China
cSchool of Medicine, Wuhan University, Wuhan 430071, China
First published on 17th November 2015
Abstract
N 6-Methyladenine (m6A) is the most abundant internal modification on mammalian mRNA. Very recently, m6A has been reported as a potentially important ‘epigenetic’ mark in eukaryotes. Until now, site-specific detection of m6A is technically very challenging. Here, we first reveal that m6A significantly hinders DNA- and RNA-directed DNA synthesis. Systematic investigations of 5′-triphosphates of a variety of 5-substituted 2′-deoxyuridine analogs in primer extension have been performed. In the current study, a quantitative analysis of m6A in the RNA or DNA context has been achieved, using Bst DNA polymerase catalyzed primer extension. Molecular dynamics study predicted that m6A in template tends to enter into and be restrained in the MGR region of Bst DNA polymerase, reducing conformational flexibility of the DNA backbone. More importantly, a site-specific determination of m6A in human ribosomal RNA (rRNA) with high accuracy has been afforded. Through a cumulative analysis of methylation alterations, we first reveal that significantly cancer-related changes in human rRNA methylation were present in patients with hepatocellular carcinoma.
Introduction
Epigenetic modifications on nucleic acids can significantly regulate the long-term gene activity and expression without any alteration in nucleotide sequence.1 Both DNA and RNA can be methylated at the N6 position of adenine (‘A’ in Fig. 1) to form N6-methyladenine (‘m6A’ in Fig. 1),2 which is one of the most important and common epigenetic markers. m6A is a prevalent modification present in the genome of bacteria3 and plays importantly regulatory roles in DNA restriction-modification systems.4 More importantly, several key reports have suggested that it may have a gene regulatory function in eukaryotes, including green alga,5 worm,6 and fly.7 Since the discovery of m6A demethylation in mammalian mRNAs by fat mass and obesity associated protein (FTO)8 and ALKBH5,9 there has been a great burst of interest in RNA epigenetics study.10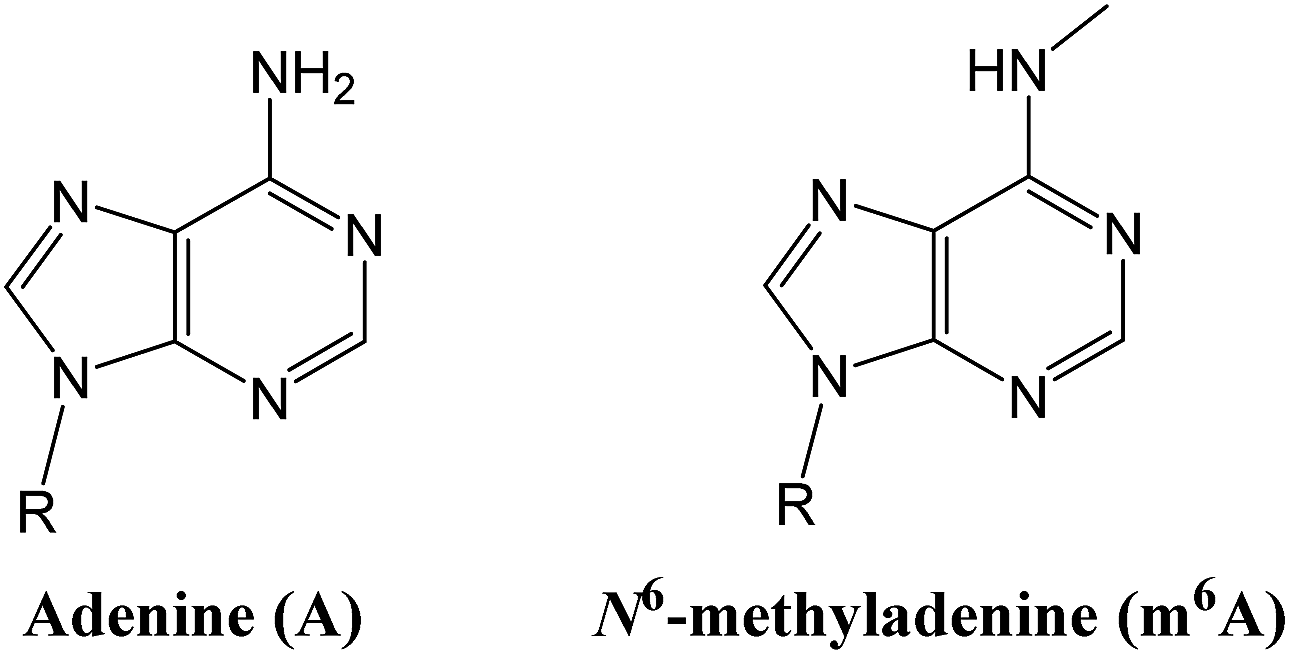 | ||
| Fig. 1 Structure illustration of adenine and N6-methyladenine (m6A). | ||
Due to the vital roles of m6A, dysregulation of RNA methylation can be associated with aberrant gene expression, which further lead to human diseases.11 In particular, some studies have shown that obesity and related diseases (type II diabetes) are associated to increased FTO activity and abnormally decreased amounts of m6A in patients.12 To the best of our knowledge, human cancers almost universally develop dysregulation of epigenetic marks, during both cancer initiation and disease progression.13 Since total RNA can be easily isolated from samples derived from cancer patients, it is well suited for further detection.14 Until now, the association of m6A modification with human cancer remains elusive, probably due to limited detection strategies. Recently, a pioneering method for m6A determination has been developed using a recombinant Thermus thermophilus DNA polymerase I (Tth pol) and specific m6A residues have been determined in human RNAs.15 However, the polymerization activities of Tth pol in RNA-directed DNA synthesis still remains to be improved.
