
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium-catalyzed regioselective addition of the ortho C–H bond in aromatic amides to the C–C double bond in α,β-unsaturated γ-lactones and dihydrofurans†
Kaname
Shibata
and
Naoto
Chatani
*
Department of Applied Chemistry, Faculty of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: chatani@chem.eng.osaka-u.ac.jp
First published on 25th September 2015
Abstract
An unprecedented C–H alkylation using α,β-unsaturated γ-lactones (butenolides) and dihydrofurans was achieved by the Rh-catalyzed reaction of benzamides. C–C bond formation occurs between the ortho-position of the benzamide derivative and the γ-position of the butenolide or the α-position of the dihydrofuran. The presence of an 8-aminoquinoline directing group is crucial for the success of the reaction. The results of deuterium labeling experiments indicate that the cleavage of the C–H bond is reversible and suggest that a migratory carbene insertion is involved as the key step.
Introduction
During the past several decades, the chelation-assisted functionalization of C–H bonds has undergone rapid development and is now used in the synthesis of a wide variety of natural products and medicinally relevant compounds.1,2 While a wide variety of such functionalizations have been developed to date, the addition of C–H bonds to alkenes represents one of the most fundamental functionalizations of C–H bonds because of its atom economy.3,4 While various alkenes can be used as coupling partners, the range of alkenes that are applicable to C–H alkylation remains limited to vinylsilanes and tert-butylethylene, which contain no allylic hydrogen, and activated alkenes, such as styrenes and α,β-unsaturated carbonyl compounds. Methodology for using various alkenes as participants in C–H alkylation is still needed. Not only the applicability of alkenes, but also the control of regioselectivity in the addition of C–H bonds to alkenes remains to be solved. Acrylic esters exclusively undergo β-addition reactions to give linear products. In reactions with styrenes, the extent of regioselectivity depends on the catalytic system being used.5 In many cases, terminal alkenes containing an allylic hydrogen are not applicable in the reaction, however, Chang and Ackermann recently reported Rh- and Ru-catalyzed alkylation of C–H bonds with various terminal alkenes, in which β-addition takes place with linear products being produced.6 We wish to report herein the first example of C–H alkylation of α,β-unsaturated γ-lactones and dihydrofurans, in which C–C bond formation occurs between the ortho-position of an aromatic amide and the γ-position of the butenolide or the α-position of the dihydrofuran, irrespective of the position of the C–C double bond, to give 5-aryl-γ-butyrolactone or 2-aryltetrahydrofuran derivatives, respectively (Scheme 1). These types of products cannot be produced by the alkylation of C–H bonds with alkyl halides. 5-Aryl-γ-butyrolactone derivatives7 and 2-aryltetrahydrofuran derivatives8 are key structural components of many biologically active and pharmaceutically important molecules, making the construction of such structures synthetically important. Our strategy for constructing 5-aryl-γ-butyrolactone and 2-aryltetrahydrofuran frameworks involves the catalytic activation of a C–H bond in a benzene ring, which then adds to the C–C double bond in a butenolide or dihydrofuran derivative. In addition, the directing group can be easily removed or elaborated to another useful functionality.9 | ||
| Scheme 1 The addition of C–H bonds across alkenes. | ||
Results and discussion
When the butenolide 2a was used as the coupling partner in the reaction of 1a, the expected product 4aa resulting from the reaction at the β-position of the lactone was not obtained, but 3aa was instead produced as the sole product (Scheme 2). After screening a number of bases, it was found that the reaction is sensitive to the nature of the base used. In general, the use of Na2CO3 or K2HPO4 gave the best results.10 To the best of our knowledge, this is the first example of C–H bond alkylation with α,β-unsaturated lactones. | ||
| Scheme 2 Rh-catalyzed C–H alkylation of 1a with butenolide 2a. | ||
We next examined the effect of the directing group.11 Among the directing groups examined, only 8-aminoquinoline gave the corresponding alkylation product. Most importantly, 2-phenylpyridine, which is an extensively used pyridine directing group in catalytic C–H functionalization reactions, was ineffective. Our research has recently focused on the utilization of an N,N′-bidentate directing chelation system in the functionalization of C–H bonds.12,13 This result also shows the potential of such a system for exploring new types of functionalizations of C–H bonds.
