
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimetallic iron–iron and iron–zinc complexes of the redox-active ONO pincer ligand†
Janice L.
Wong
a,
Robert F.
Higgins
b,
Indrani
Bhowmick
b,
David Xi
Cao
a,
Géza
Szigethy
a,
Joseph W.
Ziller
a,
Matthew P.
Shores
*b and
Alan F.
Heyduk
*a
aDepartment of Chemistry, University of California, Irvine, California 92697, USA. E-mail: aheyduk@uci.edu; Tel: +1-949-824-8806
bDepartment of Chemistry, Colorado State University, Fort Collins, Colorado 80523, USA. E-mail: matthew.shores@colostate.edu; Tel: +1-970-491-7235
First published on 8th December 2015
Abstract
A new bimetallic platform comprising a six-coordinate Fe(ONO)2 unit bound to an (ONO)M (M = Fe, Zn) has been discovered ((ONOcat)H3 = bis(3,5-di-tert-butyl-2-phenol)amine). Reaction of Fe(ONO)2 with either (ONOcat)Fe(py)3 or with (ONOq)FeCl2 under reducing conditions led to the formation of the bimetallic complex Fe2(ONO)3, which includes unique five- and six-coordinate iron centers. Similarly, the reaction of Fe(ONO)2 with the new synthon (ONOsq˙)Zn(py)2 led to the formation of the heterobimetallic complex FeZn(ONO)3, with a six-coordinate iron center and a five-coordinate zinc center. Both bimetallic complexes were characterized by single-crystal X-ray diffraction studies, solid-state magnetic measurements, and multiple spectroscopic techniques. The magnetic data for FeZn(ONO)3 are consistent with a ground state S = 3/2 spin system, generated from a high-spin iron(II) center that is antiferromagnetically coupled to a single (ONOsq˙)2− radical ligand. In the case of Fe2(ONO)3, the magnetic data revealed a ground state S = 7/2 spin system arising from the interactions of one high-spin iron(II) center, one high-spin iron(III) center, and two (ONOsq˙)2− radical ligands.
Introduction
The controlled, modular assembly of multi-metallic coordination complexes with novel structures, electronic and magnetic properties, and reactivities remains a challenge in synthetic inorganic chemistry. Well-tailored multidentate ligands have been used to great effect in the synthesis of small homo- and heterometallic clusters.1,2 Ligands that feature differentiated metal binding sites have been used to access heterobimetallic complexes with two different metal ions and with unexpected reactivity profiles.3,4 Metallocene complexes have been elaborated into multi-metallic complexes through the incorporation of ancillary metal binding groups into the cyclopentadienyl ligand.5,6 These efforts typically aim to exploit the redox properties of the metallocene in the resulting multi-metallic assembly. Our strategy takes a modular approach in which a discrete metal complex of a redox-active pincer ligand is used as a ligand for a second metal center to generate homo- and heterobimetallic complexes.The redox-active ONO pincer ligand, derived from bis(3,5-di-tert-butyl-2-phenol)amine ((ONOcat)H3; Chart 1), has received considerable attention for the preparation of coordination complexes with interesting electronic structures and unusual reactivities. A myriad of homoleptic, six-coordinate compounds of the formula M(ONO)2 have been reported in the literature for both 3d metal ions and a variety of main group metals.7–12 While many of these complexes display multiple stable oxidation states, their saturated coordination sphere limits their ability to perform inner-sphere reactivity.
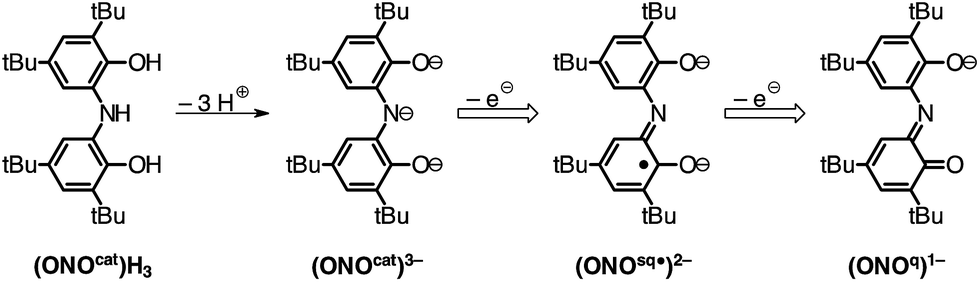 | ||
| Chart 1 Oxidation states of the ONO pincer ligand. | ||
We have discovered that the M(ONO)2 fragment itself can be used as a building block for the preparation of multimetallic complexes. The well-known complex Fe(ONO)2 can act as a chelating ligand towards a second metal center through the formation of two μ2-phenolate bridging units. New homobimetallic Fe2(ONO)3 and heterobimetallic FeZn(ONO)3 complexes have been prepared and their electronic and magnetic properties studied. The results reported here highlight both a modular method for preparing bimetallic structures and the electronic and magnetic complexity that arises from the combination of radical ligands and multiple open-shell metal ions.