Here, we report that m6A significantly hinders DNA- and RNA-directed DNA synthesis (Fig. 2) using a different polymerase, and a quantitative analysis of m6A in RNA or DNA context has been achieved. As an application of our approach, a site-specific determination of m6A in human ribosomal RNA (rRNA) of cultured tumor cells has been afforded. More importantly, different levels of methylation in human rRNA between hepatic cancerous and paracancerous tissues of patients with hepatocellular carcinoma have been identified using the new strategy. Our findings may provide new insights on DNA and RNA epigenetics of m6A.
 | ||
| Fig. 2 m6A hinders DNA- or RNA-directed DNA synthesis. | ||
Results
N 6-Methyladenine hinders DNA-dependent DNA synthesis
Since m6A in DNA may play an important regulatory role, we first conducted a study to illustrate its effects on DNA-directed DNA synthesis. Bst DNA polymerase, large fragment (Bst) is commercially available and is widely used in molecular biology applications.16 Here, it is used in the current study using DNA templates containing a site-specific A or m6A.DNA-A, DNA-m6A and primer 1 (sequences in Table S1†) were used to set up the model reaction. The dTTP or dUTP (structures in Fig. S1†) was used, and the incorporation efficiencies following different incubation times for DNA-A or DNA-m6A were compared together. In the current study, parameter RE (relative extension value) refers to normalized value of amount of reacted primer (‘primer + 1’ product) relative to the total amount of primer DNA. As shown in Fig. 3, a consistently lower RE for dTTP and dUTP opposite the m6A-template was characterized. For the same strand (Fig. S2†), a similar RE for dTTP incorporation compared with dUTP was observed through a same incubation time. Since there is a 5-substituted methyl difference between dUTP and dTTP, it raises an intriguing question about whether new uridine derivatives may affect DNA replication dynamics and enlarge incorporation discrepancies between DNA-A and DNA-m6A.
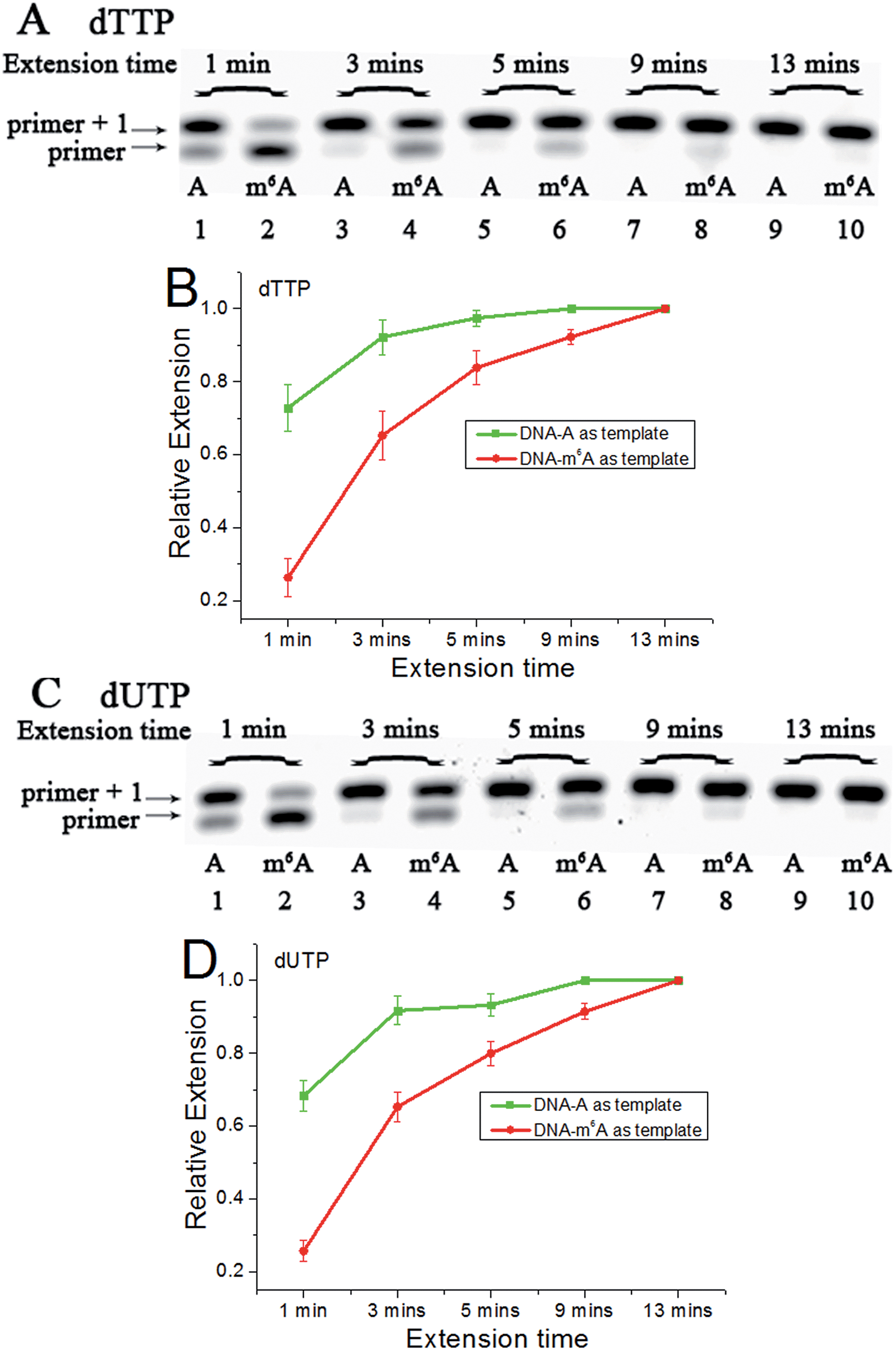 | ||
| Fig. 3 The analysis displays the difference of nucleotide incorporation opposite a templating A or m6A. Lane 1, 3, 5, 7, 9, DNA-A was used as a template; lane 2, 4, 6, 8, 10, DNA-m6A was used as a template. (A) and (C), representative gel image showing incorporation of dTTP or dUTP; (B) and (D), all data are presented as the means ± SEM from three independent experiments. | ||
Next, we used 5-hmdUTP and 5-formyl-dUTP (structures in Fig. S1†).17 The results demonstrated much lower incorporation efficiencies of both these two triphosphates opposite the DNA-m6A compared to DNA-A (Fig. S3†). Also, 5-hmdUTP and 5-formyl-dUTP were incorporated in the extended DNAs with a lower efficiency, compared with dTTP and dUTP (Fig. S4†). Inspired by the electron density sequence of 5-formyl-dUTP < 5-hmdUTP < dTTP, we hypothesised that electron-withdrawing groups would reduce the interaction between the incoming nucleotide and the opposite A or m6A.
