DOI:
10.1039/C5SC03120F
(Edge Article)
Chem. Sci., 2016,
7, 1212-1223
Oxidation triggers extensive conjugation and unusual stabilization of two di-heme dication diradical intermediates: role of bridging group for electronic communication†
Received
21st August 2015
, Accepted 26th October 2015
First published on 26th October 2015
Abstract
MauG is a diheme enzyme that utilizes two covalently bound c-type hemes to catalyse the biosynthesis of the protein-derived cofactor tryptophan tryptophylquinone. The two hemes are physically separated by 14.5 Å and a hole-hopping mechanism is proposed in which a tryptophan residue located between the hemes undergoes reversible oxidation and reduction to increase the effective electronic coupling element and enhance the rate of reversible electron transfer between the hemes in bis-Fe(IV) MauG. The present work describes the structure and spectroscopic investigation of 2e-oxidations of the synthetic diheme analogs in which two heme centers are covalently connected through a conjugated ethylene bridge that leads to the stabilization of two unusual trans conformations (U and P′ forms) with different and distinct spectroscopic and geometric features. Unlike in MauG, where the two oxidizing equivalents are distributed within the diheme system giving rise to the bis-Fe(IV) redox state, the synthetic analog stabilizes two ferric hemes, each coupled with a porphyrin cation radical, a scenario resembling the binuclear dication diradical complex. Interestingly, charge resonance-transition phenomena are observed here both in 1e and 2e-oxidised species from the same system, which are also clearly distinguishable by their relative position and intensity. Detailed UV-vis-NIR, X-ray, Mössbauer, EPR and 1H NMR spectroscopic investigations as well as variable temperature magnetic studies have unraveled strong electronic communications between two porphyrin π-cation radicals through the bridging ethylene group. The extensive π-conjugation also allows antiferromagnetic coupling between iron(III) centers and porphyrin radical spins of both rings. DFT calculations revealed extended π-conjugation and H-bonding interaction as the major factors in controlling the stability of the conformers.
Introduction
Metalloporphyrin π-cation radicals are of immense interest due to their occurrence as intermediates in the catalytic cycle of many heme containing enzymes, e.g., peroxidases,1 catalases,2 cytochrome P450 1a,3etc., and their photosynthetic reaction center.4 In spite of the common active intermediates, the reactivity differs from enzyme to enzyme. The extent of coupling between metal center and porphyrin π-cation radical may be linked to the various activities in the heme enzymes, and this has therefore led to the study of metalloporphyrin π-cation radicals and mixed valence π-cation radicals (where one electron is removed per two porphyrin rings).5,6
Numerous diheme enzymes such as MauG7,8 and bacterial diheme cytochrome c peroxidases (bCcP)9 serve as active catalysts in various important chemical transformations in biology. For instance, bCcP mediates peroxidase activity, whereby it transfers the oxidizing equivalents from H2O2 to cytochrome c or other small redox proteins.9 MauG (Fig. 1) is a terminal enzyme involved in the biosynthesis of the catalytic tryptophan tryptophylquinone (TTQ) cofactor of methylamine dehydrogenase (MADH). Although the two heme units are physically separated in both enzymes, they share electrons efficiently behaving as a single diheme unit rather than as independent heme centers. A tryptophan residue is, however, positioned in between two heme centers and has been proposed to act as a bridge in order to promote the electronic communication between the heme centers.7–9
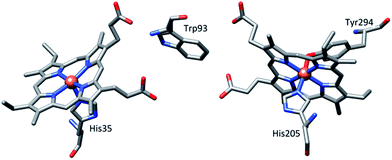 |
| | Fig. 1 Relative orientation of hemes and the intervening tryptophan residue in MauG (PDB ID code 3L4M).7a | |
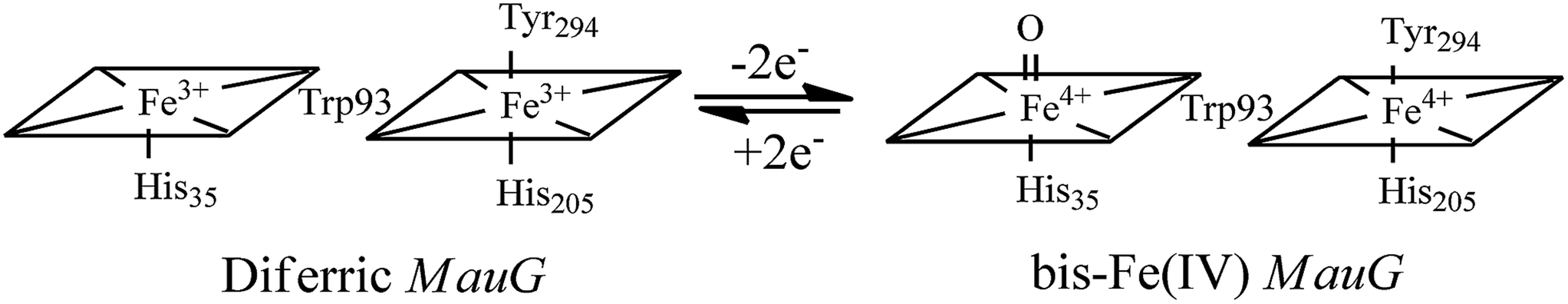
High-valent Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O intermediates are frequently invoked in the catalytic cycles of Fe-dependent oxidizing enzymes.1–3 In heme-dependent enzymes, the two-electron oxidized intermediate (compound I) consists of an Fe(IV) species (S = 1) coupled to an organic radical (S = 1/2) that is located on the porphyrin ring/axial ligand or a nearby amino acid residue (compound ES). In MauG, however, two oxidizing equivalents derived from H2O2 are distributed within the diheme system as two positive charges, giving rise to a bis-Fe(IV) redox state (Scheme 1) in which one heme is present as Fe(IV)O and the other as Fe(IV) with axial histidine and tyrosine ligation.8 As the two hemes are physically separated by 14.5 Å, a hole-hopping mechanism was proposed wherein the tryptophan residue reversibly oxidized and reduced in order to boost the effective electronic coupling element and magnify the rate of electron transfer between the heme centers in the bis-Fe(IV) MauG.8
O intermediates are frequently invoked in the catalytic cycles of Fe-dependent oxidizing enzymes.1–3 In heme-dependent enzymes, the two-electron oxidized intermediate (compound I) consists of an Fe(IV) species (S = 1) coupled to an organic radical (S = 1/2) that is located on the porphyrin ring/axial ligand or a nearby amino acid residue (compound ES). In MauG, however, two oxidizing equivalents derived from H2O2 are distributed within the diheme system as two positive charges, giving rise to a bis-Fe(IV) redox state (Scheme 1) in which one heme is present as Fe(IV)O and the other as Fe(IV) with axial histidine and tyrosine ligation.8 As the two hemes are physically separated by 14.5 Å, a hole-hopping mechanism was proposed wherein the tryptophan residue reversibly oxidized and reduced in order to boost the effective electronic coupling element and magnify the rate of electron transfer between the heme centers in the bis-Fe(IV) MauG.8
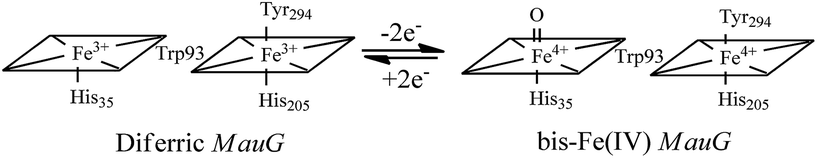 |
| | Scheme 1 Conversion of the hemes of MauG from diferric to bis-Fe(IV) redox state.8 | |
The electronic communication between the porphyrin moieties in the ground and/or excited state can be facilitated by using covalently connected linkers in conjugation with porphyrin rings. In this context, a large number of porphyrin dimers connected via conjugated linkers, viz. alkene, alkyne, imino, and azo bridges, have been studied due to their unique optical properties.10 Our recent efforts have been directed towards exploring the role of intermacrocyclic interactions in modulating various properties, viz. redox potential, spin state etc.11 However, interactions between a pair of porphyrin π-cation radicals connected through a linker remain unexplored.
In the present study, we have made synthetic analogs of the diheme centers of MauG in which two porphyrin rings have been covalently connected through a conjugated but rigid ethylene bridge which would separate the two macrocycles to the furthest extent. Step-wise oxidations up to two oxidizing equivalents have been performed using chemical oxidants. Unlike the bis-Fe(IV) state as obtained in MauG, the two electron oxidized complex stabilizes two ferric hemes, each coupled with a porphyrin cation radical, a scenario resembling the binuclear dication diradical complex. This work describes an alternative mechanism to store two oxidizing equivalents above the ferric state in dihemes. Spectroscopic investigations have unraveled strong electronic communications between two porphyrin π-cation radicals via the ethylene bridge which eventually alter the nature of such bridge. The extensive π-conjugation also allows antiferromagnetic coupling between iron(III) centers and porphyrin radical spins of both rings. DFT calculations have been employed which further support our spin-coupling model as obtained from the magnetic measurements and also provide insight into the preferential stabilization of various geometrical conformers under certain conditions.
In earlier work on monomeric metalloporphyrin π-cation radicals, spin coupling between the metal ion and the oxidized porphyrin ring in a number of derivatives that differed in metal ion, axial ligation, and/or porphyrin ligand were investigated.5,6 The inter-ring coupling was found to be closely related only to the degree of the ring overlap. In the present investigation, however, the inter-ring coupling between two monomeric Fe(III)porphyrin π-cation radicals has been demonstrated to occur only through the bridge although they are widely separated in space.
Result and discussion
Dichlorodiiron(III) ethylene bridged octaethyl porphyrin dimers have been synthesized both in cis (cis-1) and trans (trans-1) isomers using the procedures reported earlier.11j,12 Oxidation of these complexes has been performed in a step-wise manner using chemical oxidants and monitored using UV-vis-NIR spectroscopy. Scheme 2 shows the synthetic outline and list of diheme dication diradical intermediates reported here along with the abbreviations used.