Table 1 shows some representative results for the reaction of various aromatic amides with butenolide 2a. In the case of meta-substituted aromatic amides, contrary to the case when methyl acrylate was used in a similar chelation system,13a the alkylation proceeded selectively only at the less hindered position, as in 3fa–3ja, because the introduced lactone moiety is a sterically demanding group compared with the acrylate moiety. In the case of the benzamide 1o, both ortho C–H bonds underwent alkylation to give the double alkylation product 3oa in 61% isolated yield.
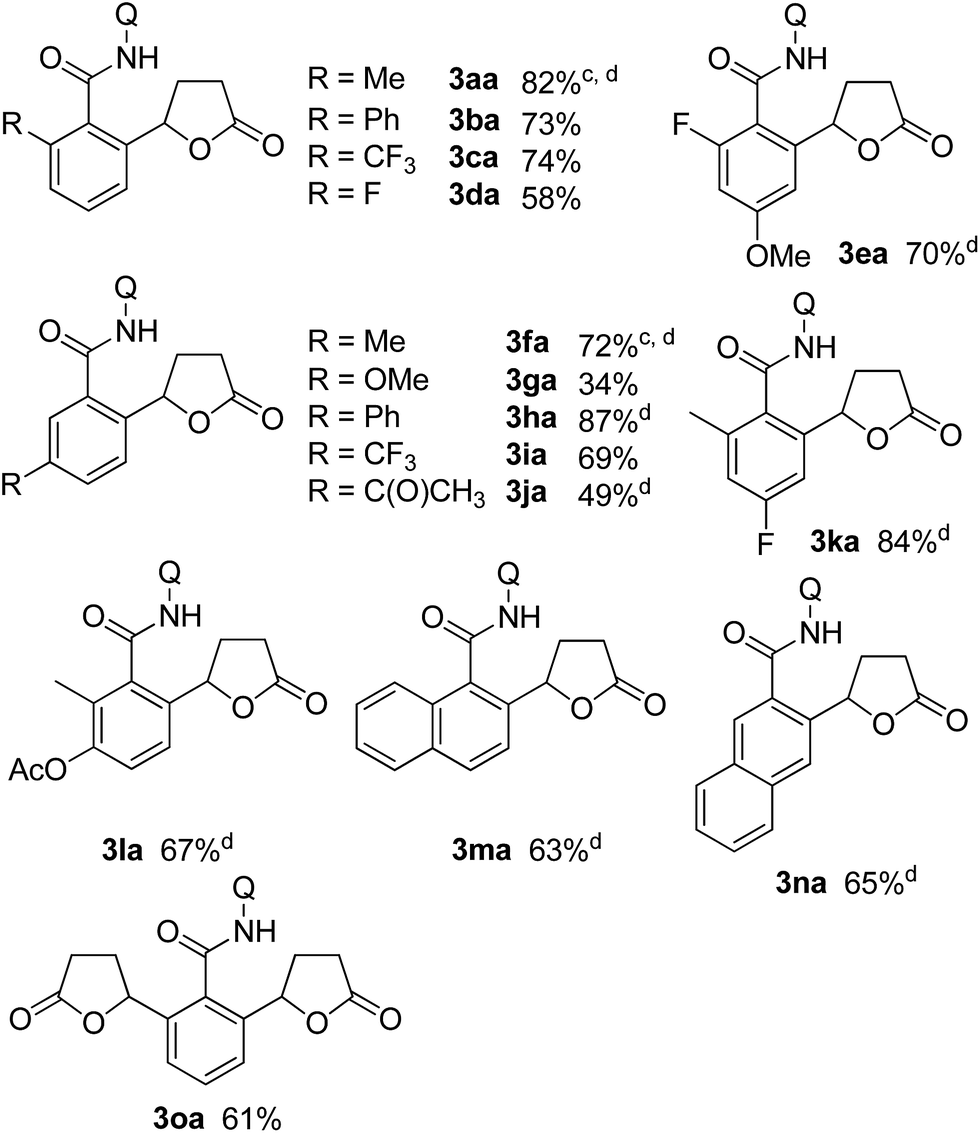
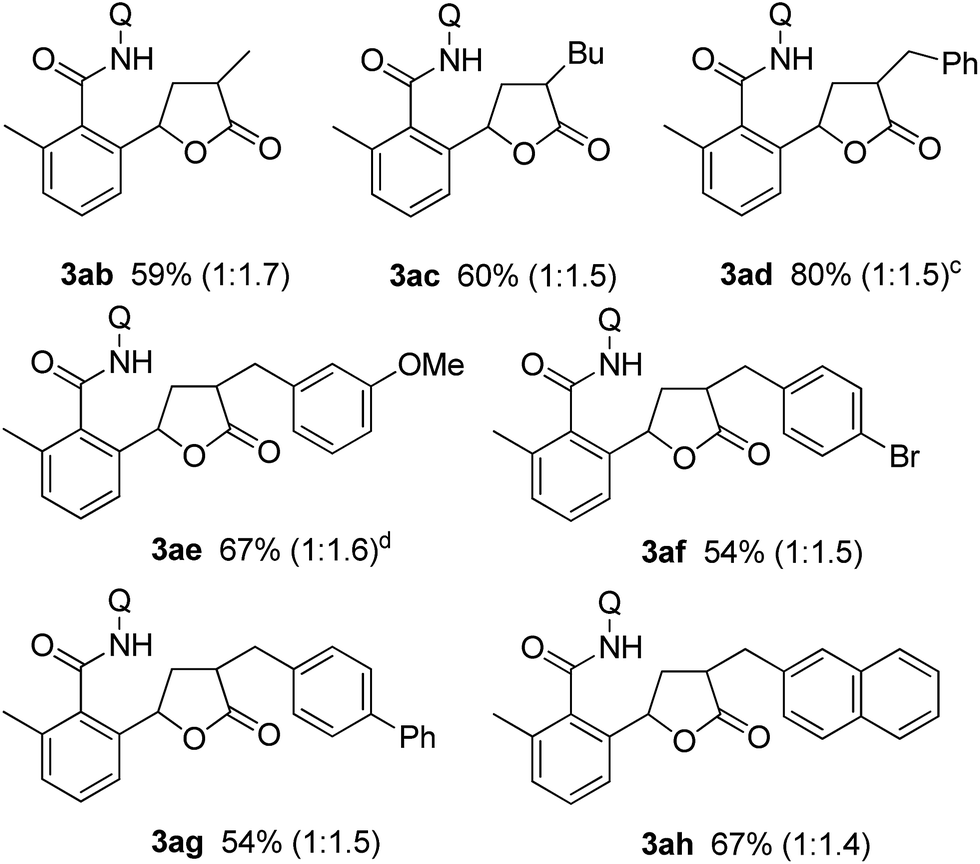
Substituted α,β-unsaturated γ-lactones 2 were also applicable to the present reaction (Table 2). Me, Bu, and benzyl substituted lactones could all be used, as in 3ab, 3ac, and 3ad. In all cases, a mixture of cis and trans isomers was produced in a comparable ratio. The ratio was constant even when the reaction was stopped after a short reaction time, suggesting that the ratio obtained is a thermodynamic ratio. Lactones bearing functional groups on the phenyl ring, such as Br and MeO, were tolerated under the reaction conditions.