Subsequently, a family of 5-halogenated uridine analogs, including 5-F-dUTP, 5-Br-dUTP and 5-Iodo-dUTP (structures in Fig. S1†) were used.18 As shown in Fig. S5 and S6,† a consistently lower incorporation of 5-halogenated dUTPs opposite DNA-m6A was characterized, compared with DNA-A. A comparable amount of 5-Br-dUTP and 5-Iodo-dUTP to dTTP was incorporated into the primer, while much less incorporation of 5-F-dUTP was observed (Fig. S6†).
We further assessed some other sequences to test the universality of our finding. For Mycobacterium tuberculosis (Mtb), corA gene is a strongest candidate methylation-affected gene,19 which is methylated at the nucleotide three base pairs downstream of the predicted sigma factor −10 site on the non-template strand while the last nucleotide of the −10 site is methylated on the template strand (Fig. S7†).19b Hence, the non-methylated sequence from −9 bp to +15 bp on the non-template strand and the one from −23 bp to −4 bp on the template strand relative to the transcriptional start site (TSS) and their methylated counterparts were synthesized and used as templates for methylation analysis (corA-non-A, corA-non-m6A, corA-temp-A and corA-temp-m6A, sequences in Table S1†). Respective FAM-labelled primers were designed and used in the following study (primer-non and primer-temp in Table S1†). As expected, a much lower incorporation of dTTP or dUTP opposite the m6A-templates was characterized, compared with their A-counterparts (Fig. S8 and S9†). The results further suggest that m6A hinders DNA-directed DNA synthesis in a sequence-independent manner.
To further evaluate the incorporation efficiency of the studied triphosphates between different templates, we next investigated other DNA polymerases, including Klenow fragment (exo-) and Klenow fragment with exonuclease activity. The following results showed that the reduction of substrate activity by adenine methylation was universal (Fig. S10 and S11†), while these two polymerases produced a decreased discrepancy compared with Bst DNA polymerase. Hence, Bst DNA polymerase was used for further m6A analysis in the following studies.
Calculations studies
In a very relevant study,20 Kool and co-workers reported the destabilizing effects of m6A in duplexes through NMR analysis and thermodynamic measurements. To gain more insights on structural basis, we investigated the conformation of m6A or A in complexes between DNA templates and Bst DNA polymerase, using molecular dynamics (MD) approach supplemented with potential of mean force (PMF) analysis.21 The starting structure was generated using the deposited crystal structure (PDB code 2BDP), which contains Bst DNA polymerase, a primer and a template.22 m6A was built using DFT (density functional theory) calculation at the B3LYP/6-311G level.23 The A that lies in the active site is manually modified to m6A. Since the minor-groove recognition (MGR) region in Bst DNA polymerase plays a key role to specifically recognize correctly paired bases, it is required to provide sequence-independent interactions with its DNA substrate.22 The most common B-DNA conformation is observed for the DNA outside the MGR region, corresponding to the characteristic C2′-endo sugar pucker. A pronounced bend in conformation take place at the star of the MGR region (Fig. S12†).22 The sugar pucker switches to the C3′-endo conformation characteristic of A-form DNA, during the replication process. However, the methyl group of m6A significantly perturbates the backbone torsion angles and sugar puckering (detailed torsional angles in Table S2, S3 and Fig. S13†). As demonstrated in Fig. 4, the MD trajectory shows that m6A tends to adopt a stubborn B-form with the characteristic C2′-endo sugar pucker. Hence, m6A in template tends to enter into and be restrained in the MGR region, reducing conformational flexibility for the DNA backbone. This would hamper the essential shift between active sites of Bst DNA polymerase and primer–template complex. As a result, it is easier to incorporate dTTP into the growing chain opposing A. Our calculation is therefore consistent to some extent with the previous study.20 | ||