Results and discussion
Synthesis and structural characterization
The bimetallic complex Fe2(ONO)3 was initially isolated in low yields as a crystalline solid upon treatment of (ONOq)FeCl2 (ref. 13) with KC8 in the absence of a strongly coordinating solvent. The bimetallic complex was identified and structurally characterized by single-crystal X-ray diffraction experiments. Purple crystals of Fe2(ONO)3, isolated by the Pasteur method, were monoclinic, falling into the space group P21/c. The structure of Fe2(ONO)3 is given as an ORTEP diagram in Fig. 1, top. The complex comprises two different iron centers. One iron, Fe(1), is five-coordinate with τ = 0.265 (eqn (1); α = ∠N(1)–Fe(1)–O(6) = 140.64(6)°; β = ∠O(1)–Fe(1)–O(2) = 156.53(6)°), indicating a geometry that is intermediate between trigonal bipyramidal (τ = 1) and square pyramidal (τ = 0).14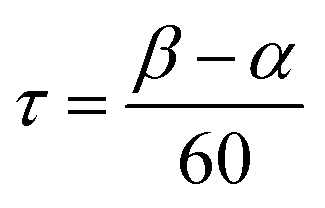 | (1) |
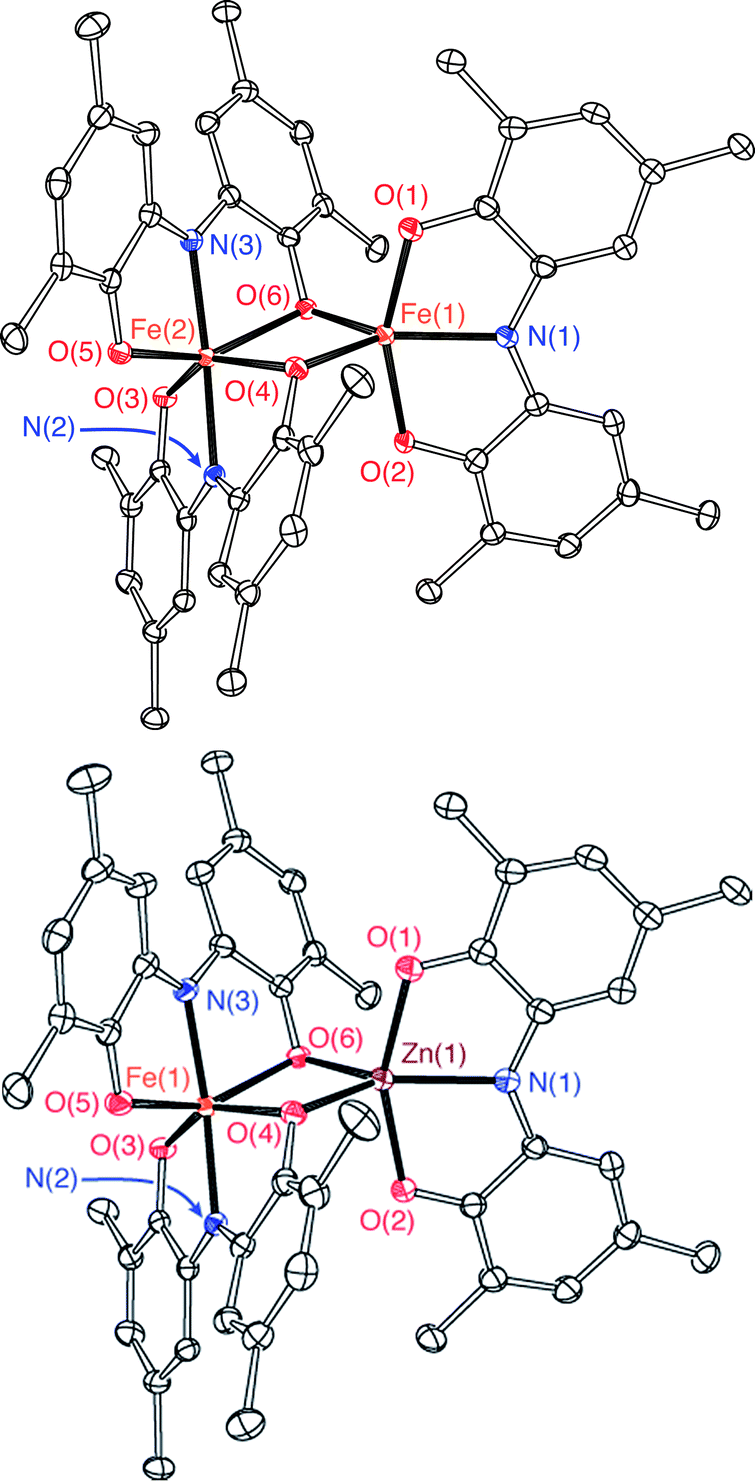 | ||
| Fig. 1 ORTEP diagrams of Fe2(ONO)3 (top) and FeZn(ONO)3 (bottom). Ellipsoids are drawn at 50% probability. Hydrogen atoms, methyl carbons of tert-butyl groups, and solvent molecules are omitted for clarity. | ||
Assignment of the metal oxidation state for the five-coordinate iron center can be made by comparing the structural metrics for Fe(1) of Fe2(ONO)3 with the previously published structures (ONOq)FeIIICl2 and (ONOcat)FeIII(py)3.13 Whereas iron–oxygen bond distances appear to be insensitive to the oxidation state of the ONO ligand, the iron–nitrogen bond distance is shorter for reduced forms of the ONO ligand. Accordingly, (ONOq)FeIIICl2 has an Fe–N distance of 2.1571(14) Å whereas (ONOcat)FeIII(py)3 has an Fe–N distance of 1.9902(18) Å. The iron–nitrogen bond distance, Fe(1)–N(1), of Fe2(ONO)3 falls between these two values at 2.0733(17) Å. Similarly, C–O and C–N bond distances within the non-bridging ONO ligand of the five-coordinate fragment are 1.316(2) and 1.374(3) Å, respectively, which fall in between the values measured for the C–O and C–N bond distances of (ONOq)FeIIICl2 (shorter bonds) and (ONOcat)FeIII(py)3 (longer bonds). These results are consistent with the (ONOsq˙)2− oxidation state for the non-bridging ONO ligand coordinated to Fe(1). Iron–oxygen bond distances to the bridging phenoxide donors are significantly shorter for the five-coordinate iron (average Fe(1)–(μ-O) = 1.976 ± 0.002 Å) than for the six-coordinate iron (average Fe(2)–(μ-O) = 2.162 ± 0.003 Å). Altogether, these data suggest that the five-coordinate iron center is best described as an [(ONOsq˙)FeIII]1+ fragment, with a six-coordinate [Fe(ONO)2]1− fragment acting as a monoanionic chelating ligand to the Fe(1) center.