| a Reaction conditions: amide 1 (0.3 mmol), lactone 2a (0.9 mmol), [RhCl(cod)]2 (0.015 mmol), K2HPO4 (0.075 mmol), toluene (1 mL), at 160 °C for 24 h. b Isolated yields. The number in parentheses denotes the ratio of cis and trans isomers. c The reaction was run for 12 h. d KOAc was used in place of K2HPO4. |
|---|
|
The 2,3-dihydrofuran 5a and the 2,5-dihydrofuran 5b also participated in the alkylation reaction (Table 3). When the reaction was carried out under two different sets of conditions, i.e. methods A and B, marginal effects were observed. Irrespective of the position of the olefin as in 5a and 5b, C–C bond formation took place at the α-position (next to the oxygen atom) of the dihydrofuran.14
| a Reaction conditions (method A): amide 1a (0.3 mmol), dihydrofuran (0.6 mmol), [RhCl(cod)]2 (0.0075 mmol), KOAc (0.075 mmol), toluene (1 mL), at 160 °C for 12 h. b Reaction conditions (method B): amide 1a (0.3 mmol), dihydrofuran (0.6 mmol), [Rh(OAc)(cod)]2 (0.0075 mmol), PivOH (0.3 mmol), toluene (1 mL), at 160 °C for 12 h. c Isolated yields. The number in parentheses denotes the ratio of cis and trans isomers. d NMR yield. |
|---|

|
The results for the reaction of meta-substituted benzamides with 5a are shown in Table 4. The reaction was carried out under the reaction conditions of method B. Irrespective of the electronic nature of the substituent, only the less hindered C–H bonds reacted.