| Fig. 4 A template containing m6A forms a stable complex with primer in the Bst DNA polymerase active site place. The structure is superposed with the template–primer (green/orange) duplex. Key residues in the MGR region of enzyme (pink) are displayed, including Tyr 714 (766), Arg 615 (668) and Gln 797 (849). The protein–DNA interface in the MGR region is stabilized by hydrogen bonds (H-bonds, violet) and the stacking interaction between the template base and the corresponding residue [Tyr 714 (766)]. The torsion angles were significantly changed by m6A in the template. | ||
Quantitating m6A in DNAs
Next, we asked whether our method could be optimized to provide quantitative analysis of m6A in DNAs. Therefore, we used 5-hmdUTP and explicitly studied the alteration of the thermal extension times, enzyme concentrations, as well as the extension temperatures, to achieve a better discrimination (Fig. S15†). With the optimized conditions in hand, we mixed known ratios of DNA-m6A with the A-containing counterpart (Table S4†) and measured the yield of 5-hmdUTP incorporation at a fixed time point. The values of RE in this reaction were inversely linearly proportional to the amount of m6A present (Fig. S16†), suggesting that this method can be used in quantitative analysis of the methylation extent at the candidate site. As shown in Fig. S17,† 5-formyl-dUTP was also successfully used by this method for methylation analysis of DNAs.N 6-Methyladenine hinders RNA-dependent DNA synthesis
Accounting for the importance of m6A in mammalian mRNAs,24 we were interested whether the extension discrepancy is present between A- and m6A-containing RNA templates. RNA-A, RNA-m6A and primer 2 (sequences in Table S1†) were used to perform this study. Through gel analysis, a consistently m6A-dependent inhibition was characterized (Fig. 5). RNA-m6A incorporates much less dTTPs or dUTPs than RNA-A. For dTTP, a significant extension could proceed beyond the site complementary to the A in RNA-A after an incubation of 3 min, while a moderate to negligible extension in RNA-m6A was characterized. Steady-state incorporation kinetics study revealed that RNA-A (Vmax/Km = 5.12 ± 0.76) is a much better substrate than RNA-m6A (Vmax/Km = 0.16 ± 0.04) for Bst DNA polymerase.25 For dUTP, much less extension opposite RNA-A compared with dTTP was observed (Fig. S18†), while RNA-m6A was not a good template for dUTP incorporation. The extension of dTTP opposite RNA-A was accomplished after an incubation of 6 min (Fig. 5), while the incorporation of 5-formyl-dUTP or 5-hmdUTP was not very efficient after even 15 min (Fig. S19 and S20†).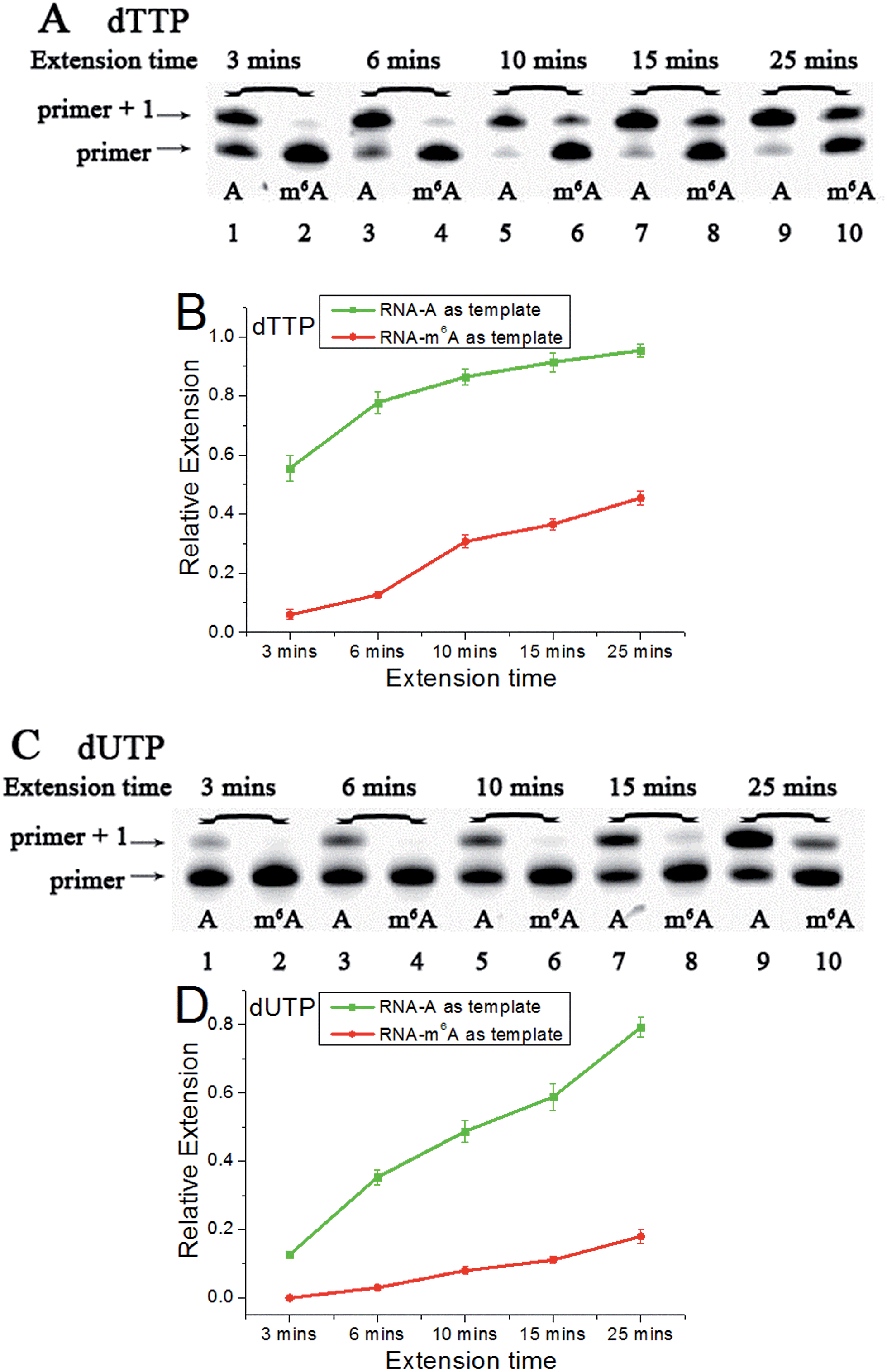 | ||
| Fig. 5 The analysis displays the difference of nucleotide incorporation opposite a templating A or m6A. Lane 1, 3, 5, 7, 9, RNA-A used as a template; lane 2, 4, 6, 8, 10, RNA-m6A used as a template. (A) and (C), representative gel image showing incorporation of dTTP or dUTP; (B) and (D), all data are presented as the means ± SEM from three independent experiments. | ||
Next, three 5-halogenated uridine analogs were also assessed and compared for extension behavior opposite the site of A or m6A. As shown in Fig. S21,† distinctive extensions were identified as functions of increasing duration. Like dTTP, 5-Iodo-dUTP or 5-Br-dUTP were efficiently incorporated into the growing chain to generate ‘primer + 1’ products. However, because of the strong electron-withdrawing group at the 5 position of 5-F-dUTP, the resulting extension is relatively minimal, as observed in PAGE analyses demonstrating a lower yield for the reaction with 5-F-dUTP than the reaction with other uridine analogs (Fig. S21A and S21B†). A consistent reactivity sequence with the previous DNA template was characterized (Fig. S22†).