Definitive assignment of the metal oxidation state for the six-coordinate iron center, Fe(2), is not possible from the structural data alone. The neutral Fe(ONO)2 monometallic unit has been characterized as an iron(III) complex containing one (ONOsq˙)2− ligand and one (ONOq)1− ligand.16 Addition of one electron to neutral Fe(ONO)2 to give the monoanion posited above (i.e. [Fe(ONO)2]1−), would then afford one of two limiting electron configurations:16 (a) assigning the added electron to the iron center would give [FeII(ONOsq˙)(ONOq)]1− or (b) assigning the added electron to the redox-active ligands would give [FeIII(ONOsq˙)2]1−. The structural data for Fe2(ONO)3 alone do not distinguish between these two possibilities. While intraligand bond distances for the two ONO ligands are the same within error, this result could stem from the symmetric [FeIII(ONOsq˙)2]1− electronic configuration or from a mixed-valence [FeII(ONOsq˙)(ONOq)]1− electronic configuration with strong delocalization (class III Robin–Day mixed valency).17,18 More insight into the electronic configuration around the six-coordinate iron center was provided by analysis of the solid-state magnetic susceptibility data and solution electronic absorption data (vide infra).
Given the novel structure and electronic properties of Fe2(ONO)3, a more direct synthetic method for accessing the complex was sought. Initial efforts were directed towards a metathesis route to prepare Fe2(ONO)3 from doubly-reduced Fe(ONO)2 and (ONOq)FeIIICl2. According to Scheme 1, Fe(ONO)2 dissolved in THF was treated with two equivalents of freshly-prepared KC8, followed by one equivalent of (ONOq)FeIIICl2. The product was isolated by extraction into toluene after removal of the THF solvent. Diffusion of acetonitrile into a toluene solution of the complex afforded the product as a dark purple microcrystalline solid in 77% yield. An alternative route to prepare Fe2(ONO)3 relies on the reaction between neutral Fe(ONO)2 and (ONOcat)FeIII(py)3. As summarized in Scheme 1, one-to-one mixtures of Fe(ONO)2 and (ONOcat)FeIII(py)3 in toluene produced the desired bimetallic complex in 88% yield. In this reaction, (ONOcat)FeIII(py)3 becomes the five-coordinate iron center after the expulsion of the pyridine ligands and the transfer of one electron to give the six-coordinate [Fe(ONO)2]1− fragment. The order of the pyridine exchange and electron transfer reactions is not known; however, previous studies indicated that pyridine dissociation from (ONOcat)FeIII(py)3 occurs readily in solution.13