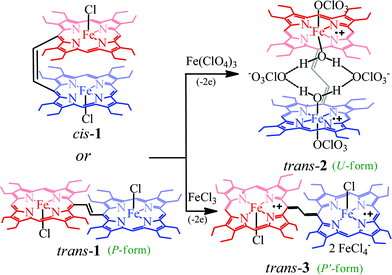 |
| | Scheme 2 Synthetic outline. | |
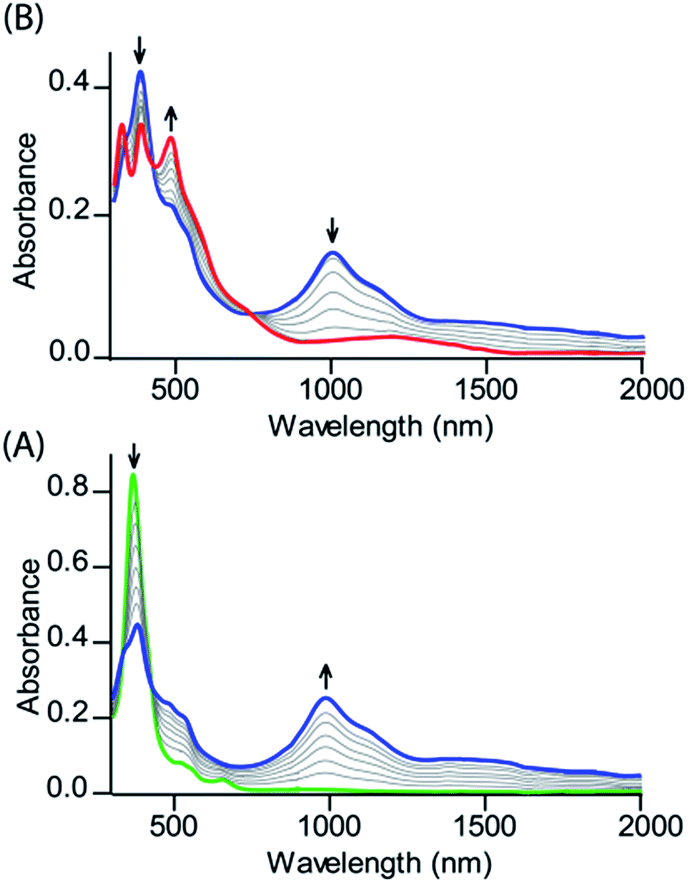
Fig. 2 shows the UV-visible spectral changes upon step-wise oxidations of cis-1 using Fe(ClO4)3 as an oxidant. Gradual addition of a CH3CN solution of Fe(ClO4)3 as an oxidizing agent in up to one equivalent to a dichloromethane solution of cis-1 leads to a sharp decrease in the Soret band intensity at 390 nm along with the appearance of a low energy band at 989 nm, which is characteristic of the intra-valence charge transfer obtained due to the formation of a mixed-valence π-cation radical dimer.6,13 The absorbance of the 989 nm band has showed a linear dependence on the concentration, ruling out the possibility of intermolecular dimerization as its origin.6 On further addition of the Fe(ClO4)3 solution in up to two equivalents, the Soret band at 390 nm decreases gradually again along with the appearance of two new bands at 326 and 477 nm, while the intensity of the absorbance at 989 nm decreases with the appearance of a new broad band near 1150 nm. The 2e-oxidized complex thus obtained has been isolated in the solid state as a dication diradical species (trans-2) in good yields and has been structurally characterized. Further addition of the oxidant showed no observable change in the absorption spectra.
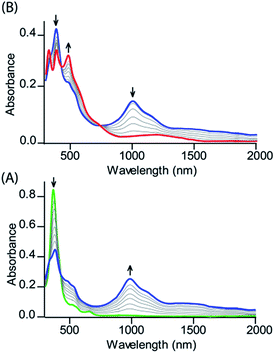 |
| | Fig. 2 UV-vis-NIR (in CH2Cl2 at 295 K) spectral change of 1.2 × 10−5 (M) solution of cis-1 upon gradual addition of (A) 0 to 1.0 eq. and (B) 1.0 to 2.0 eq. of Fe(ClO4)3. | |
Similar spectral changes have also been obtained during the oxidation of cis-1 when FeCl3 was used as an oxidant (Fig. 3). Upon one-electron oxidation, a sharp decrease in the Soret band intensity at 390 nm has been observed along with the appearance of a low energy band at 989 nm due to the formation of a mixed-valence π-cation radical dimer. Upon addition of the FeCl3 solution in up to four equivalents, the Soret band at 390 nm increased gradually and underwent a blue shift to 365 nm along with the appearance of two new bands at 317 and 488 nm in the UV-visible regions. In the NIR region, the intensity of the absorbance at 989 nm decreases while a broad band near 1210 nm appears. The two-electron oxidized complex thus obtained has also been isolated as a dication diradical species (trans-3) in the solid state and has been structurally characterized. The absorption spectrum of trans-3 displays a Soret band at 365 nm, two new bands at 317 and 488 nm, and a broad NIR band near 1210 nm. Trans-1 also produces similar spectral changes upon oxidation with Fe(ClO4)3/FeCl3 leading to the formation of trans-2/3, respectively (Fig. S1 and S2†). Gradual addition of an acetonitrile solution of Fe(ClO4)3 to a CH2Cl2 solution of trans-1 at −40 °C (Fig. S3†) also showed similar spectral changes as observed at room temperature.
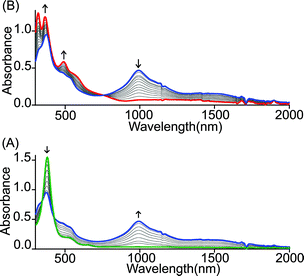 |
| | Fig. 3 UV-vis-NIR (in CH2Cl2 at 295 K) spectral change of 2.1 × 10−5 (M) solution of cis-1 upon gradual addition of (A) 0 to 2.0 and (B) 2.0 to 4.0 eq. of FeCl3. | |
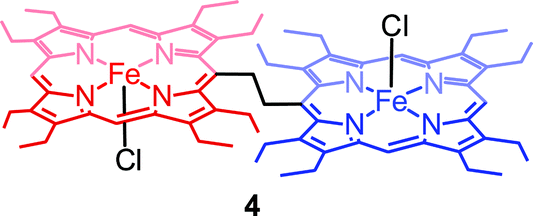
Fig. 4 shows the UV-visible spectral change upon gradual addition of a CH3CN solution of Fe(ClO4)3 into a CH2Cl2 solution of 1,2-bis[(chloro){5-(2,3,7,8,12,13,17,18-octaethylporphyrinato)}iron(III)]ethane, 4 (Chart 1), which is a highly flexible ethane-bridged analog of 1. Upon 1e-oxidation, the intensity of the Soret band decreases slightly along with a small blue-shift while a low-intense NIR band at 1100 nm is observed which, however, upon 2e-oxidation disappears completely.
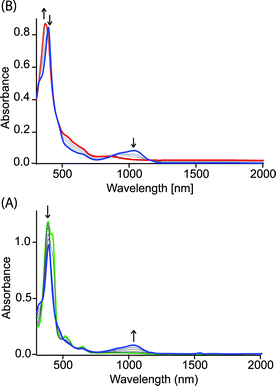 |
| | Fig. 4 UV-vis-NIR spectral change (in CH2Cl2 at 295 K) of 1.4 × 10−5 (M) solution of 4 (green line) upon gradual addition of (A) 0 to 1.0 and (B) 1.0 to 2.0 eq. of Fe(ClO4)3. | |
 |
| | Chart 1 1,2-Bis[(chloro){5-(2,3,7,8,12,13,17,18-octaethylporphyrinato)}iron(III)]ethane, 4. | |
It is interesting to compare the UV-vis-NIR spectra of 2e-oxidized complexes reported in Fig. 5. The absorption bands in the 480–500 nm, 600–900 and 1100–1300 nm regions, which are observed in both trans-2 and trans-3, are absent completely in the 2e-oxidized product of 4 and are, therefore, attributed to the extensive conjugation between two porphyrin π-cation radicals (vide infra) in the former complexes. The intensity of the Soret band has also been drastically reduced in trans-2 and trans-3 (as compared to the 2e-oxidized product of 4) which is suggestive of reduced aromaticity of the porphyrin rings in the complexes because of conjugation (vide infra).10c
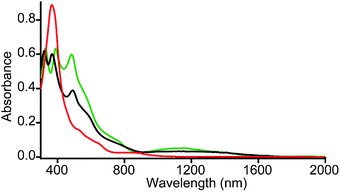 |
| | Fig. 5 UV-vis-NIR spectra (in CH2Cl2 at 295 K) of trans-2 (green line), trans-3 (black line) and 2e-oxidized product of 4 (red line). | |