Quantitating m6A in synthetic RNAs
We further performed m6A analysis of RNA mixtures. A series of artificial samples (Table S5†) were prepared. The incorporation of 5-Iodo-dUTP as a function of increasing RNA-m6A concentration is shown in Fig. S23.† The current study showed a very good concordance between the values of RE and the ratios of m6A present (Fig. S23B†), suggesting that this method is well suited for RNA methylation analysis at a specific locus. As shown in Fig. S24,† m6A analysis in RNA context was also accomplished using dTTP.Accurate determinations of m6A sites in human ribosomal RNAs
Next, we asked and set out to test directly whether this strategy could be used to determine the methylation states of different sites in rRNA from human cells. We tested the extracted total RNAs from cultured HeLa cells.26 A well-known m6A modification identified in human rRNA is located at position 1832 in the 18S subunit, and a neighboring A without any modification at position 1781 has been elucidated in a previous study.27 Also, the site at position 4984 remains unmodified in the 28S subunit. The according labeled primers targeting these sites have been used.15 Since equal amounts of RNAs have been used in the test, direct comparison of RE values could accurately reflect the methylation level. As shown in Fig. S25,† a large proportion of primer1781A (lane 2) and primer4984A (lane 10) was extended, indicating low methylation status of human rRNAs at these sites. In contrast, almost no extended products was observed with primer1832 mA (lane 4), indicating complete methylation status of human rRNAs at the 1832 site. These results are well consistent with the known methylation states of the studied nucleotides.15,27Next, we proceed to determine the methylation status of more A sites in human rRNAs. It has been reported that there is only one m6A between positions 4189 and 4190 in human 28S rRNA, while the other one remains unmodified.28 Two primers with a same length were used to target the probed nucleotides.15 As shown in Fig. S25,† almost all primer4189 were extended to get longer products in the reaction (lane 6), while no extended products were characterized for primer4190 in this analysis (lane 8). This could be interpreted as that human 28S RNA is predominantly methylated at position 4190, while not at position 4189. Our result was entirely consistent with the previous work by another group.28b Analysis of cultured MCF-7 tumor cells revealed a very similar m6A modification pattern (Fig. S26†).
rRNA methylation analysis of clinical specimens
To demonstrate the reliability and make practical application of this method, m6A analysis of rRNA in clinical specimens was performed. Fresh hepatic cancerous or paracancerous tissue from a same patient with hepatocellular carcinoma were collected, and total RNAs were extracted using standard methods. The same primers were used to probe the aforementioned sites. Based on these results (Fig. 6 and S27†), human rRNA in both hepatic cancerous and paracancerous tissues was methylated at position 1832 in the 18S subunit and at position 4190 in 28S subunit. However, this quantitative comparative analysis revealed a higher value of RE for hepatic cancerous tissue at positions 4189 and 4984 in 28S subunit, indicating lower levels of RNA methylation at these sites. By contrast, a smaller value of RE for hepatic cancerous tissue at position 1781 in the 18S subunit was observed, indicating a higher level of RNA methylation at this site.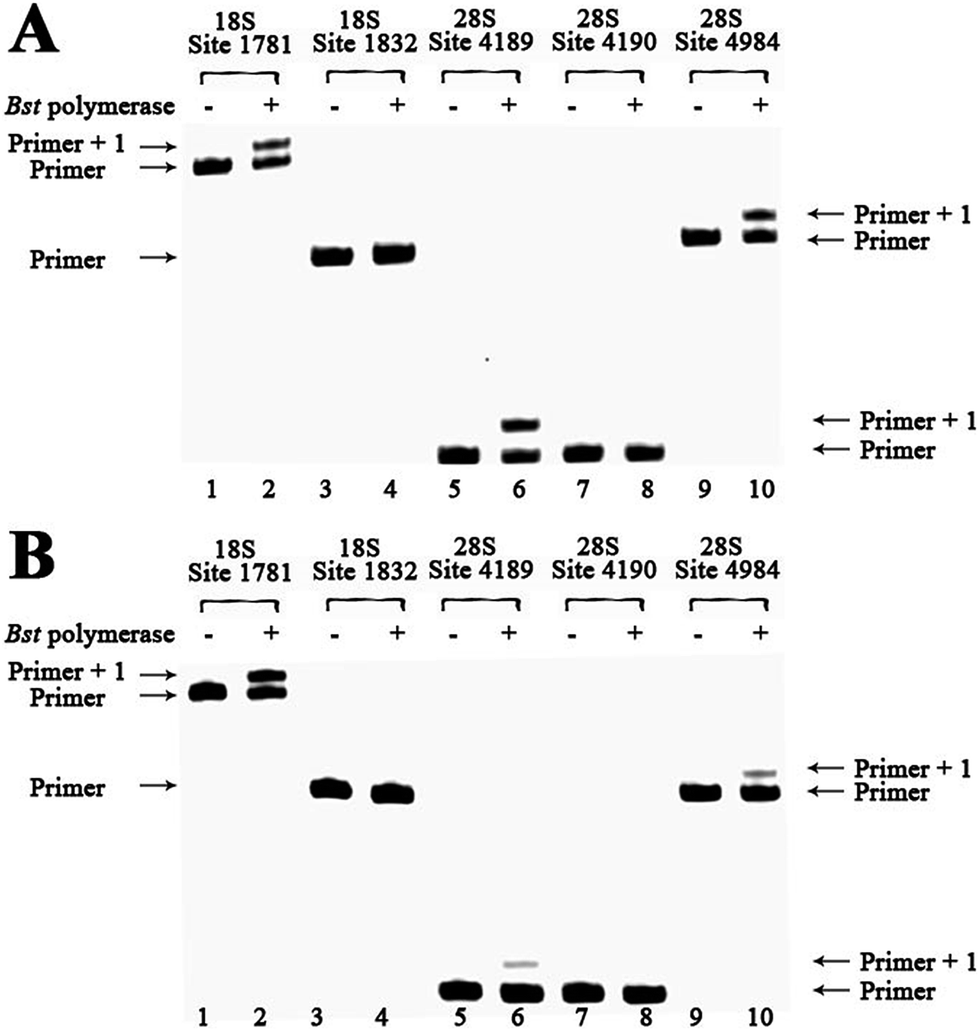 | ||