 | ||
| Scheme 1 Synthesis of Fe2(ONO)3 by metathesis and redox pathways. | ||
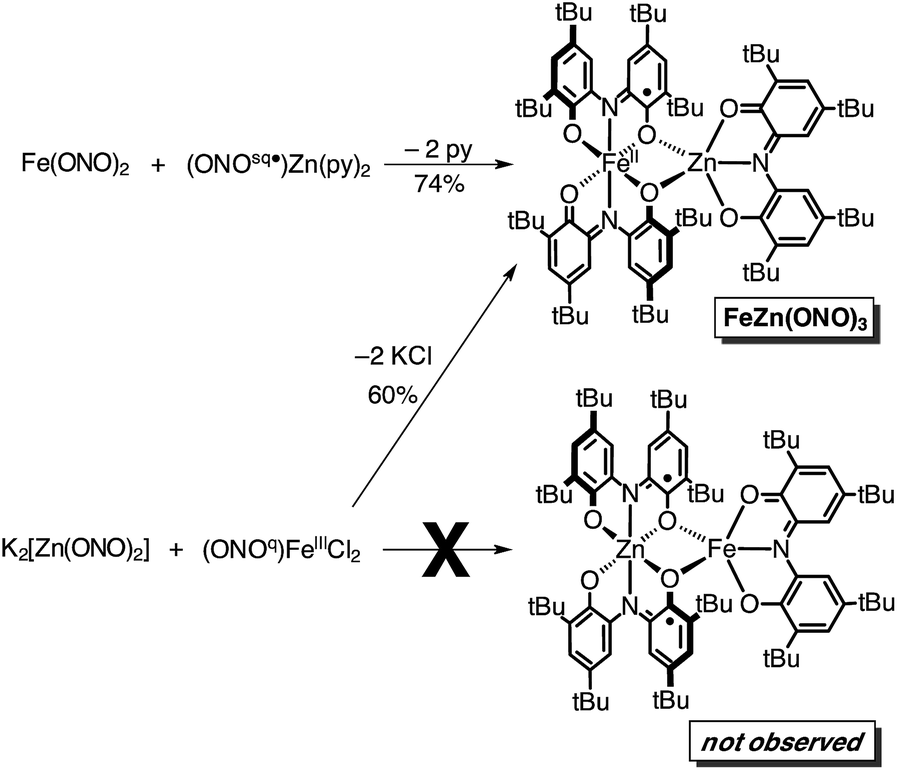
In an attempt to elaborate the bimetallic motif of Fe2(ONO)3, and in an attempt to generate a derivative with a simpler electronic structure, we targeted analogous zinc complexes. Efforts to prepare the all-zinc complex, i.e., Zn2(ONO)3, were not successful. Alternatively, the new heterobimetallic complex FeZn(ONO)3, which contains a six-coordinate iron and a five-coordinate zinc, was prepared in high yields by the reaction of (ONOsq˙)Zn(py)2 with Fe(ONO)2 in toluene as summarized in Scheme 2. The new zinc synthon, (ONOsq˙)Zn(py)2, was prepared in 71% yield from zinc acetate and one equivalent of K2(ONOsq˙) in neat pyridine (eqn (2)). Attempts to prepare the complementary heterobimetallic complex containing a six-coordinate zinc and a five-coordinate iron, either from Zn(ONO)2 and (ONOcat)FeIII(py)3 or from [Zn(ONO)2]2− and (ONOq)FeIIICl2, produced only the isomer with a six-coordinate iron and five-coordinate zinc.
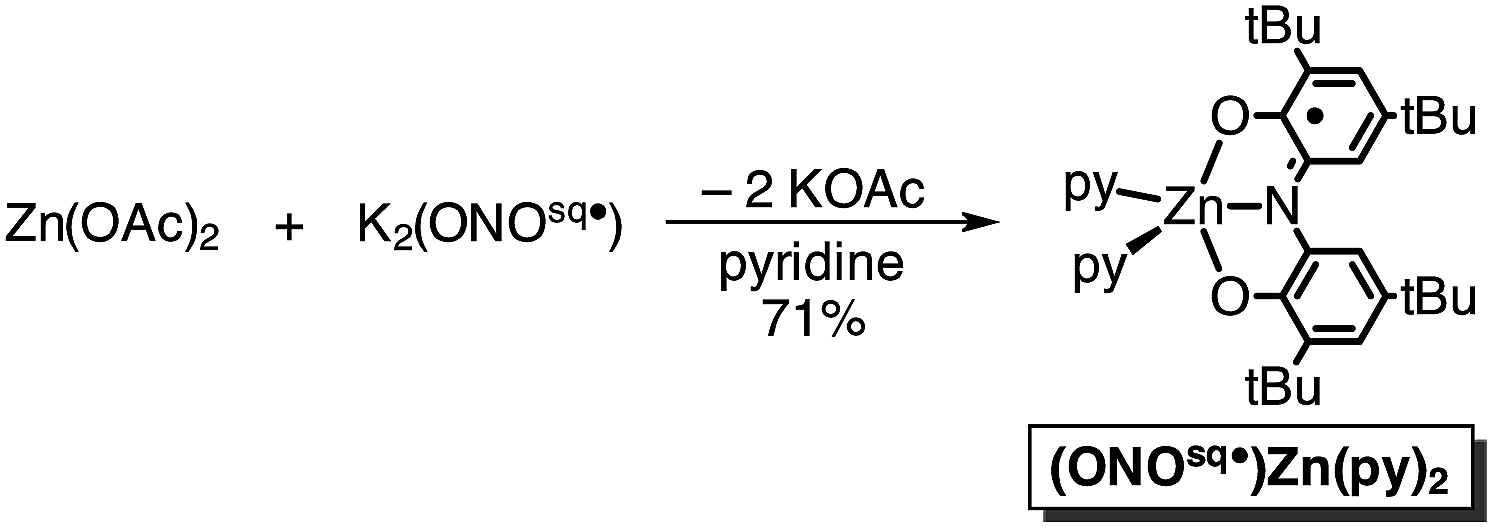 | (2) |
 | ||
| Scheme 2 Synthesis of FeZn(ONO)3. | ||
Both the monometallic (ONOsq˙)ZnII(py)2 complex and the heterobimetallic FeZn(ONO)3 complex were characterized by single-crystal X-ray diffraction studies. The structure of (ONOsq˙)ZnII(py)2 is presented as an ORTEP diagram in Fig. 2. The complex crystallizes in the P21 space group and the asymmetric unit contains two (ONOsq˙)ZnII(py)2 molecules, one pyridine molecule, and one acetonitrile molecule. Unlike the square-planar geometry determined for (ONOsq˙)ZnII(NEt3),19 the structure of (ONOsq˙)ZnII(py)2 is five-coordinate with a geometry that lies in between square pyramidal and trigonal bipyramidal.20 Intraligand C–O and C–N bond distances of 1.307(5) Å and 1.358(5) Å, respectively, are consistent with the (ONOsq˙)2− oxidation state of the redox-active ligand. The zinc–nitrogen bond distances to the pyridine ligands all fall in the 2.06–2.07 Å range. Consistent with a zinc(II) coordinated to an (ONOsq˙)2− ligand radical, the solution EPR spectrum of (ONOsq˙)ZnII(py)2 at 77 K shows a broad, isotropic signal at g = 2.00, consistent with an S = 1/2 spin system (see ESI†).