To gain insights into the reaction mechanism, deuterium labeling experiments were carried out in the absence of alkene (Scheme 3a). Irrespective of the reaction conditions (method A: KOAc and method B: PivOH), a significant amount of H/D exchange occurred even within a short reaction time (15 min), and the exchange occurred only at the ortho position (Scheme 3a), indicating that the cleavage of the C–H bond is reversible and does not appear to be the rate-determining step. The reaction of 1a in toluene-d8 was carried out. No deuterium atom was incorporated into the product, indicating that in the H/D exchange reaction, the proton source is not the solvent, but rather, that the proton comes from the NH bond in the substrate (scheme not shown). When the reaction was carried out in the presence of dihydrofuran 5a (Scheme 3b), alkylation product 6aa-d was obtained in 7% and 32% NMR yield, depending on the additive used, along with recovery of the starting material 1a-d7. Curiously and unexpectedly, a deuterium atom was incorporated into the THF ring only at the α-position and no deuterium atoms were detected at any of the other positions in the THF ring by 1H NMR. Both the ortho-carbon and hydrogen atom in the benzamide attach to the α-carbon of the THF ring. A similar result was obtained even when 2,5-dihydrofuran (5b) was used as the coupling partner (Scheme 3c). To exclude the possibility that H/D exchange occurs at the α-position of the THF ring under the reaction conditions employed, 6aa was reacted with CD3COOD at 160 °C in the presence of a rhodium complex. However, no deuterium was introduced into 6aa (scheme not shown). These results suggest that the ortho C–H bond appears to undergo a migratory carbene insertion, as discussed later.
 | ||
| Scheme 3 Deuterium labeling experiments. | ||
Based on our previously reported studies13a,b and the results obtained in the present work, a proposed mechanism for the reaction is shown in Scheme 4. The coordination of the quinoline nitrogen in the aromatic amide 1 to give a Rh(I)X species, followed by the oxidative addition of an N–H bond, gives complex A.15 The insertion of 5a into the H–Rh bond in A gives complex B, which, after the elimination of HX, affords the carbene complex C.16 The migratory insertion of the ortho C–H bond to a carbene moiety in the complex C through the oxidative addition of the ortho C–H bond followed by α-hydride migration gives D,17,18 which undergoes reductive elimination followed by protonation to give the final product with regeneration of the Rh(I) species. An alternative mechanism for generating the complex A involves the coordination of a quinoline nitrogen to the Rh(I) center, a ligand exchange to generate the Rh(I) complex E with the concomitant generation of HX, followed by the reaction of the complex E with HX. The ortho C–H bond of complex E then undergoes a reversible oxidative addition to the rhodium center to form the cyclometalated Rh–H complex F, the formation of which accounts for the reversibility of the cleavage of the C–H bond at the ortho position of the benzamide.19 As shown in Scheme 3b, deuterium was only incorporated at the α-position of the THF ring of the product. The proposed mechanism involving the formation of the intermediate carbene complex C is consistent with the deuterium labeling data shown in Scheme 3b, although no direct experimental evidence for this exisst. To better understand the details of the reaction mechanism, more experiments including DFT calculations will be needed.
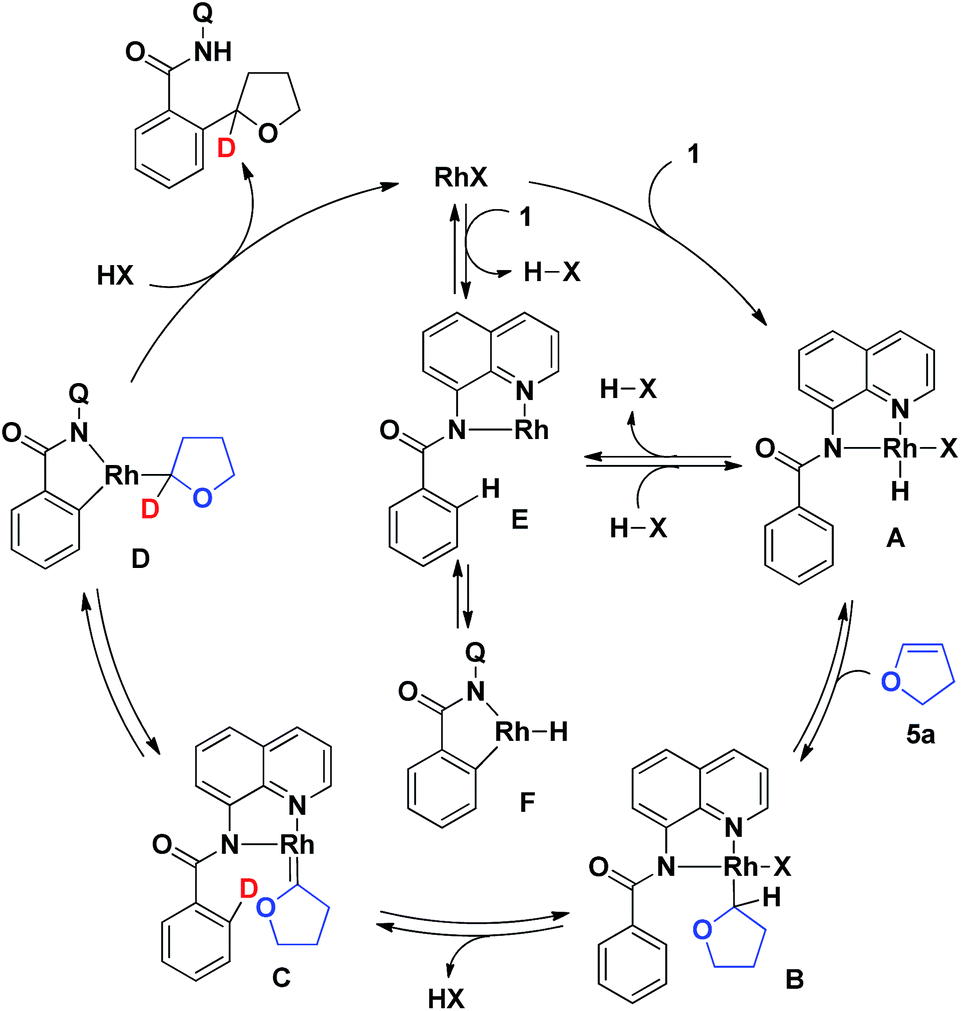 | ||
| Scheme 4 Proposed mechanism. | ||
Scheme 5 shows the potential synthetic utility of the C–H bond alkylation reaction. The treatment of 3ha under acidic conditions gave the isobenzofuran-1(3H)-one derivative 7 in 85% isolated yield. The present protocol was also applicable to the preparation of highly substituted and/or functionalized 5-aryl-γ-butyrolactone derivatives. When the lactone 1f was used in the reaction, the mono-alkylated product 3fa was exclusively formed, as shown in Table 1. The remaining hindered ortho-C–H bond in 3fa could be successfully alkylated with methyl acrylate in the presence of a rhodium catalyst13a to give compound 8, which contains three different adjacent carbonyl functional groups on the benzene ring.
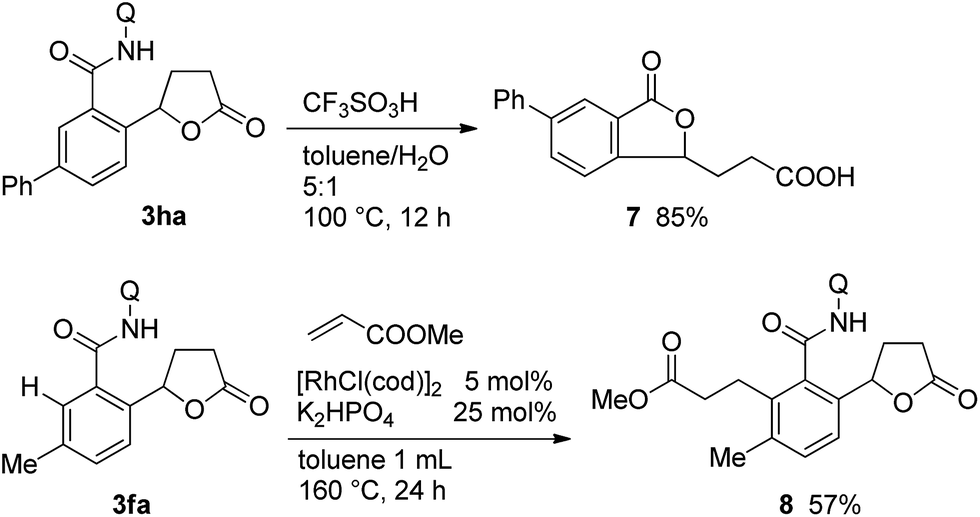 | ||
| Scheme 5 Synthetic applications. | ||
Conclusion
In summary, this reaction represents the first example of C–H alkylation with butenolides, in which C–C bond formation occurs between the ortho-position of an aromatic amide and the γ-position of a butenolide derivative. In addition, dihydrofurans can also be used in the alkylation reaction, in which case, C–C bond formation occurs between the ortho-position of the aromatic amide and the α-position of the dihydrofuran, irrespective of the position of the C–C double bond. The use of an 8-aminoquinoline moiety as a directing group is crucial for the success of the reaction. In fact, the reaction with 2-phenylpyridine, which has been used extensively as a substrate for a wide variety of functionalizations of C–H bonds, did not succeed. The functionalization of C–H bonds using an N,N′-bidentate directing group began to appear in the literature only in the last ten years, since Daugulis reported the Pd(II)-catalyzed arylation of C–H bonds in aliphatic amides in 2005.20 Since then, it has been shown that various transition metal complexes can be used in the N,N′-bidentate chelation system.12 As more mechanistic information emerges, new and more exciting advances can be anticipated.21Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Molecular Activation Directed toward Straightforward Synthesis” from the Ministry of Education, Culture, Sports, Science and Technology, and by JST Strategic Basic Research Programs “Advanced Catalytic Transformation Program for Carbon Utilization (ACT-C)” from Japan Science and Technology Agency. K. S. expresses his special thanks for a JSPS Research Fellowship for Young Scientists. We also thank the Instrumental Analysis Center, Faculty of Engineering, Osaka University, for assistance with the MS and HRMS.Notes and references
- For recent reviews on chelation-assisted functionalization of C–H bonds, see: (a) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (b) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef CAS PubMed; (c) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (d) F. Mo, J. R. Tabor and G. Don, Chem. Lett., 2014, 43, 264 CrossRef CAS; (e) M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843 RSC; (f) N. Kuhl, N. Schröder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443 CrossRef CAS; (g) S. D. Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461 CrossRef; (h) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC; (i) G. Qiu and J. Wu, Org. Chem. Front., 2015, 2, 169 RSC.
- (a) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC; (b) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (c) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (d) D. Y.-K. Chen and S. W. Youn, Chem.–Eur. J., 2012, 18, 9452 CrossRef CAS PubMed; (e) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (f) Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66 CrossRef CAS PubMed.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS.
- (a) F. Kakiuchi and S. Murai, Acc. Chem. Res., 2002, 35, 826 CrossRef CAS PubMed; (b) J. R. Andreatta, B. A. McKeown and T. B. Gunnoe, J. Organomet. Chem., 2011, 696, 305 CrossRef CAS; (c) S. Pan and T. Shibata, ACS Catal., 2013, 3, 704 CrossRef CAS; (d) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed; (e) F. Kakiuchi, T. Kochi and S. Murai, Synlett, 2014, 25, 2390 CrossRef CAS.
- For selected recent examples, see: (a) K. Gao and N. Yoshikai, J. Am. Chem. Soc., 2011, 133, 400 CrossRef CAS PubMed; (b) W.-C. Shih, W.-C. Chen, Y.-C. Lai, M.-S. Yu, J.-J. Ho, G. P. A. Yap and T.-G. Ong, Org. Lett., 2012, 14, 2046 CrossRef CAS PubMed; (c) S. Pan, N. Ryu and T. Shibata, J. Am. Chem. Soc., 2012, 134, 17474 CrossRef CAS PubMed; (d) G. E. M. Crisenza, N. G. McCreanor and J. F. Bower, J. Am. Chem. Soc., 2014, 136, 10258 CrossRef CAS PubMed.
- (a) J. KwakJ, Y. Ohk, Y. Jung and S. Chang, J. Am. Chem. Soc., 2012, 134, 17778 CrossRef PubMed; (b) M. Schinkel, I. Marek and L. Ackermann, Angew. Chem., Int. Ed., 2013, 52, 3977 CrossRef CAS PubMed.
- (a) P. V. Ramachandran, D. Pratihar, H. N. G. Nair, M. Walters, S. Smith, M. T. Yip-Schneider, H. Wu and C. M. Schmidt, Bioorg. Med. Chem. Lett., 2010, 20, 6620 CrossRef CAS PubMed; (b) P. V. Ramachandran, D. R. Nicponski, H. N. G. Nair, M. A. Helppi, P. D. Gagare, C. M. Schmidt and M. T. Yip-Schneider, Bioorg. Med. Chem. Lett., 2013, 23, 6911 CrossRef CAS PubMed; (c) P. V. Ramachandran and D. R. Nicponski, Chem. Commun., 2014, 50, 15216 RSC; (d) C. K. Tan, J. C. Er and Y.-Y. Yeung, Tetrahedron Lett., 2014, 55, 1243 CrossRef CAS.
- (a) C. Lee, H. Kim and Y. Kho, J. Nat. Prod., 2002, 65, 414 CrossRef CAS PubMed; (b) B. B. Messanga, S. F. Kimbu, B. L. Sondengam and B. Bodo, Phytochemistry, 2002, 59, 435 CrossRef CAS PubMed; (c) C. Kraft, K. Jenett-Siems, I. Köhler, B. Tofern-Reblin, K. Siems, U. Bienzle and E. Eich, Phytochemistry, 2002, 60, 167 CrossRef CAS PubMed.
- For reported examples of the conversion of an 8-aminoquinoline moiety to other functional groups, see Scheme S1 in the ESI.†.
- See Table S1 in the ESI.†.
- See Fig. S1 in the ESI.†.
- For reviews on the transition-metal-catalyzed functionalization of C–H bonds assisted by a bidentate directing group, see: (a) M. Corbet and F. de Campo, Angew. Chem., Int. Ed., 2013, 52, 9896 CrossRef CAS PubMed; (b) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (c) L. C. Misal Castro and N. Chatani, Chem. Lett., 2015, 44, 410 CrossRef; (d) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (e) R. K. Rit, M. R. Yadav, K. Ghosh and A. K. Sahoo, Tetrahedron, 2015, 71, 4450 CrossRef CAS.
- For our recent papers on the Rh-catalyzed functionalization of C–H bonds assisted by an 8-aminoquinoline group, see: (a) K. Shibata and N. Chatani, Org. Lett., 2014, 16, 5148 CrossRef CAS PubMed; (b) K. Shibata, T. Yamaguchi and N. Chatani, Org. Lett., 2015, 17, 3584 CrossRef CAS PubMed; See also, (c) A. Yokota and N. Chatani, Chem. Lett., 2015, 44, 902 CrossRef CAS; (d) T. Kubo, Y. Aihara and N. Chatani, Chem. Lett., 2015, 44, 1365 CrossRef.
- While a single example has been reported, Nishimura quite recently reported the Ir-catalyzed hydroarylation of 2-arylpyridines with 5a, in which the reaction took place at the α-position of 5a. Y. Ebe and T. Nishimura, J. Am. Chem. Soc., 2015, 137, 5899 CrossRef CAS PubMed.
- For oxidative addition of N–H bonds to a Rh(I) complex, see: E. Vélez, M. P. Betoré, M. A. Casado and V. Polo, Organometallics, 2015, 34, 3959 CrossRef , and references cited therein.
- (a) M. T. Whited, Y. Zhu, S. D. Timpa, C.-H. Chen, B. M. Foxman, O. V. Ozerov and R. H. Grubbs, Organometallics, 2009, 28, 4560 CrossRef CAS; (b) J. Meiners, A. Friedrich, E. Herdtweck and S. Schneider, Organometallics, 2009, 28, 6331 CrossRef CAS; (c) N. J. Brookes, M. T. Whited, A. Ariafard, R. Stranger, R. H. Grubbs and B. F. Yates, Organometallics, 2010, 29, 4239 CrossRef CAS; (d) J. E. V. Valpuesta, E. Álvarez, J. López-Serrano, C. Maya and E. Carmona, Chem.–Eur. J., 2012, 18, 13149 CrossRef CAS PubMed.
- For migratory C–H insertion into a metal carbene complex, see: (a) N. D. Jones, G. Lin, R. A. Gossage, R. McDonald and R. G. Cavell, Organometallics, 2003, 22, 2832 CrossRef CAS; (b) T. Cantat, M. Demange, N. Mézailles, L. Ricard, Y. Jean and P. L. Floch, Organometallics, 2005, 24, 4838 CrossRef CAS; (c) H. Heuclin, X. F. L. Goff and N. Mézailles, Chem.–Eur. J., 2012, 18, 16136 CrossRef CAS PubMed.
- For chelation-assisted functionalization of C–H bonds via metal carbene migratory insertion, see: (a) W.-W. Chan, S.-F. Lo, Z. Zhou and W.-Y. Yu, J. Am. Chem. Soc., 2012, 134, 13565 CrossRef CAS PubMed; (b) X. Yu, S. Yu, J. Xiao, B. Wan and X. Li, J. Org. Chem., 2013, 78, 5444 CrossRef CAS PubMed; (c) T. K. Hyster, K. E. Ruhl and T. Rovis, J. Am. Chem. Soc., 2013, 135, 5364 CrossRef CAS PubMed; (d) Z. Shi, D. C. Koester, M. Boultadakis-Arapinis and F. Glorius, J. Am. Chem. Soc., 2013, 135, 12204 CrossRef CAS PubMed; (e) F. Hu, Y. Xia, F. Ye, Z. Liu, C. Ma, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 1364 CrossRef CAS PubMed; (f) H.-W. Lam, K.-Y. Man, W.-W. Chan, Z. Zhou and W.-Y. Yu, Org. Biomol. Chem., 2014, 12, 4112 RSC; (g) J. Jeong, P. Patel, H. Hwang and S. Chang, Org. Lett., 2014, 16, 4598 CrossRef CAS PubMed; (h) Y. Xia, Z. Liu, S. Feng, Y. Zhang and J. Wang, J. Org. Chem., 2015, 80, 223 CrossRef CAS PubMed; (i) S. Sharma, S. H. Han, S. Han, W. Ji, J. Oh, S.-Y. Lee, J. S. Oh, Y. H. Jung and I. S. Kim, Org. Lett., 2015, 17, 2852 CrossRef CAS PubMed; (j) X.-G. Liu, S.-S. Zhang, J.-Q. Wu, Q. Li and H. Wang, Tetrahedron Lett., 2015, 56, 4093 CrossRef CAS; (k) I. E. Iagafarova, D. V. Vorobyeva, A. S. Peregudov and S. N. Osipov, Chem. Commun., 2015, 51, 4950 Search PubMed; (l) Y. Liang, K. Yu, B. Li, S. Xu, H. Song and B. Wang, Chem. Commun., 2014, 50, 6130 RSC.
- For a paper on the base-catalyzed H/D exchange of H–Rh species in D2O, see: K. Lemma, A. Ellern and A. Bakac, Inorg. Chem., 2003, 42, 3662 CrossRef CAS PubMed . See also A. Giuseppe, R. Castalenas, J. Perez-Torrente, F. J. Lahoz and L. A. Oro, Chem.–Eur. J., 2014, 20, 8391 CrossRef PubMed.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed.
- A. R. Kapdi, Dalton Trans., 2014, 43, 3021 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc03110a |
| This journal is © The Royal Society of Chemistry 2016 |