| Fig. 6 Methylation analysis of 18S and 28S rRNA for hepatic cancerous or paracancerous tissue from a same patient with hepatocellular carcinoma. dTTP is incorporated for analysis. Lanes 1, 3, 5, 7 and 9, control samples without addition of Bst DNA polymerase; lanes 2, 4, 6, 8 and 10, 0.1 U Bst DNA polymerase was used. (A) Analysis of hepatic cancerous tissues of patient 1 with hepatocellular carcinoma; (B) analysis of hepatic paracancerous tissues of the same patient. | ||
We further used this method to determine rRNA methylation levels at the aforementioned sites within 19 more patients with hepatocellular carcinoma (Fig. S28–S46†). For proof-of-principle, all of the hepatic cancerous tissues from different patients were grouped together and a paracancerous pool was built for comparison. Multivariate statistical analysis was performed to analyze the obtained data using two-sample Hotelling's T-squared test. The low P value (P = 0.0096) provides evidence that there is a significant methylation difference between hepatic cancerous and paracancerous tissues. To further test the methylation difference, Wilcoxon signed-rank test was performed on single locus. As shown in Fig. 7, significantly cancer-related changes were observed in these two pools. For positions 4189 and 4984 (28S RNA), values of RE in hepatic cancerous tissues demonstrate a bimodal distribution, with some having “normal” values (relative to paracancerous tissue) and a second cluster with evidently high values. These results also suggest that the medians and averages of RE at positions 4189 and 4984 (28S RNA) in cancerous tissues are obviously higher than that of the paracancerous control, and while an opposite trend was observed at position 1781 (18S rRNA). Moreover, a bimodal distribution for values of RE was observed in these two groups of tissues, indicating a high heterogeneity of methylation at position 1781 (18S rRNA).
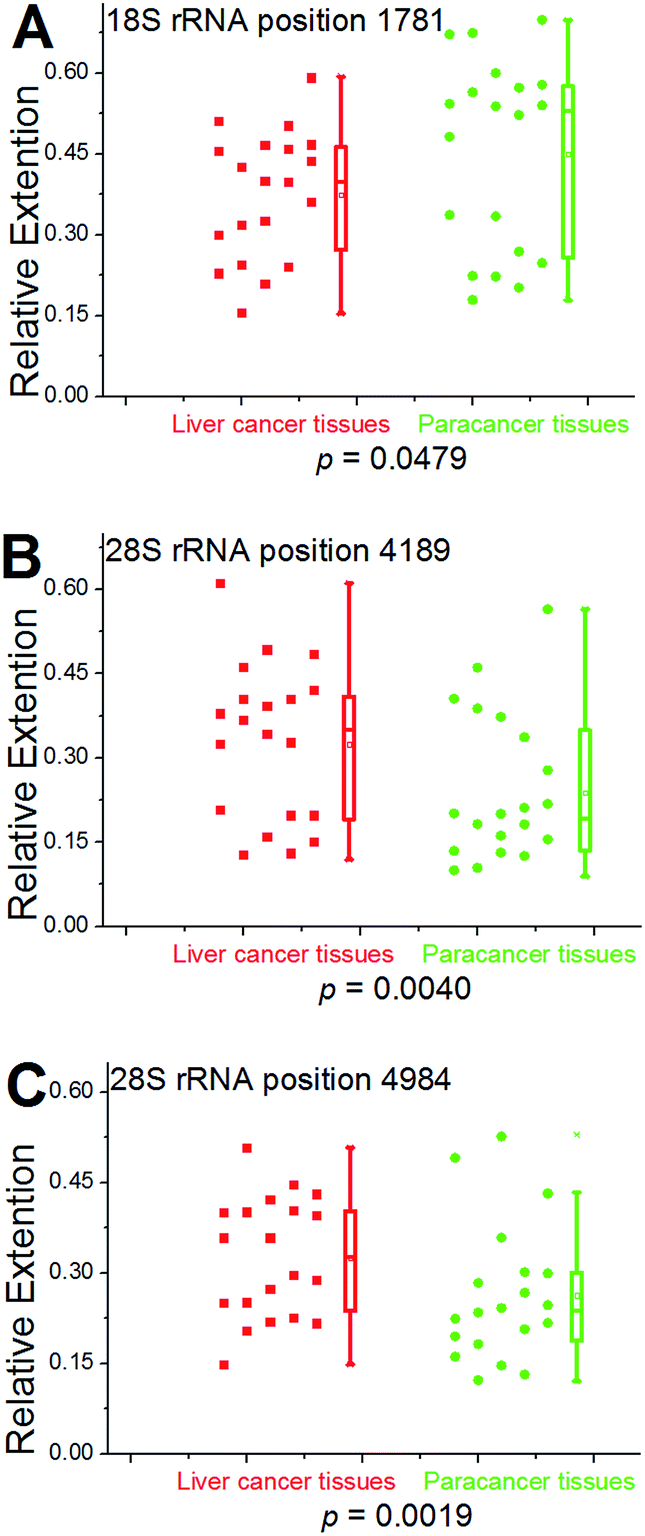 | ||
| Fig. 7 Statistical analysis for 18S and 28S rRNA methylation in clinical specimens. Left scatter plots indicate the RE values of all tested cases obtained from hepatic cancerous or paracancerous tissues in patients with hepatocellular carcinoma. Right box plots represent the distribution of the data. The median value is identified by a line inside the box. The length of the box represents the interquartile range. The P-value is calculated by Wilcoxon rank-sum test. | ||
Discussion
It is very interesting to think about whether m6A in an organism can affect central dogma's processes, including DNA replication and RNA reverse-transcription.29 In the present study, we first demonstrate that in vitro DNA synthesis is hindered by the presence of m6A. Recently, m6A has been reported to be present in eukaryotic genomic DNAs.5–7 Importantly, several m6A-binding proteins have been revealed by different groups.10c,30 As a potentially stable base, m6A may affect the folding of chromatin and local replication activity through selective recruitment of m6A-binding proteins. Our study therefore proposes a possibility that DNA replication forks may stall at m6A sites and such stalling may help the cell avoid mutations.In our study, we used a variety of uridine and 5-substituted uridine analogs involving a primer extension by Bst DNA polymerase, and a good discrimination between A- and