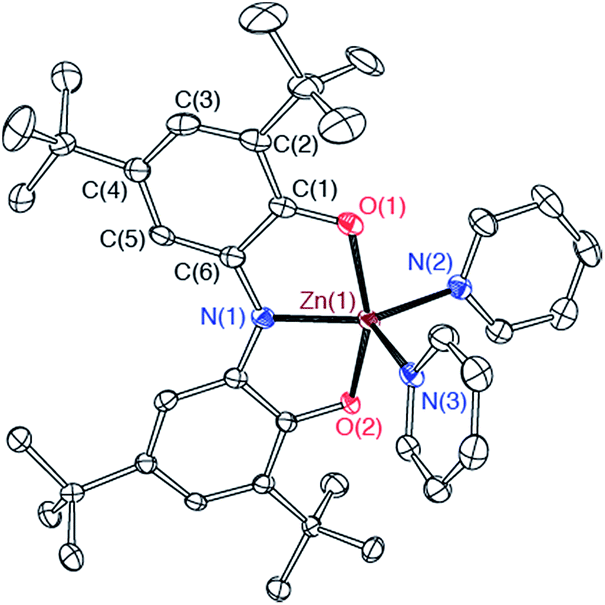 | ||
| Fig. 2 ORTEP diagram of one crystallographically unique (ONOsq˙)ZnII(py)2 molecule. Ellipsoids are drawn at 50% probability. Hydrogen atoms and pyridine and acetonitrile solvent molecules are omitted for clarity. | ||
Single-crystal X-ray diffraction experiments performed on FeZn(ONO)3 confirm a solid-state structure analogous to Fe2(ONO)3 but with zinc occupying the five-coordinate metal site. Fig. 1, bottom, shows the structure of FeZn(ONO)3 as an ORTEP diagram. The Fe⋯Zn distance of 3.05 Å is too long for a metal–metal bond (the sum of their covalent radii is 234 pm). The five coordinate zinc center again lies between trigonal bipyramidal and square pyramidal geometries with τ = 0.252.21 The (ONO) ligand of the five-coordinate zinc center has contracted C–O bond distances of 1.266(3) Å and C–N bond distances of 1.342(3) Å, indicative of a monoanionic (ONOq)1− ligand. Congruously, the C–C bond distances of this ONO ligand show strong cyclohexadiene character in the ligand backbone. For charge-balance considerations, the six-coordinate iron fragment must again act as a monoanionic chelating ligand towards the zinc center, with the same two possible oxidation states, either (a) [FeII(ONOsq˙)(ONOq)]1− or (b) [FeIII(ONOsq˙)2]1−. Again in this case, the structural data does not distinguish between these two possibilities; however, bond distances within the [Fe(ONO)2]1− fragments of Fe2(ONO)3 and FeZn(ONO)3 show a high degree of similarity.
To provide further evidence for the identities of the metal ions in the five- and six-coordinate sites of FeZn(ONO)3, the complex was decomposed in neat pyridine as summarized in eqn (3). The 1H NMR spectrum of samples of FeZn(ONO)3 heated in pyridine-d5 revealed the presence of Fe(ONO)2 while no Zn(ONO)2 was observed. Similarly, an EPR spectrum of the reaction mixture displayed the isotropic, S = 1/2 signal indicating the formation of (ONOsq˙)ZnII(py)2, whereas no resonances associated with (ONOcat)FeIII(py)3 were observed. These results are consistent with iron occupying the six-coordinate site and zinc in the five-coordinate site as proposed by the single-crystal data. For comparison, heating samples of Fe2(ONO)3 in pyridine-d5 resulted in the generation of paramagnetic 1H NMR resonances for Fe(ONO)2 and EPR resonances for (ONOcat)FeIII(py)3.
 | (3) |
Magnetism and electronic structure
In an effort to better elucidate the distribution of redox and spin states in Fe2(ONO)3 and FeZn(ONO)3, solid-state magnetic susceptibility experiments were carried out. Given the closed-shell electron configuration of zinc(II) in FeZn(ONO)3, the magnetic behavior of the heterobimetallic complex was considered first. Fig. S7† shows χMT values for FeZn(ONO)3 between 2 K and 300 K. As the temperature decreases from 300 K, χMT decreases linearly from a value of 2.52 cm3 K mol−1 to a value of 1.59 cm3 K mol−1 at 10 K, consistent with temperature independent paramagnetism (TIP). Below 10 K, χMT drops abruptly to a value of 1.19 cm3 K mol−1 at 2 K, suggestive of magnetic anisotropy and/or antiferromagnetic coupling.Given that the zinc and non-bridging ONO ligand can be treated as a closed-shell [ZnII(ONOq)]1+ fragment, two spin models were considered for modeling the magnetic susceptibility of FeZn(ONO)3. The first model (a) contained a high-spin FeII ion, a single (ONOsq˙)2− ligand, and a single (ONOq)1− ligand. The second model (b) contained a high-spin FeIII ion and two (ONOsq˙)2− ligands. In both scenarios, strong antiferromagnetic coupling between unpaired electrons on the iron and unpaired electrons on the ONO ligand(s) would generate an S = 3/2 total spin ground state and a theoretical χMT value of 1.875 cm3 K mol−1 with g = 2, consistent with the experimental value. The S = 3/2 system was modeled using julX22 to give the fit shown in Fig. 3 based on the following parameters: g = 1.84, axial anisotropy with D = −2.96 cm−1, TIP = 2.91 × 10−3 cm3 mol−1 (subtracted), and intermolecular coupling with Θ = −0.058 cm−1. A g value less than 2.0 is consistent with previous reports on the magnetic properties of Fe(ONO)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8,16 and consistent with the 4 K EPR spectrum of FeZn(ONO)3 shown in Fig. S3.† The TIP value is large but reasonable considering the accessibility of other electronic isomers and the proximity of spin excited states with larger S values.23,24 Further supporting this model, the magnetization trends toward saturation between 2–3 μB as the field is increased to 5 T at 1.8 K (Fig. S8†), consistent with S = 3/2 where g < 2 and significant axial anisotropy is observed. The PHI program25 was used to estimate the extent of antiferromagnetic coupling between the unpaired electrons on the iron and (ONOsq˙)2− ligand(s) for the two spin models. As shown in Fig. S11,† the most reasonable parameters were found for model (a) that includes a high-spin FeII ion and only one (ONOsq˙)2− ligand (J ≅ −260 cm−1). Notably model (b), which includes a high-spin FeIII and two (ONOsq˙)2− ligands, did not give realistic exchange coupling parameters (see ESI†).
8,16 and consistent with the 4 K EPR spectrum of FeZn(ONO)3 shown in Fig. S3.† The TIP value is large but reasonable considering the accessibility of other electronic isomers and the proximity of spin excited states with larger S values.23,24 Further supporting this model, the magnetization trends toward saturation between 2–3 μB as the field is increased to 5 T at 1.8 K (Fig. S8†), consistent with S = 3/2 where g < 2 and significant axial anisotropy is observed. The PHI program25 was used to estimate the extent of antiferromagnetic coupling between the unpaired electrons on the iron and (ONOsq˙)2− ligand(s) for the two spin models. As shown in Fig. S11,† the most reasonable parameters were found for model (a) that includes a high-spin FeII ion and only one (ONOsq˙)2− ligand (J ≅ −260 cm−1). Notably model (b), which includes a high-spin FeIII and two (ONOsq˙)2− ligands, did not give realistic exchange coupling parameters (see ESI†).
 | ||
| Fig. 3 Temperature dependence of the dc magnetic susceptibility data for FeZn(ONO)3 (blue squares) and Fe2(ONO)3 (red circles) collected at an applied field of 1000 Oe; data have been corrected for TIP; the lines represent best fits to the models depicted in the inset schemes (julX).22 | ||
Whereas the magnetic susceptibility data alone cannot definitively differentiate between the “high-spin FeII + one (ONOsq˙)2−” and “high-spin FeIII + two (ONOsq˙)2−” electronic isomers, comparison of the electronic absorption spectra for FeZn(ONO)3 and Fe(ONO)2 lends support for the former arrangement. Fig. S5† shows the UV-vis-NIR absorption spectra of FeZn(ONO)3 and Fe(ONO)2. Both spectra show similar molar absorptivities for a band near 400 nm, which has been attributed to a semiquinonate-based transition.26 Since Fe(ONO)2 has been characterized as having one (ONOsq˙)2− ligand, it follows that FeZn(ONO)3 has only one (ONOsq˙)2− ligand. From the combined structural, magnetic, and electronic absorption data, we conclude that the FeZn(ONO)3 complex contains a high-spin ferrous ion and one bridging semiquinonate ligand. Thus, the ground state electron distribution in FeZn(ONO)3 can be assigned as FeII(ONOsq˙)(ONOq)ZnII(ONOq), which is represented as a Lewis structure in Scheme 2.
For the diiron complex Fe2(ONO)3, the replacement of the diamagnetic (ONOq)ZnII fragment with a paramagnetic (ONOsq˙)FeIII fragment significantly changes the magnetic properties of the complex. The magnetization dependence on reduced field for Fe2(ONO)3, shown in Fig. 4, shows non-superimposable isofield lines and saturation of the magnetization at 5.48 μB in a 5 T magnetic field. Together, these data support an S = 7/2 ground state with significant axial magnetic anisotropy and g < 2. Attempts to fit the magnetization vs. applied magnetic field data to various spin state and anisotropy models using ANISOFIT27 are summarized in the ESI:† the best fit has the parameters S = 7/2, g = 1.95, D = 2.40 cm−1 and |E| = 0.00177 cm−1. Again, the low g value is consistent with the EPR spectrum of Fe2(ONO)3 collected at 4 K (Fig. S4†), which shows signals at g = 2.02, 1.96 and 1.86.
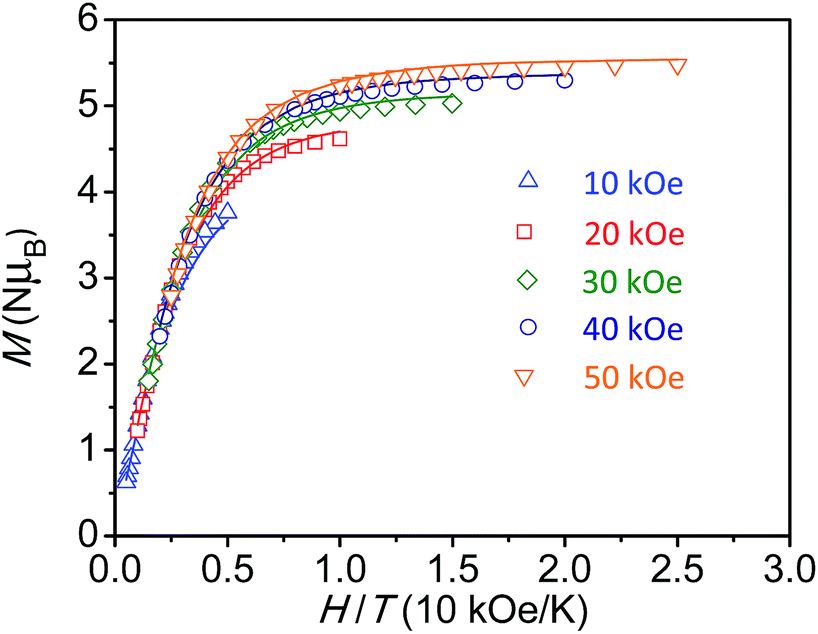 | ||
| Fig. 4 Reduced field dependence of magnetization for Fe2(ONO)3; lines represent best fits of the data to an S = 7/2 ground state model, as determined by ANISOFIT.27 | ||
The magnetic susceptibility data for Fe2(ONO)3 support an electronic configuration that is best described as FeII(ONOsq˙)(ONOq)FeIII(ONOsq˙). This configuration is represented as a Lewis structure in Scheme 1, and it preserves the same [FeII(ONOsq˙)(ONOq)]− six-coordinate unit that was postulated above for FeZn(ONO)3. Consistent with two different iron centers, the Mössbauer spectrum of Fe2(ONO)3 (Fig. S6†) shows two overlapping doublet signals.28 Fig. S12† shows the temperature-dependent magnetic susceptibility data for Fe2(ONO)3 at an applied field of 1000 Oe. At ambient temperature, χMT has a value of 7.00 cm3 K mol−1, which decreases to 5.76 cm3 K mol−1 at 100 K, then begins an upturn, achieving a maximum value of 6.25 cm3 K mol−1 at 10 K. Below 10 K, χMT decreases rapidly to 5.69 cm3 K mol−1 at 2.5 K. Attempts to model this data using unconstrained interactions among four spin centers (two iron centers and two (ONOsq˙)2− ligands) failed to fit the data in a reasonable way. Instead, a simpler model was developed that explicitly conserves the six-coordinate, S = 3/2, FeII(ONOsq˙)(ONOq) fragment from the FeZn(ONO)3 complex and combines it with a five-coordinate, S = 2, (ONOsq˙)FeIII fragment suggested by the single-crystal structural data. The resulting fit to the χMT data for Fe2(ONO)3 is shown in Fig. 3 and provided the following parameters: g3/2 = 1.84, g2 = 2.10, D = 5.15 cm−1, |E| = 0.04 cm−1, J = +1.57 cm−1, and TIP = 2.59 × 10−3 cm3 mol−1 (subtracted). Further support for this model can be found in the EPR and electronic absorption spectra. In the UV-vis spectrum, we note that the diiron complex exhibits molar absorptivity values nearly twice as large as FeZn(ONO)3 near 400 nm (Fig. S5†), consistent with the presence of two ligand radicals that are not strongly coupled to one another. Other models considered (see ESI†) did not fit with the combined structural, spectroscopic and magnetic data.
Conclusions
Formal reduction of Fe(ONO)2 affords a “ligand” with an electronic structure that is best described as [FeII(ONOsq˙)(ONOq)]−, which can act as a monoanionic chelating ligand, allowing the construction of bimetallic complexes with interesting redox and magnetic properties. The [FeII(ONOsq˙)(ONOq)]− unit coordinates to a second metal center through the formation of two μ2-phenoxide oxygen atoms. For the iron–zinc complex, FeZn(ONO)3, the complex is best described as FeII(ONOsq˙)(ONOq)ZnII(ONOq) with an S = 3/2 ground state. In this bimetallic complex, the juxtaposition of (ONOq)− and (ONOsq˙)2− ligands on the six-coordinate iron center sets up an example of ligand-based mixed-valency. In the case of the diiron complex, Fe2(ONO)3, the complex is best described as FeII(ONOsq˙)(ONOq)FeIII(ONOsq˙) with an S = 7/2 ground state. The diiron complex is unusual in that it is an example of a double mixed-valence complex: in addition to the same ligand-based mixed valency, the juxtaposition of iron(II) and iron(III) centers with different coordination geometries affords metal-based mixed-valence character.17 The modular nature of these molecules suggests a new strategy for the synthesis of multi-metallic systems with intriguing redox and magnetic properties.Acknowledgements
This work was supported by the National Science Foundation (CHE-1152543 to AFH and CHE-1363274 to MPS) and by Colorado State University. The authors thank Raúl Hernández Sánchez for collection of Mössbauer data and Jason Jones for assistance with EPR data collection.Notes and references
- (a) A. R. Fout, Q. Zhao, D. J. Xiao and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 16750–16753 CrossRef CAS PubMed; (b) T. M. Powers, N. X. Gu, A. R. Fout, A. M. Baldwin, R. Hernández Sánchez, D. M. Alfonso, Y.-S. Chen, S.-L. Zheng and T. A. Betley, J. Am. Chem. Soc., 2013, 135, 14448–14458 CrossRef CAS PubMed.
- (a) J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed; (b) J. S. Kanady, P. H. Lin, K. M. Carsch, R. J. Nielsen, M. K. Takase, W. A. Goddard and T. Agapie, J. Am. Chem. Soc., 2014, 136, 14373–14376 CrossRef CAS PubMed.
- (a) B. P. Greenwood, G. T. Rowe, C. H. Chen, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2009, 132, 44–45 CrossRef PubMed; (b) J. P. Krogman, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2011, 133, 14582–14585 CrossRef CAS PubMed.
- (a) S. J. Tereniak, R. K. Carlson, L. J. Clouston, V. G. Young, E. Bill, R. Maurice, Y. S. Chen, H. J. Kim, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2013, 136, 1842–1855 CrossRef PubMed; (b) L. J. Clouston, R. B. Siedschlag, P. A. Rudd, N. Planas, S. Hu, A. D. Miller, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2013, 135, 13142–13148 CrossRef CAS PubMed.
- (a) P. L. Diaconescu, Acc. Chem. Res., 2010, 43, 1352–1363 CrossRef CAS PubMed; (b) X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264–11267 CrossRef CAS PubMed.
- A. Shafir and J. Arnold, Organometallics, 2003, 22, 567–575 CrossRef CAS.
- A. Y. Girgis and A. L. Balch, Inorg. Chem., 1975, 14, 2724 CrossRef CAS.
- C. G. Pierpont, S. K. Larsen and S. R. Boone, Pure Appl. Chem., 1988, 60, 1331 CrossRef CAS.
- L. G. Ranis, K. Werellapatha, N. J. Pietrini, B. A. Bunker and S. N. Brown, Inorg. Chem., 2014, 53, 10203–10216 CrossRef CAS PubMed.
- S. Bruni, A. Caneschi, F. Cariati, C. Delfs, A. Dei and D. Gatteschi, J. Am. Chem. Soc., 1994, 116, 1388–1394 CrossRef CAS.
- C. Camacho-Camacho, G. Merino, F. J. Martinez-Martinez, H. Noth and R. Contreras, Eur. J. Inorg. Chem., 1999, 1021–1027 CrossRef CAS.
- S. Shekar and S. N. Brown, Organometallics, 2013, 32, 556–564 CrossRef CAS.
- J. L. Wong, R. H. Sánchez, J. G. Logan, R. A. Zarkesh, J. W. Ziller and A. F. Heyduk, Chem. Sci., 2013, 4, 1906–1910 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. V. Rijn and G. C. Verschoor, Dalton Trans., 1984, 1349–1356 RSC.
- F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple bonds between metal atoms, Springer Science, New York, NY, 2005 Search PubMed.
- C. L. Simpson, S. R. Boone and C. G. Pierpont, Inorg. Chem., 1989, 28, 4379–4385 CrossRef CAS.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247–422 CrossRef CAS.
- Non-ordered orientation of the Fe2(ONO)3 molecule in the crystal would also result in similar intraligand bond lengths for the ONO ligands.
- P. Chaudhuri, M. Hess, K. Hildenbrand, E. Bill, T. Weyhermueller and K. Wieghardt, Inorg. Chem., 1999, 38, 2781–2790 CrossRef CAS PubMed.
- The asymmetric unit contains two crystallographically unique molecules. For one molecule τ = 0.305 (α = ∠N(1)–Zn(1)–N(2) = 139.3(1)°; β = ∠O(1)–Zn(1)–O(2) = 157.6(1)°) and for the second molecule τ = 0.183 (α = ∠N(1)–Zn(1)–N(2) = 146.9(1)°; β = ∠O(1)–Zn(1)–O(2) = 158.3(1)°).
- α = ∠N(1)–Zn(1)–O(2) = 140.57(7)°; β = ∠O(1)–Zn(1)–O(2) = 155.71(6)°.
- E. Bill, julX (version 1.41), Max Planck Institute for Bioinorganic Chemistry, Mulheim an der Ruhr, Germany, 2008 Search PubMed.
- A. Bakac, Physical Inorganic Chemistry: Reactions, Processes, and Applications, John Wiley & Sons Inc., Hoboken, New Jersey, 2010, pp. 74–75 Search PubMed.
- A. S. Attia, B. J. Conklin, C. W. Lange and C. G. Pierpont, Inorg. Chem., 1996, 35, 1033 CrossRef CAS PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164 CrossRef CAS PubMed.
- E. Evangelio, M.-L. Bonnet, M. Cabanas, M. Nakano, J.-P. Sutter, A. Dei, V. Robert and D. Ruiz-Molina, Chem.–Eur. J., 2010, 16, 6666 CrossRef CAS PubMed.
- M. P. Shores, J. J. Sokol and J. R. Long, J. Am. Chem. Soc., 2002, 124, 2279 CrossRef CAS PubMed.
- δ(1) = 0.59 mm s−1 and ΔEQ(1) = 1.60 mm s−1 and δ(2) = 0.49 mm s−1 and ΔEQ(2) = 1.81 mm s−1. Similar Mössbauer parameters have been reported for monomeric iron complexes of the ONO pincer ligand. See ref. 13 and 16.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental procedures and magnetic measurements and models. CCDC 1417565–1417567. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03006d |
| This journal is © The Royal Society of Chemistry 2016 |