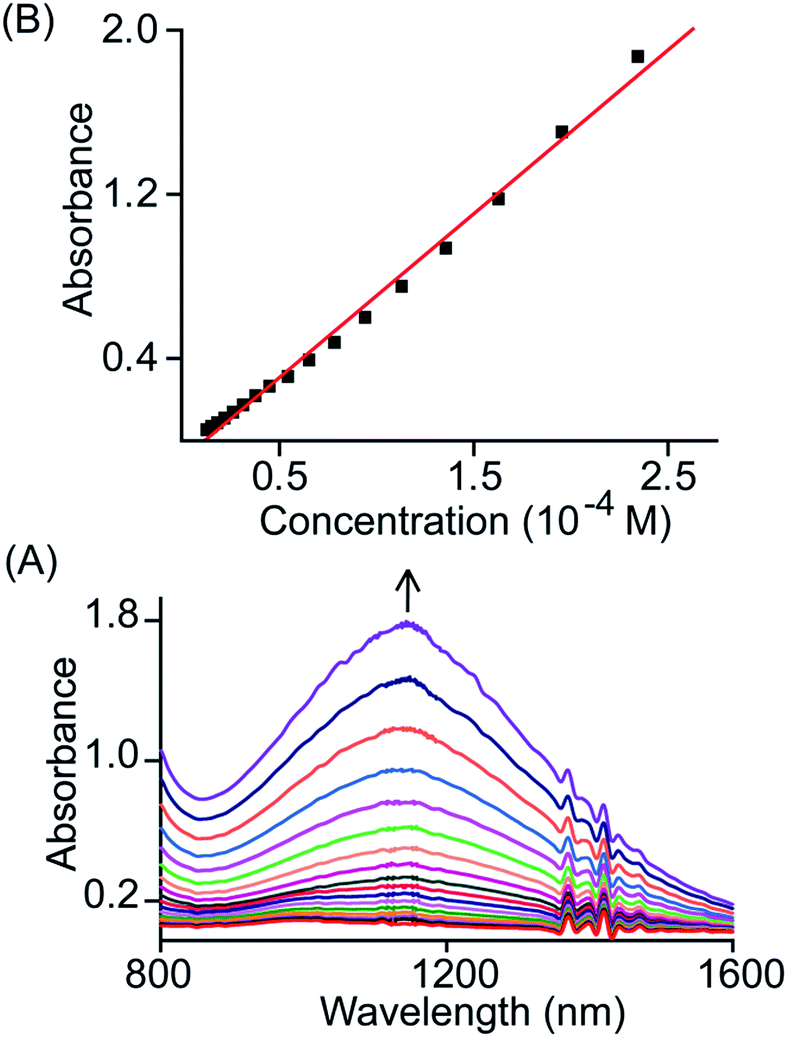
The broad NIR band that is observed at 1150 and 1210 nm for trans-2 and trans-3 can be attributed to the charge resonance (CR) stabilization of the spins and charges in the binuclear dication diradical complex. The CR stabilization energy (ΔECR) originates from exchange interactions between the molecular orbitals of each monomer and subsequent delocalization of the spin and charge over a greater number of atoms.6b,8,13 During the 1e-oxidation process (vide supra), a broad and slightly more intense NIR band at ∼1000 nm was obtained which is also attributed to charge resonance phenomena of the mixed-valence π-cation radical dimer.6,8 Interestingly, CR-transition phenomena are observed here in both 1e and 2e-oxidized complexes from the same system while the NIR bands are also clearly distinguishable by their relative position and intensity. Despite the inherent energy penalty caused by the electrostatic repulsion in the dication complex, the CR stabilization energy of the dication diradical species is larger than that of the mixed-valence π-cation radical dimer of the same system, which results in a blue-shift of the CR band.6b,8,13 In contrast, a red-shift of CR bands is observed when moving our attention from 1e to 2e-oxidation, and this is due to multiple factors related to charge delocalization through bridging ligand, intramolecular coupling between iron–radical and radical–radical spins (vide infra) etc. Through the bridging ethylene group, both porphyrin macrocycles exhibit substantial conjugation which eventually alters the nature of the bridge (ethylene to exo-methylene connectivity) resulting the stabilization of two unusual trans conformers (U and P′ type, vide infra). Moreover, the NIR bands in the absorption spectra of trans-2 and trans-3 are found to be linearly correlated with the concentration (Fig. 6 and S4†). Such Beer's law concentration dependence further supports the intramolecular origin of the CR bands.5,6 The MauG-catalyzed reaction proceeds through a bis-Fe(IV) intermediate which has been shown to exhibit CR phenomena. The bis-Fe(IV) state is an electronic equivalent of two Fe(III)porphyrin π-cation radicals with each radical spin coupled to the Fe(III) atom. Although there is large separation between two heme centers, such spin/charge resonance stabilization is facilitated by the presence of a Trp93 residue which bridges between heme centers.8
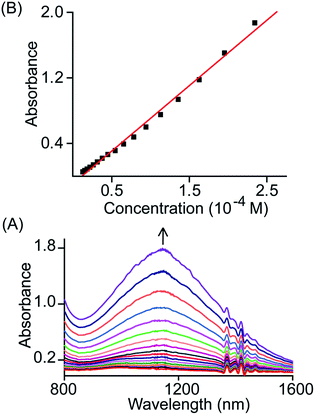 |
| | Fig. 6 (A) Gradual change in the absorbance of the NIR band (in CH2Cl2 at 295 K) of trans-2 with change in concentration and (B) plot of absorbance of the NIR band as a function of concentration. | |
The formation of porphyrin π-cation radicals is often signalled by strong IR bands.5,6 Fig. S5† displays selected portions of the IR spectra of trans-2 and trans-3 along with their unoxidized analogs, cis-1 and trans-1. The oxidized species, trans-2 and trans-3, show π-cation radical IR marker bands at 1546, 1598 and 1544, 1595 cm−1, which are absent in the unoxidized complexes. The ∼1550 cm−1 band is due to the asymmetric Cα–Cmeso stretching mode while the ∼1600 cm−1 band is due to the Cβ–Cβ stretch. Similar IR marker bands have also been reported earlier in octaethylporphyrinate and β-alkyl substituted porphyrinate π-cation radical derivatives.5,6 Thus, the IR spectral data further confirms trans-2 and trans-3 as porphyrin π-cation radicals. The molar conductances at 295 K of trans-2 (41 Ω−1 cm2 mol−1) and trans-3 (42 Ω−1 cm2 mol−1) in dichloromethane are similar to the values observed for 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 electrolytes in solution.14
:2 electrolytes in solution.14
Crystallographic characterizations
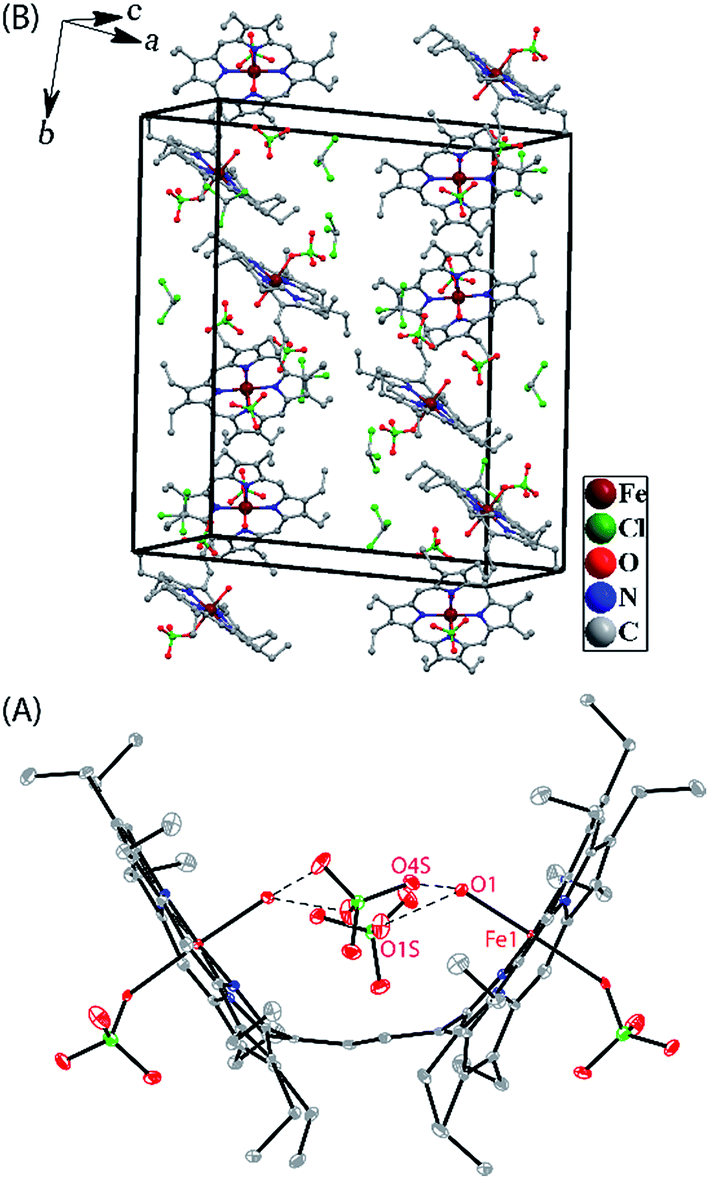
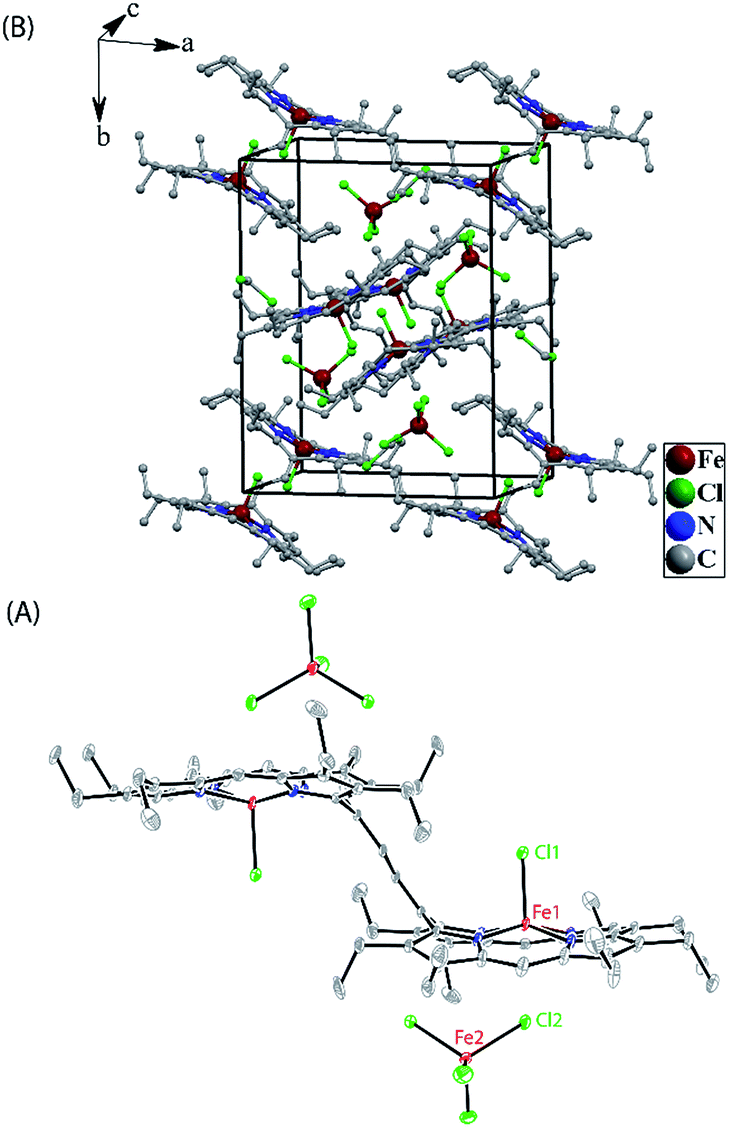
Dark needle crystals of trans-2 and trans-3 were grown via slow diffusion of hexane into chloroform and dichloromethane solutions of the complexes in air at room temperature. Perspective views of the complexes along with the molecular packing are depicted in Fig. 7 and 8. Table 1 lists the crystallographic data and data collection parameters of the complexes reported here. Both molecules crystallize in the monoclinic crystal system, however, trans-2 does so with a P2/c space group while trans-3 crystallizes with a P21/n space group. Iron centers are in six-coordinate geometry with water and perchlorate axial ligands for trans-2 and five-coordinate geometry with chloride ion as axial ligand in trans-3. The porphyrin cores of the complexes showed considerable doming. Table S1† shows the selected bond distances and angles while Table 2 compares the structure and geometrical parameters of trans-2 and trans-3 along with their unoxidized complex, trans-1.
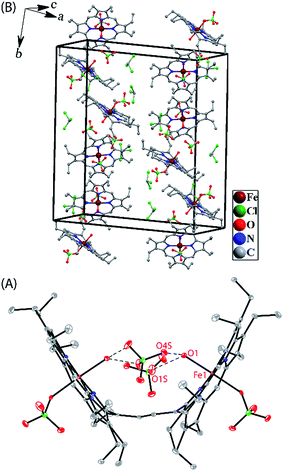 |
| | Fig. 7 (A) A perspective view (at 100 K) of trans-2 showing 50% thermal contours (H atoms have been omitted for clarity). (B) Diagram illustrating the packing of trans-2 in the unit cell (H-atoms have been omitted for clarity). | |
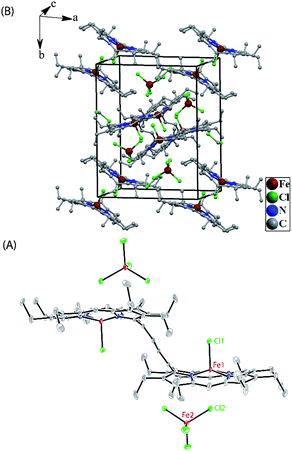 |
| | Fig. 8 (A) A perspective view (at 100 K) of trans-3 showing 50% thermal contours (H atoms have been omitted for clarity). (B) Diagram illustrating the packing of trans-3 in the unit cell (H-atoms have been omitted for clarity). | |
Table 1 Crystallographic data and data collection parameters
|
|
trans-2 |
trans-3 |
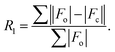
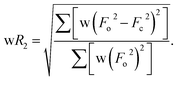
|
| Formula |
C78H96Cl16Fe2N8O18 |
C76H92Cl14Fe4N8 |
|
T (K) |
100(2) |
100(2) |
| Formula weight |
2112.52 |
1837.27 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P2/c |
P21/n |
|
a, Å |
31.467(5) |
15.165(5) |
|
b, Å |
9.589(5) |
18.236(5) |
|
c, Å |
30.770(5) |
15.623(5) |
|
α, deg |
90 |
90 |
|
β, deg |
91.556(5) |
103.044(5) |
|
γ, deg |
90 |
90 |
|
V, Å3 |
9281(5) |
4209(2) |
|
Z
|
4 |
2 |
|
d
calcd, g cm−3 |
1.512 |
1.450 |
|
μ, mm−1 |
0.842 |
1.166 |
|
F(000) |
4352 |
1892 |
| Crystal size |
0.26 × 0.20 × 0.16 mm3 |
0.22 × 0.16 × 0.12 mm3 |
| No. of unique data |
17277 |
7814 |
| Completeness to theta = 25.00° |
99.9% |
99.9% |
| No. of parameters refined |
1166 |
496 |
| GOF on F2 |
1.031 |
1.032 |
|
R
1
[I > 2σ(I)] |
0.0679 |
0.0489 |
|
R
1
(all data) |
0.1108 |
0.0680 |
| wR2b (all data) |
0.1791 |
0.1332 |
| Largest diff. peak and hole |
1.324 and −1.101 e Å−3 |
0.910 and −0.745 e Å−3 |
Table 2 Selected structural parameters
|
|
trans-1a |
trans-2 |
trans-3 |
| Molecule-I |
Molecule-II |
|
Taken from ref. 11h.
Average value.
Displacement of iron from the least-squares plane of the C20N4 porphyrinato core.
Average displacement of atoms from the least-squares plane of C20N4 porphyrinato core.
Inter-planar angle between the least-squares plane of the C20N4 porphyrinato core and the C4 plane of the bridging ethylene group (see Fig. 9A).
|
| Fe–Np (Å)b |
2.055(6) |
2.027(4) |
2.034(4) |
2.045(3) |
| Fe–O(H2) (Å) |
— |
2.093(3) |
2.095(3) |
— |
| Fe–O(ClO3) (Å) |
— |
2.187(3) |
2.178(3) |
— |
| Fe–Cl (Å) |
2.241(2) |
— |
— |
2.2249(11) |
| ΔFe24 (Å)c |
0.48 |
0.10 |
0.04 |
0.50 |
| Δ24 (Å)d |
0.21 |
0.20 |
0.16 |
0.26 |
| Fe⋯Fe (Å) |
9.84 |
8.33 |
8.31 |
9.32 |
|
Θ (°)e |
65.79 |
56.59 |
57.13 |
55.82 |
| C20–C37 (Å) |
1.478(9) |
1.374(6) |
1.371(6) |
1.364(4) |
| C37–C37A (Å) |
1.312(13) |
1.435(9) |
1.433(9) |
1.432(7) |
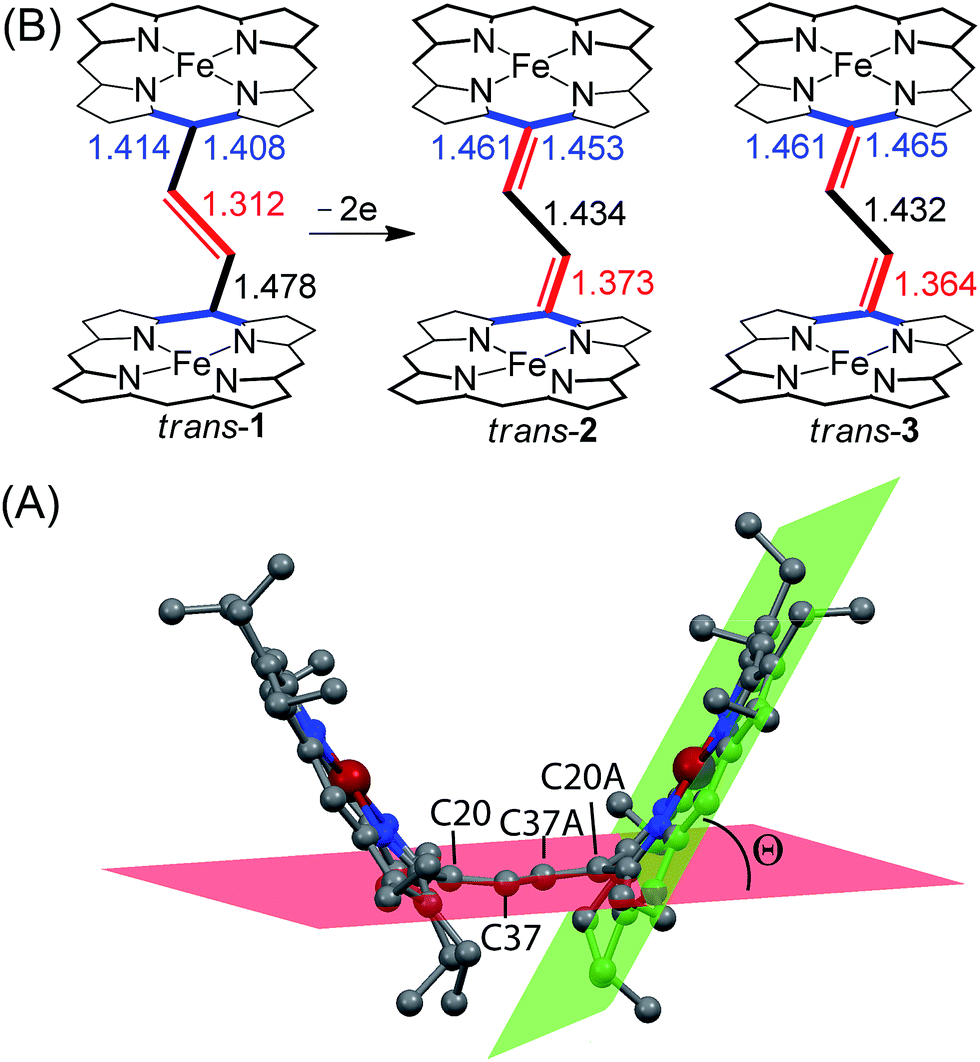
The average Fe–Np distances in the two types of complexes are very different: 2.027(4) and 2.034(4) Å for cores I and II in trans-2 and 2.045(3) Å in trans-3 which are characteristic of admixed high-spin and high-spin states of iron.11,15,16 The Fe–Cl bond distance of 2.2249(11) Å in trans-3 is shorter than the 2.241(2) Å observed in the unoxidized complex, trans-1. The slight contraction of the Fe–Cl bond distance is consistent with the increased positive charge in the oxidized complex. The displacement of iron(III) from the mean plane of the C20N4 porphyrinato core (Fe⋯Ctp) in trans-3 is 0.50 Å which is slightly larger than the 0.48 Å observed in trans-1.11h However, the Fe⋯Ctp distances in the six-coordinate complex, trans-2, are 0.10 and 0.04 Å for cores I and II, respectively, and iron is displayed in the direction of the axial H2O ligand. Although the ligand field strength of ClO4− is greater than for H2O according to the magnetochemical series,17 this is outweighed by the strong H-bonding interactions between the ligated H2O and ClO4− counterions which hold the structure in place.
The most striking feature of trans-2 is the unusual bowl shaped conformation (U-form) of the bisporphyrin unit which is stabilized by four H-bonding interactions [O⋯O: 2.761(5), 2.931(5), 2.926(5) and 2.767(5) Å] between the coordinated H2O molecules and the ClO4− counterions. The inter-planar angles (Θ) between the plane of the ethylene bridge and that of the porphyrin rings are 56.59° and 57.13° in trans-2 (Table 2) as compared to 65.79° obtained for trans-1. Although steric interactions with the β-ethyl substituents in trans-1 tend to prevent a coplanar arrangement of the porphyrin rings and the ethylene bridge, the extensive conjugation in the dication diradical species (trans-2) imposes a better coplanarity. The extended conjugation between two porphyrin units through the bridging ethylene moiety is manifested in the alteration of the C20–C37 and C37–C37A bond distances. The C20–C37 bond lengths of 1.374(6) (molecule-I) and 1.371(6) Å (molecule-II) in trans-2 are close to the CC distance, while the C37–C37A bond lengths of 1.435(9) (molecule-I) and 1.433(9) Å (molecule-II) become close to that of a C–C single bond. This results in a situation where the ethylene bridge (C–CHCH–C) transforms into an exo-methylene (CCH–CHC) connectivity between two porphyrin rings (Fig. 9B).
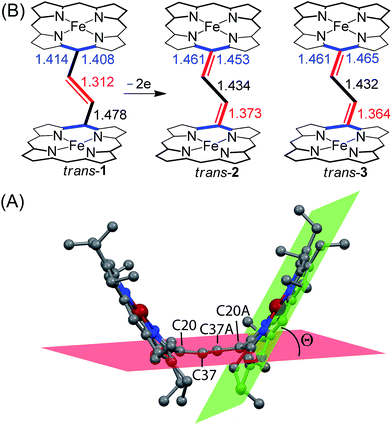 |
| | Fig. 9 (A) Diagram illustrating the angle Θ between the least-squares plane of C20N4 porphyrinato core (green) and the calculated C4 plane of the bridging ethylene group (red) of trans-2 as a representative case. (B) Selected bond distances (in Å) between oxidized and unoxidized complexes have been compared. H-atoms, axial ligands and counterions have been omitted for clarity. | |
The X-ray structure of trans-3 is similar to that of trans-1 in terms of relative orientation of the two porphyrin rings; however, they differ remarkably in several structural and geometrical parameters. The C20–C37 and C37–C37A bond distances of 1.364(4) and 1.432(7) Å and the inter-planar angle (Θ) of 55.82° in trans-3 are similar to those of trans-2, which is indicative of strong intramolecular electronic conjugation between the two rings through the ethylene bridge. The alteration of the C20–C37 and C37–C37A bond distances leads to a single bond connecting two exo-methylene groups at C20 of the two porphyrin rings. Scheme 3 describes the three possible conformations for the trans isomer. In one conformation, both porphyrins are completely orthogonal to the alkene (normal P-type), while in other two the macrocycles are almost in plane with the bridged ethylene moiety and the rings are either cofacial (U-type) or anti (P′-type) with respect to each other.10f,g The 2e-oxidized complexes, trans-2 and trans-3, do not stabilize in the normal P-type conformation but in the unusual U and P′-conformations, which facilitates extended conjugation between the two radical cores. This is the first structural evidence of the U and P′ forms in β-alkyl substituted bisporphyrins. Although the P′-form has been previously observed in a few β-unsubstituted bisporphyrins,10c–e no such conformers could be stabilized in β-alkyl substituted bisporphyrins due to steric interactions from the extreme closeness of the alkyl groups.
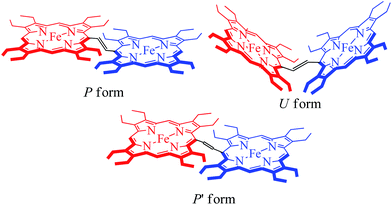 |
| | Scheme 3 Different conformations of trans ethylene-bridged Fe(III)bisporphyrin framework. | |
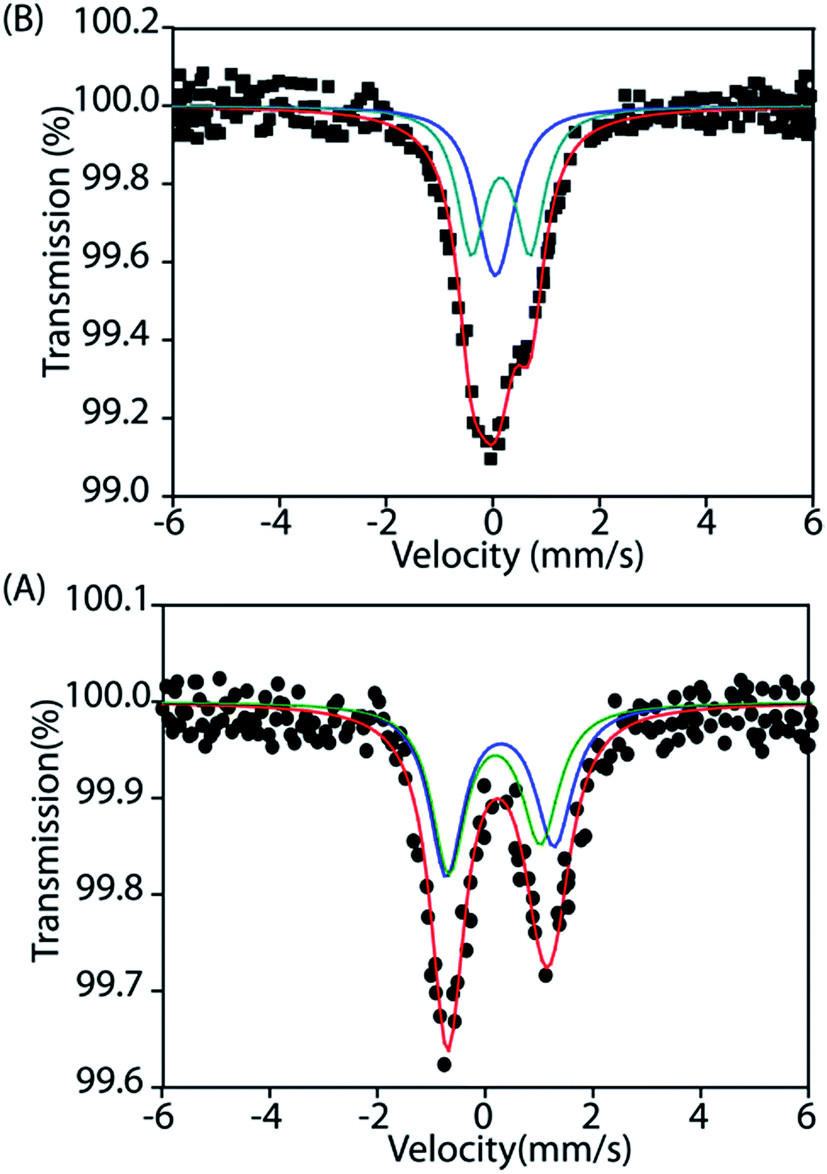
Mössbauer
To gain further insight into the electronic structure, we studied both trans-2 and trans-3 by Mössbauer spectroscopy. Fig. 10 shows the solid state Mössbauer spectra of the oxidized complexes at 295 K. The Mössbauer spectra provide compelling evidence that oxidation has occurred at the porphyrin ring only. trans-2 shows the presence of two quadrupole-split doublets [δ (ΔEq): 0.39 (2.0) and 0.33 (1.7) mm s−1], suggesting the presence of two admixed high-spin Fe(III) centers11,15,16 in the molecule as also observed in the X-ray structure (vide supra). In contrast, trans-3 displays two quadrupole-split doublets, one of which has parameters comparable to the tetrachloroferrate anion18a [δ = 0.19 mm s−1, ΔEq = 0.23 mm s−1] while the other represents a high-spin iron(III) centre [δ = 0.29 mm s−1, ΔEq = 1.1 mm s−1].
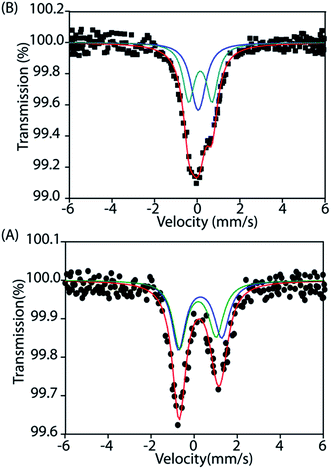 |
| | Fig. 10 Mössbauer spectra of (A) trans-2 and (B) trans-3 at 295 K. | |
Magnetic measurements
The magnetic susceptibility of trans-2 and trans-3 has been measured between 5 to 300 K using two different applied magnetic fields of 0.1 and 1 T which provide very similar results.
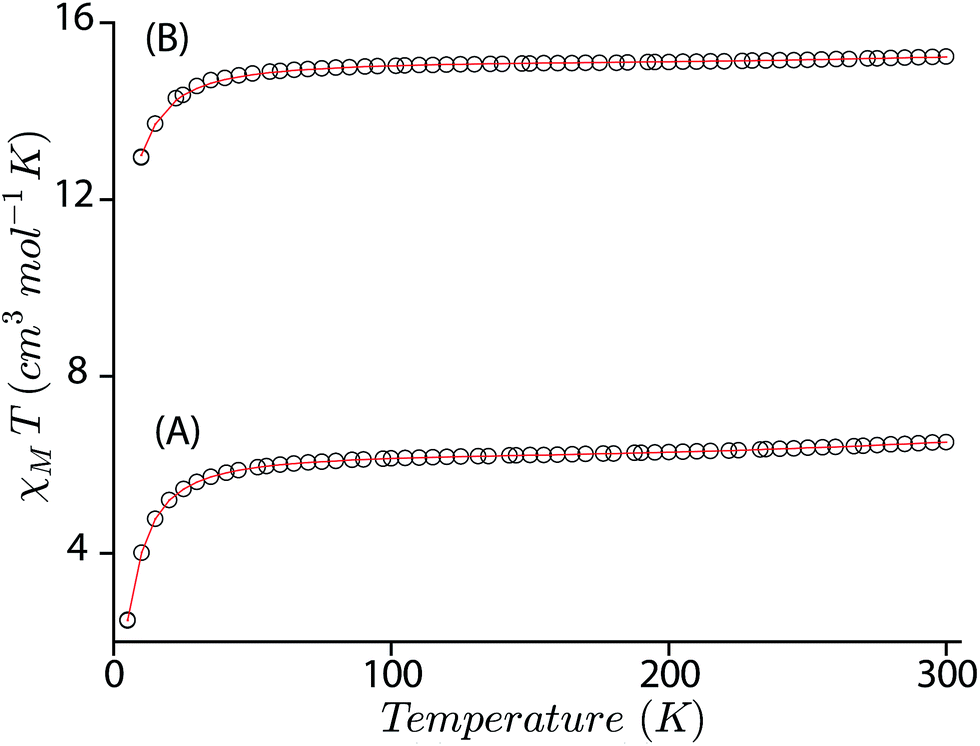
In an attempt to obtain a quantitative description of the spin–spin interaction that led to the observed magnetic moment, the magnetic susceptibility data were fitted using the software PHI.19 Acceptable fits (Fig. 11) have been obtained using a model which included three pairs of interactions: (a) intramolecular coupling between the iron and π-cation radical spin (JFe–r), (b) intramolecular coupling between two iron spins (JFe–Fe), and (c) intramolecular coupling between the two radical spins of the bisporphyrin (Jr–r). Each iron(III) center was treated as high spin (S = 5/2) with a g value set at 2.0 and the presence of small residual mononuclear iron(III) impurities was also taken into consideration. The parameters obtained from the fits are: JFe–r = −124.36 cm−1, JFe–Fe = −0.05 cm−1, Jr–r = −16.67 cm−1, D = 4.0 cm−1 for trans-2, JFe–r = −149.88 cm−1, JFe–Fe = −0.13 cm−1, Jr–r = −8.41 cm−1, and D = 6.0 cm−1 for trans-3. The ruffling of the porphyrin ring allows strong antiferromagnetic coupling between the porphyrin π-cation radical and the iron center, which is otherwise forbidden with planar macrocycles.5d The extensive π-conjugation between two porphyrin units of the bisporphyrin, through the bridging ethylene group, allows antiferromagnetic coupling between the two radical spins of the two porphyrin units although the cores are well separated and not cofacial enough to have any through space interactions.
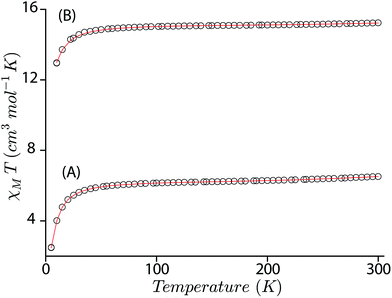 |
| | Fig. 11
Χ
M
T versus T plots for (A) trans-2, and (B) trans-3. The solid lines are best fits using the values given in the text. | |
1H NMR
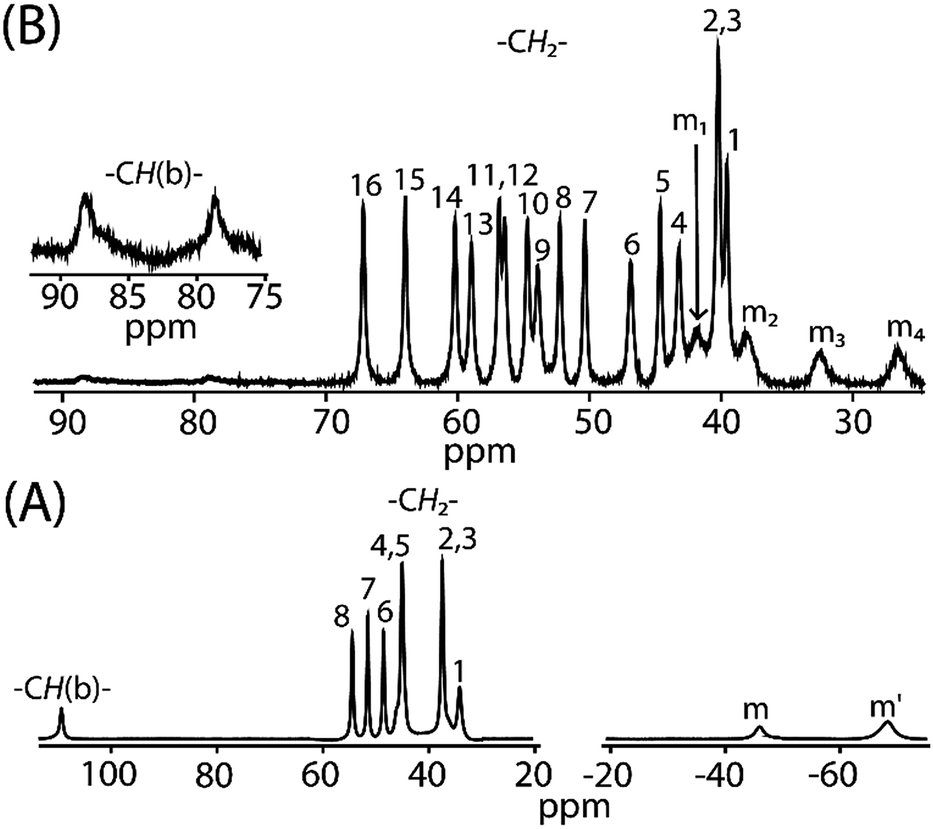
Fig. 12 compares the 1H NMR spectra between cis-1 and trans-2. For trans-3 the signals are too broad to be detected. The 1H NMR spectra of cis-1 shows the presence of eight methylene proton resonances between 34 to 55 ppm, two upfield shifted meso signals at −68.9 and −46.2 ppm in 2:1 intensity ratio, and a highly downfield shifted bridging signal at 110 ppm. However, sixteen methylene and four meso signals were obtained upon two electron oxidation of cis-1 to form trans-2, which confirms the presence of two inequivalent iron(III) centers as also evident from the X-ray structure and Mössbauer spectra of the complex in the solid state. The most conspicuous features in the 1H NMR spectra of trans-2 are the wide range of methylene resonances (39.5 to 67.1 ppm) and highly downfield shifted meso proton signals (at 26.5, 32.4, 37.9 and 41.8 ppm) which are attributed to the presence of porphyrin π-cation radicals and also to the increase in coordination number (from five to six).20 Mulliken spin densities have also been calculated and reveal a negative spin density on the meso and bridging carbon atoms (Fig. S6†), hence the downfield shifts of the corresponding proton signals which also indicates the presence of the antiferromagnetic coupling of the iron spin with the S = 1/2 porphyrin radical as observed in the variable temperature magnetic measurements.20a,b Moreover, bridging protons are found at 78.6 and 88.3 ppm which are extremely broad due to the extensive delocalization of the radical through the bridging group as also evident in the alteration of the C20–C37 and C37–C37A bond lengths (vide supra). The temperature dependence of the proton signals follows the Curie law (Fig. S7†) which suggests the presence of a single spin state throughout the temperature range.
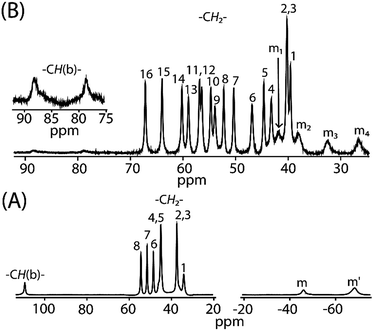 |
| | Fig. 12
1H NMR spectra (in CDCl3 at 295 K) of (A) cis-1 and (B) trans-2. Here, –CH(b)– marks the bridging protons and m, m′, m1–m4 represents meso proton signals. | |
EPR
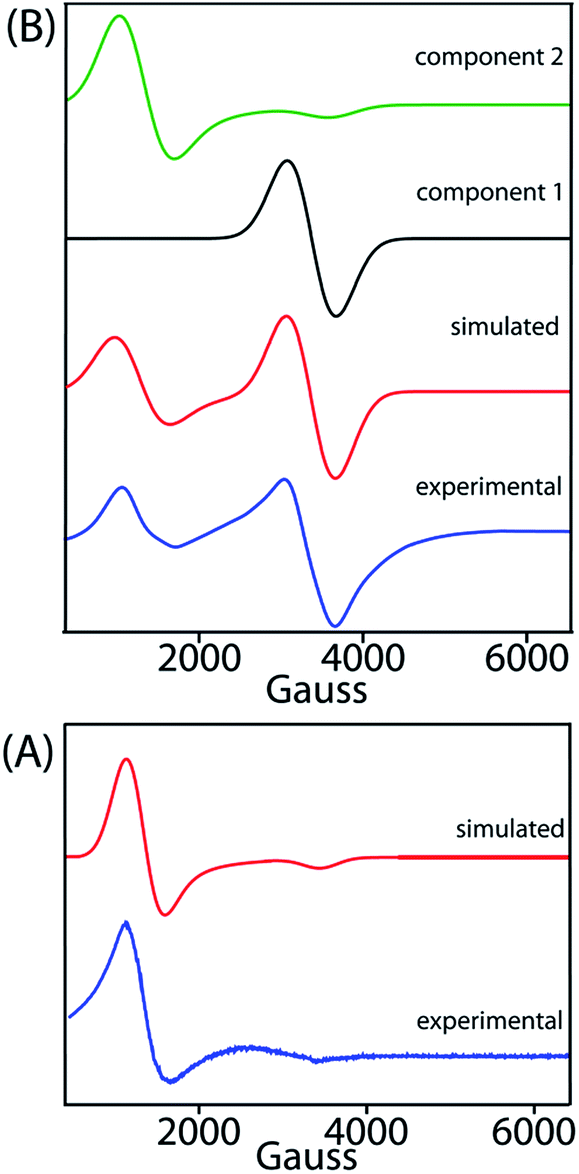
X-band EPR spectra have been recorded at 77 K for trans-2 and trans-3 both in solid and solution phases and Fig. 13 displays the experimental and simulated spectra while Table S2† lists the simulation parameters. Simulation of the spectra of trans-2 yields g⊥ = 5.470 and g∥ = 1.945 which is consistent with admixed high spin iron(III) as also observed from the Mössbauer data and X-ray structure of the complex in the solid state. Trace B shows the EPR spectra of trans-3 which is composed of two distinct features: a single isotropic line (g = 2.016) characteristic of an unresolved 6S1 state indicating spin S = 1/2 and a +1/2 ↔ −1/2 transition, consistent with a [FeCl4]− anion,18b and a second minor component with g⊥ = 5.750 and g∥ = 1.942, indicative of a high-spin iron(III). Moreover, the EPR signals of the 2e-oxidised complexes reported here are weak due to relatively weak intramolecular coupling between iron–radical (JFe–r) and radical–radical (Jr–r) spins (vide supra), although stronger coupling would result in no observable signals.
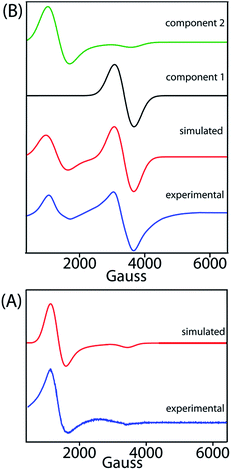 |
| | Fig. 13 X-band EPR spectra of (A) trans-2 and (B) trans-3 in CH2Cl2 (at 77 K). Blue and red lines represent experimental and simulated spectra, respectively. The black and green lines represent the two components used in the simulation of the EPR spectra of trans-3. | |
MauG is an enzyme unique in several respects. In heme-dependent enzymes, the two-electron oxidized intermediate (compound I) consists of an Fe(IV) species coupled to an organic radical that is located on the porphyrin ring/axial ligand or a nearby amino acid residue (compound ES).1–3 However, the two oxidizing equivalents derived from H2O2 are distributed within the diheme system as two positive charges, giving rise to the bis-Fe(IV) redox state in MauG.8 This is electronically equivalent to two ferric hemes, each coupled with a porphyrin cation radical, a scenario resembling the binuclear dication diradical complex as observed in the 2e-oxidized complexes of trans-2 and trans-3 reported here. Although the spins and charges are delocalized throughout the diheme system and are stabilized via a series of possible charge resonance structures (responsible for the observed NIR band), the most stable form is a binuclear dication diradical complex which is found spectroscopically as the most stable conformer both in the solid state and in solution.
Molecular modeling
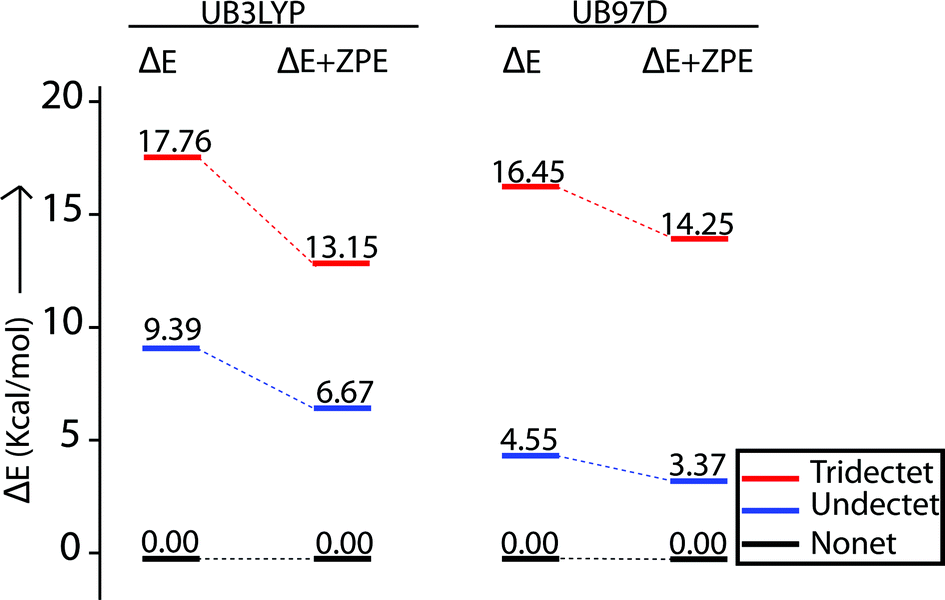
A series of DFT calculations have been carried out to get more insights into the electronic structure of the oxidized species. Trans-2 has been optimized in three possible spin multiplicities: nonet (9) state (considering antiferromagnetic interactions between the π-cation radical and the iron(III) spin), undectet (11) state (considering antiferromagnetic coupling between the two π-cation radical centers) and tridectet (13) state (considering ferromagnetic interactions between the cation radical and iron(III) spin). However, the interactions between the two iron(III) centers of trans-2 in all these states have been considered to be ferromagnetic in nature. Since we are dealing with a large computational model, we have chosen to stick with a pure high-spin state (S = 5/2) for the iron(III) centers, although experimental data suggest a minor contribution from the intermediate spin (S = 3/2) in trans-2. Fig. S8 and S9† show the optimized geometries of trans-2 with varying spin states in unrestricted B3LYP21 and B97D22 (includes dispersion correction) functional, while Fig. 14 schematically represents their relative energies. Clearly, the nonet state is energetically more stable than the undectet state, and the tridectet state is the least stable. This is also in good agreement with the experimental results.
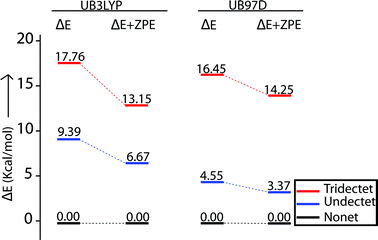 |
| | Fig. 14 Relative spin-state energies of nonet (9), undectet (11) and tridectet (13) states of trans-2 as calculated using unrestricted B3LYP and B97D functionals in DFT. All the ΔE and ΔE + ZPE values are relative to nonet (9) state. | |
In an attempt to find the probable reason behind such stabilization of the U form over the others, we have further optimized the molecule in different conformations, namely the P′ and P forms, both in the presence and absence of counterions. Fig. S10–S15† represent the optimized geometries while Fig. 15 shows the relative energies of different conformations both in the presence and absence of counterions. For trans-2, the U form is more stable than the P′ and P forms by 16.34 kcal mol−1 and 30.27 kcal mol−1, respectively. However, in the absence of counterions, the P′ form becomes the most stable one, being 5.63 kcal mol−1 lower in energy than the U-form, while the P-form is similar (with a difference of −0.6 kcal mol−1) in energy to the U-form. This proves that H-bonding interactions between the axial H2O and the ClO4− counterion are responsible for the unusual stabilization of the U conformer which is otherwise strained. This is also evident in the stabilization of the P′ conformer for trans-3 where there is no H-bonding interaction with the counterion (FeCl4−). Both the HOMO and the LUMO (Fig. 16 and S16†) of trans-2 were found to have significant coefficients on the bridging ethylene group for the U and P′ forms, suggesting a substantial conjugation through the bridge, while the P form shows no such conjugation which is also in accord with the experiment. Although metallo-OEP radicals are known to have an a1u type HOMO, trans-2 is best described as a mixture of both a1u and a2u type orbitals (Fig. 16).23 While the positions of infrared marker bands of porphyrin π-cation radicals are characteristics of an a1u-type symmetry, the presence of a2u type is clearly evident in the large shifts of the meso proton signals in the 1H NMR (vide supra). Thus, computational studies clearly support the experimental observations.
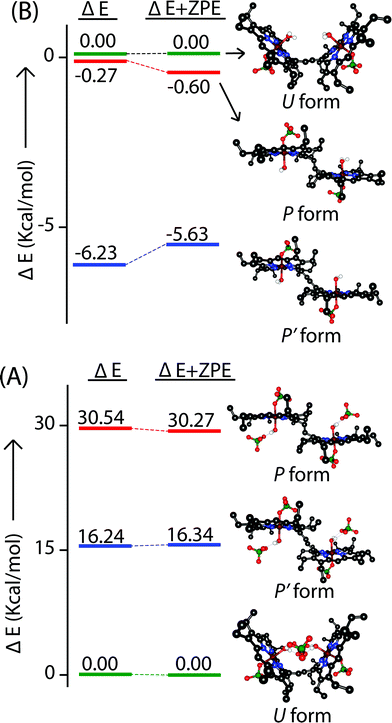 |
| | Fig. 15 Relative energies of the U, P, and P′ form of nonet (9) states of (A) trans-2 and (B) trans-2 (without counterion) as calculated using unrestricted B3LYP in DFT. All the ΔE and ΔE + ZPE values are relative to the U form. | |
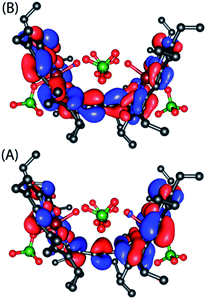 |
| | Fig. 16 (A) HOMO and (B) LUMO of U form of trans-2 as obtained from the unrestricted B3LYP optimized geometry using nonet (9) state. | |
Summary
We have reported here the structural and spectroscopic evidence for two interacting Fe(III) porphyrin π-cation radicals that are covalently connected through an ethylene bridge and thus are too widely separated to have any through space interaction. 2e-oxidation of the diiron(III) porphyrin dimer leads to the conversion of the cis isomer to trans along with the stabilization of two unusual conformations (U and P′-type) which have widely different and distinct spectroscopic and geometric features and are different from the normal trans conformer (P-type). Through the bridging ethylene group, both porphyrin macrocycles exhibit notable conjugation resulting in effective coupling between iron and porphyrin radical spins of both rings. In fact, oxidation leads to a change in identity of the bridge which further highlights the key role played by the bridge in communicating the electronic properties between two rings. The conformers can be considered as a real supramolecule rather than two interacting discrete Fe(III) porphyrin π-cation radicals. DFT calculations further support the spin coupling model obtained from magnetic studies suggesting the nonet state of trans-2 as the energetically favorable one. It also revealed that the extended π-conjugation and H-bonding interactions are the major factors in controlling the stability of the unusual conformers in the oxidized complexes.
Acknowledgements
This work is dedicated to Prof. Samaresh Mitra on the occasion of his 75th birthday.
We thank IIT Kanpur for providing all the facilities and support. The Council of Scientific and Industrial Research (CSIR), New Delhi, and Science and Engineering Research Board (SERB), India are gratefully acknowledged for financial support. D. S. and S. D. thank University Grant Commission (UGC), India and A. K. and S. B. thank the Council of Scientific and Industrial Research (CSIR), New Delhi for their fellowship.
References
-
(a) T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed;
(b)
P. R. Ortiz de Montellano, in Biocatalysis Based on Heme Peroxidases, ed. E. Torres and M. Ayala, Springer, Berlin, 2010, pp. 79–107 Search PubMed;
(c) G. L. Berglund, G. H. Carlsson, A. T. Smith, H. Szöke, A. Henriksen and J. Hajdu, Nature, 2002, 417, 463–468 CrossRef CAS PubMed.
-
(a) M. Alfonso-Prieto, X. Biarnés, P. Vidossich and C. Rovira, J. Am. Chem. Soc., 2009, 131, 11751–11761 CrossRef CAS PubMed;
(b) P. Chelikania, I. Fitab and P. C. Loewen, Cell. Mol. Life Sci., 2004, 61, 192–208 CrossRef PubMed;
(c) M. T. Green, J. Am. Chem. Soc., 2001, 123, 9218–9219 CrossRef CAS PubMed.
-
(a)
P. R. Ortiz de Montellano, Cytochrome P450: structure, mechanism and biochemistry, Plenum, Kluwer, New York, 3rd edn, 2005 Search PubMed;
(b) A. B. McQuarters, M. W. Wolf, A. P. Hunt and N. Lehnert, Angew. Chem., Int. Ed., 2014, 53, 4750–4752 CrossRef CAS PubMed;
(c) P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed;
(d) J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS PubMed;
(e) I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2277 CrossRef CAS PubMed;
(f) S. Shaik, D. Kumar, S. P. de Visser, A. Altun and W. Thiel, Chem. Rev., 2005, 105, 2279–2328 CrossRef CAS PubMed;
(g) B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed;
(h) L. Ji, A. S. Faponle, M. G. Quesne, M. A. Sainna, J. Zhang, A. Franke, D. Kumar, R. van Eldik, W. Liu and S. P. de Visser, Chem.–Eur. J., 2015, 21, 9083–9092 CrossRef CAS PubMed.
-
(a) P. Jordan, P. Fromme, H. T. Witt, O. Klukas, W. Saenger and N. Krau, Nature, 2001, 411, 909–917 CrossRef CAS PubMed;
(b) K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed;
(c) Z. Liu, H. Yan, K. Wang, T. Kuang, J. Zhang, L. Gui, X. An and W. Chang, Nature, 2004, 428, 287–292 CrossRef CAS PubMed;
(d) A. A. Pascal, Z. Liu, K. Broess, B. van Oort, H. van Amerongen, C. Wang, P. Horton, B. Robert, W. Chang and A. Ruban, Nature, 2005, 436, 134–137 CrossRef CAS PubMed;
(e) B. Loll, J. Kern, W. Saenger, A. Zouni and J. Biesiadka, Nature, 2005, 438, 1040–1044 CrossRef CAS PubMed;
(f) A. Amunts, O. Drory and N. Nelson, Nature, 2007, 447, 58–63 CrossRef CAS PubMed.
-
(a) M. Li, T. J. Neal, G. R. A. Wyllie, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 2010, 49, 8078–8085 CrossRef CAS PubMed;
(b) T. J. Neal, S. J. Kang, I. Turowska-Tyrk, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 2000, 39, 872–880 CrossRef CAS PubMed;
(c) S. Hu and T. G. Spiro, J. Am. Chem. Soc., 1993, 115, 12029–12034 CrossRef CAS;
(d) S. Nakashima, H. Ohya-Nishiguchi, N. Hirota, H. Fujii and I. Morishima, Inorg. Chem., 1990, 29, 5207–5211 CrossRef CAS.
-
(a) M. Li, T. J. Neal, G. R. A. Wyllie, A. G. Oliver, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 2011, 50, 9114–9121 CrossRef CAS PubMed;
(b) A. Takai, C. P. Gros, J. M. Barbe, R. Guilard and S. Fukuzumi, Chem.–Eur. J., 2009, 15, 3110–3122 CrossRef CAS PubMed;
(c) K. E. Brancato-Buentello, S.-J. Kang and W. R. Scheidt, J. Am. Chem. Soc., 1997, 119, 2839–2846 CrossRef CAS;
(d) W. R. Scheidt, K. E. Brancato-Buentello, H. Song, K. V. Reddy and B. Cheng, Inorg. Chem., 1996, 35, 7500–7507 CrossRef CAS.
-
(a) L. M. R. Jensen, R. Sanishvili, V. L. Davidson and C. M. Wilmot, Science, 2010, 327, 1392–1394 CrossRef CAS PubMed;
(b) Y. Wang, M. E. Graichen, A. Liu, A. R. Pearson, C. M. Wilmot and V. L. Davidson, Biochemistry, 2003, 42, 7318–7325 CrossRef CAS PubMed;
(c) W. S. McIntire, D. E. Wemmer, A. Chistoserdov and M. E. Lidstrom, Science, 1991, 252, 817–824 CrossRef CAS PubMed;
(d) Y. Ling, V. L. Davidson and Y. Zhang, J. Phys. Chem. Lett., 2010, 1, 2936–2939 CrossRef CAS PubMed;
(e) X. Li, M. Feng, Y. Wang, H. Tachikawa and V. L. Davidson, Biochemistry, 2006, 45, 821–828 CrossRef CAS PubMed.
-
(a) J. Geng, I. Davis and A. Liu, Angew. Chem., Int. Ed., 2015, 54, 3692–3696 CrossRef CAS PubMed;
(b) J. Geng, K. Dornevil, I. Davis and A. Liu, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9639–9644 CrossRef CAS PubMed;
(c) X. Li, R. Fu, S. Lee, C. Krebs, V. L. Davidson and A. Liu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8597–8600 CrossRef CAS PubMed.
-
(a) G. S. Pulcu, K. E. Frato, R. Gupta, H. R. Hsu, G. A. Levine, M. P. Hendrich and S. J. Elliott, Biochemistry, 2012, 51, 974–985 CrossRef CAS PubMed;
(b) J. Seidel, M. Hoffmann, K. E. Ellis, A. Seidel, T. Spatzal, S. Gerhardt, S. J. Elliott and O. Einsle, Biochemistry, 2012, 51, 2747–2756 CrossRef CAS PubMed;
(c) C. F. Becker, N. J. Watmough and S. J. Elliott, Biochemistry, 2009, 48, 87–95 CrossRef CAS PubMed;
(d) G. W. Pettigrew, A. Echalier and S. R. Pauleta, J. Inorg. Biochem., 2006, 100, 551–567 CrossRef CAS PubMed;
(e) A. Echalier, C. F. Goodhew, G. W. Pettigrew and V. Fülöp, Structure, 2006, 14, 107–117 CrossRef CAS PubMed.
-
(a) M. Son, Y. M. Sung, S. Tokuji, N. Fukui, H. Yorimitsu, A. Osuka and D. Kim, Chem. Commun., 2014, 50, 3078–3080 RSC;
(b) L. Rintoul, S. R. Harper and D. P. Arnold, Phys. Chem. Chem. Phys., 2013, 15, 18951–18964 RSC;
(c) O. Locos, B. Bašić, J. C. McMurtrie, P. Jensen and D. P. Arnold, Chem.–Eur. J., 2012, 18, 5574–5588 CrossRef CAS PubMed;
(d) L. J. Esdaile, P. Jensen, J. C. McMurtrie and D. P. Arnold, Angew. Chem., Int. Ed., 2007, 46, 2090–2093 CrossRef CAS PubMed;
(e) M. J. Frampton, H. Akdas, A. R. Cowley, J. E. Rogers, J. E. Slagle, P. A. Fleitz, M. Drobizhev, A. Rebane and H. L. Anderson, Org. Lett., 2005, 7, 5365–5368 CrossRef CAS PubMed;
(f) M. Chachisvilis, V. S. Chirvony, A. M. Shulga, B. Källebring, S. Larsson and V. Sundström, J. Phys. Chem., 1996, 100, 13857–13866 CrossRef CAS;
(g) M. Chachisvilis, V. S. Chirvony, A. M. Shulga, B. Källebring, S. Larsson and V. Sundström, J. Phys. Chem., 1996, 100, 13867–13873 CrossRef CAS;
(h) V. S.-Y. Lin, S. G. DiMagno and M. J. Therien, Science, 1994, 264, 1105–1111 CAS.
-
(a) D. Sil and S. P. Rath, Dalton Trans., 2015, 44, 16195–16211 RSC;
(b) M. A. Sainna, D. Sil, D. Sahoo, B. Martin, S. P. Rath, P. Comba and S. P. de Visser, Inorg. Chem., 2015, 54, 1919–1930 CrossRef CAS PubMed;
(c) D. Sil, F. S. T. Khan and S. P. Rath, Inorg. Chem., 2014, 53, 11925–11936 CrossRef CAS PubMed;
(d) S. Bhowmik, S. Dey, D. Sahoo and S. P. Rath, Chem.–Eur. J., 2013, 19, 13732–13744 CrossRef CAS PubMed;
(e) S. K. Ghosh, S. Bhowmik, D. Sil and S. P. Rath, Chem.–Eur. J., 2013, 19, 17846–17859 CrossRef CAS PubMed;
(f) S. Bhowmik, S. K. Ghosh and S. P. Rath, Chem.–Eur. J., 2012, 18, 13025–13037 CrossRef CAS PubMed;
(g) S. Bhowmik, S. K. Ghosh and S. P. Rath, Chem. Commun., 2011, 47, 4790–4792 RSC;
(h) S. Bhowmik, D. Sil, R. Patra and S. P. Rath, J. Chem. Sci., 2011, 123, 827–837 CrossRef CAS;
(i) S. K. Ghosh and S. P. Rath, J. Am. Chem. Soc., 2010, 132, 17983–17985 CrossRef CAS PubMed;
(j) S. K. Ghosh, R. Patra and S. P. Rath, Inorg. Chim. Acta, 2010, 363, 2791–2799 CrossRef CAS;
(k) S. K. Ghosh, R. Patra and S. P. Rath, Inorg. Chem., 2010, 49, 3449–3460 CrossRef CAS PubMed;
(l) S. K. Ghosh, R. Patra and S. P. Rath, Inorg. Chem., 2008, 47, 10196–10198 CrossRef CAS PubMed;
(m) S. Dey and S. P. Rath, Dalton Trans., 2014, 43, 2301–2314 RSC.
- T. E. Clement, D. J. Nurco and K. M. Smith, Inorg. Chem., 1998, 37, 1150–1160 CrossRef CAS PubMed.
-
(a) S. V. Lindeman, S. V. Rosokha, D. L. Sun and J. K. Kochi, J. Am. Chem. Soc., 2002, 124, 843–855 CrossRef CAS PubMed;
(b) S. V. Rosokha, D. L. Sun and J. K. Kochi, J. Phys. Chem. A, 2002, 106, 2283–2292 CrossRef CAS;
(c) D. L. Sun, S. V. Rosokha, S. V. Lindeman and J. K. Kochi, J. Am. Chem. Soc., 2003, 125, 15950–15963 CrossRef CAS PubMed;
(d) J. M. Lü, S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2003, 125, 12161–12171 CrossRef PubMed.
- N. J. Hill, W. Levason, M. C. Popham, G. Reid and M. Webster, Polyhedron, 2002, 21, 445–455 CrossRef CAS.
-
(a) R. Weiss, A. Gold and J. Terner, Chem. Rev., 2006, 106, 2550–2579 CrossRef CAS PubMed;
(b) M. Nakamura, Coord. Chem. Rev., 2006, 250, 2271–2294 CrossRef CAS.
-
(a) D. Sahoo, M. G. Quesne, S. P. de Visser and S. P. Rath, Angew. Chem., Int. Ed., 2015, 54, 4796–4800 CrossRef CAS PubMed;
(b) R. Patra, S. Bhowmik, S. K. Ghosh and S. P. Rath, Dalton Trans., 2010, 39, 5795–5806 RSC;
(c) R. Patra, A. Chaudhary, S. K. Ghosh and S. P. Rath, Inorg. Chem., 2010, 49, 2057–2067 CrossRef CAS PubMed;
(d) R. Patra and S. P. Rath, Inorg. Chem. Commun., 2009, 12, 515–519 CrossRef CAS;
(e) R. Patra, A. Chaudhary, S. K. Ghosh and S. P. Rath, Inorg. Chem., 2008, 47, 8324–8335 CrossRef CAS PubMed;
(f) D. Sahoo and S. P. Rath, Chem. Commun., 2015, 51, 16790–16793 RSC.
- C. A. Reed and F. Guiset, J. Am. Chem. Soc., 1996, 118, 3281–3282 CrossRef CAS.
-
(a) D. Wyrzykowski, A. Pattek-Janczyk, T. Maniecki, K. Zaremba and Z. Warnke, Thermochim. Acta, 2006, 443, 72–77 CrossRef CAS;
(b) D. Wyrzykowski, R. Kruszyński, J. Kłak, J. Mroziński and Z. Warnke, Inorg. Chim. Acta, 2007, 360, 3354–3360 CrossRef CAS.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
-
(a) A. Ikezaki, Y. Ohgo and M. Nakamura, Coord. Chem. Rev., 2009, 253, 2056–2069 CrossRef CAS;
(b) S. Kouno, A. Ikezaki, T. Ikeue and M. Nakamura, J. Biol. Inorg. Chem., 2011, 105, 718–721 CAS;
(c)
F. A. Walker, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2010, vol. 6, pp. 1–137 Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS;
(c) P. J. Stevens, F. J. Devlin, C. F. Chabalowski and M. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef.
- S. J. Grimme, Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
-
(a) T. Vangberg, R. Lie and A. Ghosh, J. Am. Chem. Soc., 2002, 124, 8122–8130 CrossRef CAS PubMed;
(b) H. Hirao, S. Shaik and P. M. Kozlowski, J. Phys. Chem. A, 2006, 110, 6091–6099 CrossRef CAS PubMed;
(c) R. J. Cheng, P. Y. Chen, T. Lovell, T. Liu, L. Noodleman and D. A. Case, J. Am. Chem. Soc., 2003, 125, 6774–6783 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Text, figures and tables depicting synthetic procedures, detailed experimental procedures, characterisation data including UV-vis-NIR, FT-IR, DFT. CCDC 1052187 and 1052188. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03120f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence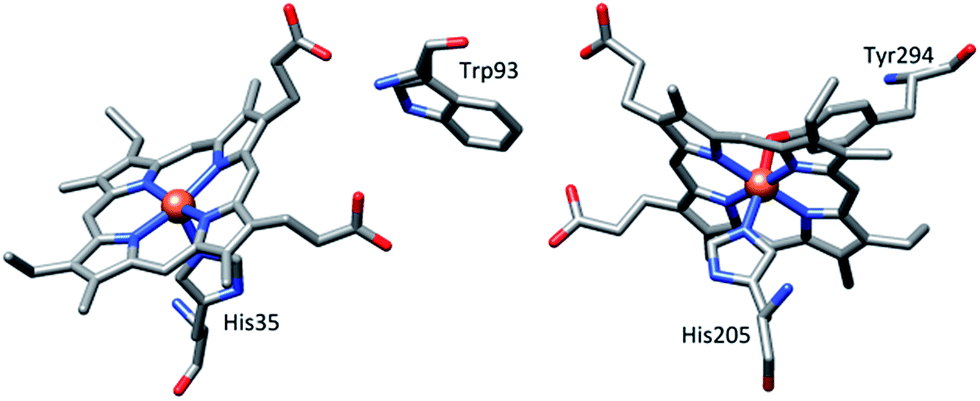




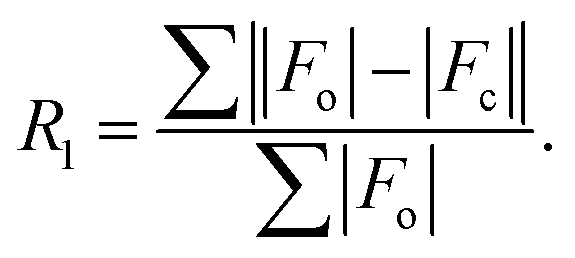 b
b