m6A-containing sequences (DNAs and RNAs) was achieved through a simple procedure. Moreover, the MD simulations demonstrate that the torsion angles for backbone conformations and puckered forms of the sugar ring vary quite significantly after methylation. This is accompanied by a monotonic widening of the minor groove. The model implies a loss of conformational freedom of binding interfaces between Bst DNA polymerase and the primer–template complex containing m6A, while this conformational flexibility is necessary for the processive movement of the enzyme.22 To some extent, m6A may cause processive Bst DNA polymerases to stall, thus leading to a block of the replication process.
Although m6A has been identified for a long time,31 there is an increasing demand to develop reliable and more efficient methods to unambiguously determine the exact position of this modification in RNA contexts,32 especially for analysing clinical specimens. To the best of our knowledge, the use of promoter methylation level as tumor biomarker has been intensively studied during the past decades.33 Since RNA is the downstream products of DNA during gene transcription, its methylation level can be potentially a more accurate tumor biomarker. To accomplish such a goal, we successfully used our method to determine the methylation status of specific sites in human rRNAs, using total RNAs extracted from cultured cells or clinical specimens. Evident differences in the RE values between the two groups are characterized and the results are statistically significant. The current study reveals that the human hepatic cancerous and paracancerous tissues are mainly methylated at position 1832 (18S RNA) and at position 4190 (28S RNA). Even more importantly, obviously lower levels of RNA methylation were characterized for hepatic cancerous tissues at positions 4189 and 4984 in the 28S subunit. At position 1781 in 18S subunit, a significantly higher level of methylation was observed in the cancerous pool, compared to the paracancerous control.
Experimental section
Detection of m6A in human rRNAs
For each reaction, 0.5 μg total RNA and each primer at 100 nM were used. This reaction was performed in 1× ThermoPol™ Reaction Buffer. A 20 μL sample was incubated in a water-bath at 45 °C for 1 h. The same protocol described in ESI† for RNA-directed DNA synthesis was used.Statistical analysis
Statistical analysis of rRNA methylation data was performed using the SPSS 19.0 software (SPSS Inc.). The methylation differences at each position between the two pools were tested with the Wilcoxon rank-sum test. Differences were considered to be significant for P < 0.05.Conclusions
Most importantly, we first reveal that significantly cancer-related changes in human rRNA methylation were present in patients with hepatocellular carcinoma. Although further evidence is needed to consolidate the connection between m6A and hepatic cancer, the methylation status of ‘A’ in target rRNA can potentially be treated as a novel tumor biomarker. Our findings can help advance understanding the function of this highly important modification in human disease.Acknowledgements
The authors thank the National Basic Research Program of China (973 Program) (2012CB720600, 2012CB720603, 2012CB720605), the National Science Foundation of China (No. 21432008, 91413109, 21372182, 21202126).Notes and references
- R. Jaenisch and A. Bird, Nat. Genet., 2003, 33, 245–254 CrossRef CAS PubMed
.
- D. Wion and J. Casadesus, Nat. Rev. Microbiol., 2006, 4, 183–192 CrossRef CAS PubMed
- G. Jia, Y. Fu and C. He, Trends Genet., 2013, 29, 108–115 CrossRef CAS PubMed
- G. Fang, D. Munera, D. I. Friedman, A. Mandlik, M. C. Chao, O. Banerjee, Z. Feng, B. Losic, M. C. Mahajan, O. J. Jabado, G. Deikus, T. A. Clark, K. Luong, I. A. Murray, B. M. Davis, A. Keren-Paz, A. Chess, R. J. Roberts, J. Korlach, S. W. Turner, V. Kumar, M. K. Waldor and E. E. Schadt, Nat. Biotechnol., 2012, 30, 1232–1239 CrossRef CAS PubMed
- Y. Fu, G. Z. Luo, K. Chen, X. Deng, M. Yu, D. Han, Z. Hao, J. Liu, X. Lu, L. C. Dore, X. Weng, Q. Ji, L. Mets and C. He, Cell, 2015, 161, 879–892 CrossRef CAS PubMed
- E. L. Greer, M. A. Blanco, L. Gu, E. Sendinc, J. Liu, D. Aristizabal-Corrales, C. H. Hsu, L. Aravind, C. He and Y. Shi, Cell, 2015, 161, 868–878 CrossRef CAS PubMed
- G. Zhang, H. Huang, D. Liu, Y. Cheng, X. Liu, W. Zhang, R. Yin, D. Zhang, P. Zhang, J. Liu, C. Li, B. Liu, Y. Luo, Y. Zhu, N. Zhang, S. He, C. He, H. Wang and D. Chen, Cell, 2015, 161, 893–906 CrossRef CAS PubMed
-
(a) G. Jia, Y. Fu, X. Zhao, Q. Dai, G. Zheng, Y. Yang, C. Yi, T. Lindahl, T. Pan, Y. G. Yang and C. He, Nat. Chem. Biol., 2011, 7, 885–887 CrossRef CAS PubMed
- G. Zheng, J. A. Dahl, Y. Niu, P. Fedorcsak, C. M. Huang, C. J. Li, C. B. Vagbo, Y. Shi, W. L. Wang, S. H. Song, Z. Lu, R. P. Bosmans, Q. Dai, Y. J. Hao, X. Yang, W. M. Zhao, W. M. Tong, X. J. Wang, F. Bogdan, K. Furu, Y. Fu, G. Jia, X. Zhao, J. Liu, H. E. Krokan, A. Klungland, Y. G. Yang and C. He, Mol. Cell, 2013, 49, 18–29 CrossRef CAS PubMed
-
(a) K. D. Meyer, Y. Saletore, P. Zumbo, O. Elemento, C. E. Mason and S. R. Jaffrey, Cell, 2012, 149, 1635–1646 CrossRef CAS PubMed
- T. A. Cooper, Circ. Res., 2012, 111, 1491–1493 CrossRef CAS PubMed
-
(a) A. Klungland and J. A. Dahl, Curr. Opin. Genet. Dev., 2014, 26, 47–52 CrossRef CAS PubMed
- M. V. Brock, C. M. Hooker, E. Ota-Machida, Y. Han, M. Guo, S. Ames, S. Glockner, S. Piantadosi, E. Gabrielson, G. Pridham, K. Pelosky, S. A. Belinsky, S. C. Yang, S. B. Baylin and J. G. Herman, N. Engl. J. Med., 2008, 358, 1118–1128 CrossRef CAS PubMed
- B. Lee, J. Mazar, M. N. Aftab, F. Qi, J. Shelley, J. L. Li, S. Govindarajan, F. Valerio, I. Rivera, T. Thurn, T. A. Tran, D. Kameh, V. Patel and R. J. Perera, J. Mol. Diagn., 2014, 16, 615–626 CrossRef CAS PubMed
- E. M. Harcourt, T. Ehrenschwender, P. J. Batista, H. Y. Chang and E. T. Kool, J. Am. Chem. Soc., 2013, 135, 19079–19082 CrossRef CAS PubMed
- S. J. Johnson, J. S. Taylor and L. S. Beese, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3895–3900 CrossRef CAS PubMed
- T. Tian, S. Peng, H. Xiao, Y. Long, B. Fu, X. Zhang, S. Guo, S. Wang, X. Zhou, S. Liu and X. Zhou, Chem. Commun., 2013, 49, 10085–10087 RSC
- I. V. Kutyavin, Biochemistry, 2008, 47, 13666–13673 CrossRef CAS PubMed
-
(a) R. Srivastava, K. P. Gopinathan and T. Ramakrishnan, J. Bacteriol., 1981, 148, 716–719 CAS
- C. Roost, S. R. Lynch, P. J. Batista, K. Qu, H. Y. Chang and E. T. Kool, J. Am. Chem. Soc., 2015, 137, 2107–2115 CrossRef CAS PubMed
- D. Case, T. Darden, T. Cheatham III, C. Simmerling, J. Wang, R. Duke, R. Luo, R. Walker, W. Zhang and K. Merz, AMBER 12, University of California, San Francisco, 2012, vol. 1, p. 3 Search PubMed.
- J. R. Kiefer, C. Mao, J. C. Braman and L. S. Beese, Nature, 1998, 391, 304–307 CrossRef CAS PubMed
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- A. Heinrichs, Nat. Struct. Mol. Biol., 2014, 21, 117 Search PubMed
- M. F. Goodman, S. Creighton, L. B. Bloom, J. Petruska and T. A. Kunkel, Crit. Rev. Biochem. Mol. Biol., 1993, 28, 83–126 CrossRef CAS PubMed
- J. R. Masters, Nat. Rev. Cancer, 2002, 2, 315–319 CrossRef CAS PubMed
- B. E. Maden, Prog. Nucleic Acid Res. Mol. Biol., 1990, 39, 241–303 CAS
-
(a) B. E. Maden, J. Mol. Biol., 1988, 201, 289–314 CrossRef CAS PubMed
-
(a) T. Pan, Trends Biochem. Sci., 2013, 38, 204–209 CrossRef CAS PubMed
-
(a) X. L. Ping, B. F. Sun, L. Wang, W. Xiao, X. Yang, W. J. Wang, S. Adhikari, Y. Shi, Y. Lv, Y. S. Chen, X. Zhao, A. Li, Y. Yang, U. Dahal, X. M. Lou, X. Liu, J. Huang, W. P. Yuan, X. F. Zhu, T. Cheng, Y. L. Zhao, X. Wang, J. M. Rendtlew Danielsen, F. Liu and Y. G. Yang, Cell Res., 2014, 24, 177–189 CrossRef CAS PubMed
-
(a) Y. Fu, D. Dominissini, G. Rechavi and C. He, Nat. Rev. Genet., 2014, 15, 293–306 CrossRef CAS PubMed
-
(a) A. Y. Golovina, M. M. Dzama, K. S. Petriukov, T. S. Zatsepin, P. V. Sergiev, A. A. Bogdanov and O. A. Dontsova, Nucleic Acids Res., 2014, 42, e27 CrossRef CAS PubMed
-
(a) M. Yang and J. Y. Park, Methods Mol. Biol., 2012, 863, 67–109 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc02902c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |