
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolation, structure and reactivity of a scandium boryl oxycarbene complex†
Baoli
Wang
a,
Xiaohui
Kang
ab,
Masayoshi
Nishiura
a,
Yi
Luo
*b and
Zhaomin
Hou
*ab
aOrganometallic Chemistry Laboratory and RIKEN Center for Sustainable Resource Science, RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. E-mail: houz@riken.jp
bState Key Laboratory of Fine Chemicals, School of Pharmaceutical Science and Technology, Dalian University of Technology, Dalian 116024, China. E-mail: luoyi@dlut.edu.cn
First published on 16th September 2015
Abstract
The reaction of a half-sandwich scandium boryl complex 1 with CO (1 atm) afforded a novel boryl oxycarbene complex 2. The structure of 2 was characterized by 1H, 13C and 11B NMR, X-ray diffraction, and DFT analysis. Further reaction of 2 with CO (1 atm) yielded a phenylamido- and boryl-substituted enediolate complex 3 through C–C bond formation between CO and the carbene unit in 2 and cleavage and rearrangement of the Si–N bond in the silylene-linked Cp–amido ligand. Upon heating, insertion of the carbene atom into a methine C–H bond in the boryl ligand of 2 took place to give an alkoxide complex 4. The reactions of 2 with pyridine and 2-methylpyridine led to insertion of the carbene atom into an ortho-C–H bond of pyridine and into a methyl C–H bond of 2-methylpyridine, respectively. The reaction of 2 with ethylene yielded a borylcyclopropyloxy complex 7 through cycloaddition of the carbene atom to ethylene.
Introduction
Carbon monoxide (CO) is an important C1 building block in chemical industry, as it can be used for the production of synthetic lubrication oils and fuels via Fischer–Tropsch reactions.1 So far, extensive studies on the reaction of CO with transition metal alkyls and hydrides have been reported in relevance to the Fischer–Tropsch process.1–3 The reaction of early transition-metal (including lanthanide and actinide) alkyls (or hydrides) with CO usually gives η2-acyl (or formyl) species that shows carbene-like characteristics in reactivity such as intramolecular 1,2-hydrogen migration, dimerization, and ketene formation.3–5 The analogous reactions of silyl, amido and phosphido complexes of some early transition metals with CO were also reported.6 In spite of extensive studies in this area, structurally characterized carbene-like species (or oxycarbene complexes) remains scarce. In 1980, Marks and co-workers reported that the reaction of a sterically demanding bis(pentamethylcyclopentadienyl) thorium neopentyl complex [(C5Me4)2Th{CH2C(CH3)3}Cl] with 1 equivalent of CO could afford a structurally characterizable oxycarbene complex [(C5Me5)2Th{η2-OCCH2C(CH3)3}Cl].4a This is perhaps the only precedent of a well-defined oxycarbene complex.Metal boryl complexes have received much attention in the last few decades because of their important roles in various chemical transformations.7–9 In this context, the reactions of metal boryl compounds with metal carbonyl complexes were recently reported, such as the nucleophilic addition of [(THF)2Li{B(NDippCH)2}] (Dipp = 2,6-diisopropylphenyl) to [Fe(CO)5] and [Cr(CO)6] as well as the intramolecular migratory addition of a boryl ligand to a carbonyl group in [(CO)4Co{B(NDippCH)2}].10a,b The reaction of metal carbonyl complexes such as K[(η5-C5H5)M(CO)3] (M = Mo, W) with B2(NMe2)2I2 to give oxycarbyne complexes was also reported.10c–e In contrast, the reaction of gaseous CO with metal boryl compounds remains much less extensively explored.9b
In 2011, we reported the reaction of a bis(amidinate)-ligated rare-earth boryl complex [{(Me3SiCH2)C(NiPr)2}2Sc{B(NDippCH)2}] with gaseous CO (1 atm), which afforded a double CO insertion product. This reaction was proposed to proceed through a scandium borylacyl (or carbene) intermediate, but the isolation of such an acyl (or carbene) species was not achieved.9b More recently, we found that a half-sandwich structure unit with a silylene-linked Cp–anilido ligand could serve as a useful platform for the isolation and transformation of rare-earth boryl species such as [Me2Si(C5Me4)(NPh)Sc{B(NDippCH)2}(μ-Cl)Li(THF)3] (1).9c In this paper, we report the isolation and structural characterization of a boryl oxycarbene complex [Me2Si(C5Me4)(NPh)Sc{η2-OCB(NDippCH)2}(THF)] (2) formed by reaction of the half-sandwich scandium boryl complex 1 with CO. The diverse reactivity of the boryl oxycarbene complex 2, such as intra- and intermolecular sp2 and sp3 C–H bond insertion, cyclopropanation with ethylene, and C–C bond formation with another molecule of CO is also described.
Results and discussion
Isolation and structure of a scandium boryl oxycarbene complex
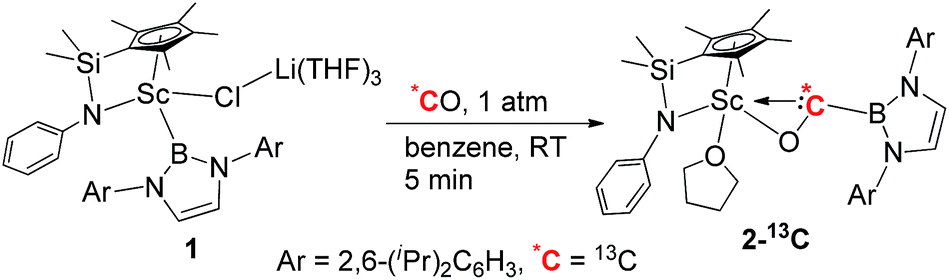
When the half-sandwich scandium boryl (1) was exposed to a CO atmosphere (1 atm) at room temperature in benzene-d6, the insertion of CO into the Sc–boryl bond took place rapidly, selectively yielding the corresponding scandium borylacyl (or oxycarbene) complex 2 in 87% yield as dark blue crystals within 5 min (Scheme 1). The (THF)3LiCl adduct in 1 is dissociated in this reaction. The reaction of 1 with 13C-enriched CO afforded the 13C-labeled analogue 2-13C (eqn (1)). The 13C NMR spectrum of 2 (or 2-13C) in benzene-d6 gave a singlet at δ 427.4 assignable to the CO group. This signal is considerably downfield shifted than those of reported transition-metal acyl complexes (δ 214.4–322.9),11 and even lower than that of the thorium oxycarbene complex [(C5Me5)2Th{η2-OCCH2C(CH3)3}Cl] (δ 360.2),4a clearly demonstrating the presence of a carbene species. The 11B{H} NMR of 2 in benzene-d6 showed a singlet at δ 16.9, which is 6.4 ppm up-field shifted from that of a cobalt Fischer-type boryl oxycarbene complex [(OC)5Cr{C(OEt)B(NDippCH)2}] (δ 23.3).10a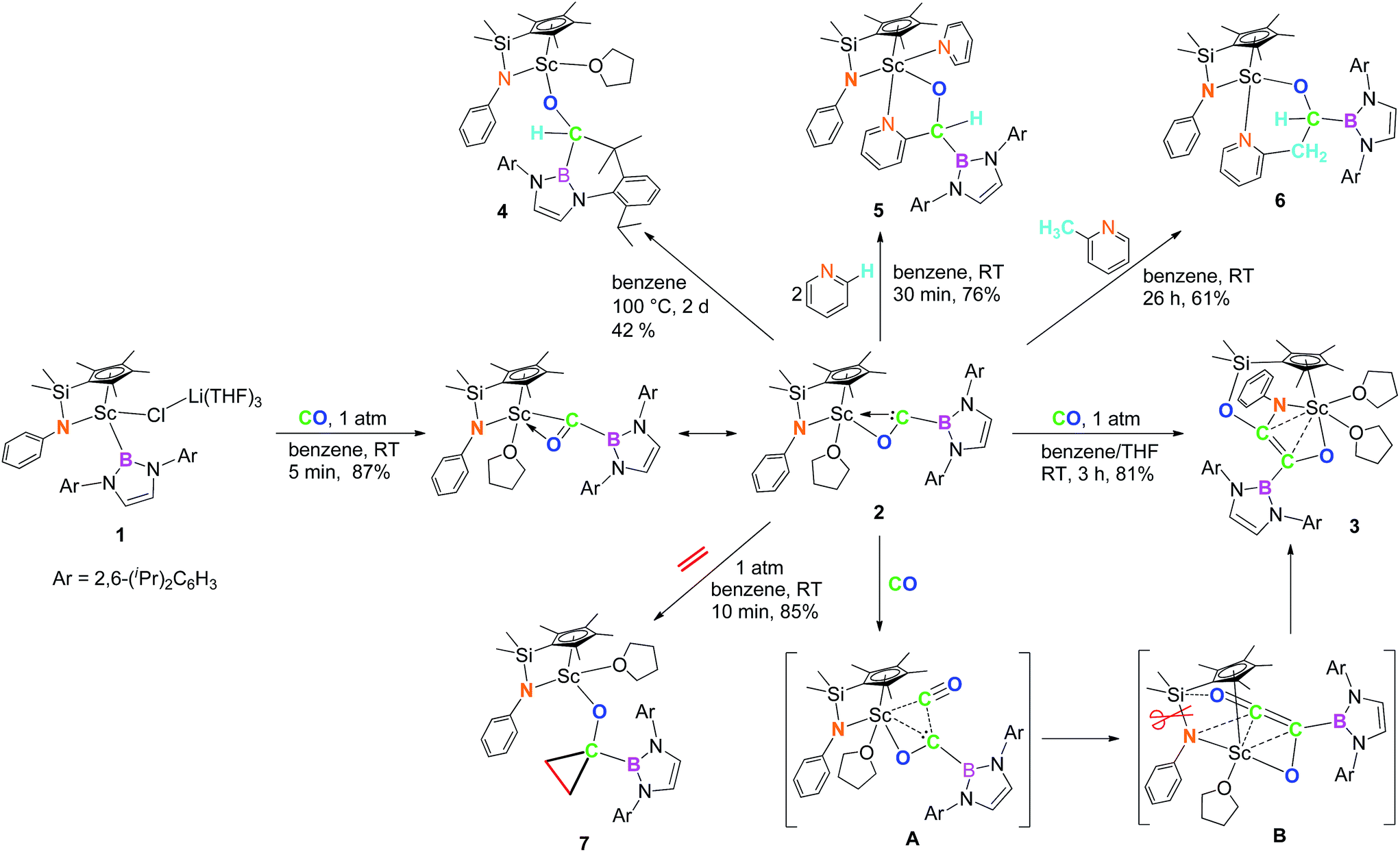 | ||
| Scheme 1 Synthesis and reactivity of the boryl oxycarbene scandium complex 2. | ||
Single crystals of 2 suitable for X-ray diffraction studies were obtained by recrystallization from a mixed hexane–benzene solution at −30 °C. An X-ray diffraction study revealed that the Sc atom is bonded to the CO unit in a η2-fashion (Fig. 1). The Sc–O1 bond distance (2.114(2) Å) is significantly shorter than that of the Sc–C1 bond (2.194(2) Å), similar to what was observed in the thorium oxycarbene complex [(C5Me5)2Th{η2-OCCH2C(CH3)3}Cl] (Th–O 2.37(2) Å, Th–C 2.44(2) Å).4a The C1–O1 bond length (1.266(3) Å) in 2 is longer than that in [(C5Me5)2Th{η2-OCCH2C(CH3)3}Cl] (1.18(3) Å),4a suggesting that the η2-CO unit in 2 is better considered as a carbene moiety than an acyl group.
 | ||
| Fig. 1 ORTEP drawing of 2 with thermal ellipsoids at the 30% level except for the 2,6-(iPr)2C6H3 groups in the boryl unit. Hydrogen atoms and the Me groups on the Cp ring have been omitted for clarity. Selected bond lengths (Å) and angles (°): Sc1–N1 2.130(2), Sc1–O1 2.114(2), Sc1–O2 2.199(2), Sc1–C1 2.194(2), C1–O1 1.266(3), C1–B1 1.577(3); Sc1–C1–O1 69.48(12), Sc1–O1–C1 76.40(12), Sc1–C1–B1 172.11(16). | ||
In order to gain a better understanding about the nature of bonding of the boryl oxycarbene unit in 2, DFT studies at the M06 level were carried out.12 The calculated structure showed excellent agreement with the crystallographic structure, especially for the bond lengths of the Sc1–C1 (2.17 Å vs. 2.194(2) Å) and Sc1–O1 (2.12 Å vs. 2.114(2) Å) bonds.
The C1–O1 stretching frequency of 2 is difficult to assign experimentally due to overlapping bands with those of the boryl moiety. The computed C1–O1 stretching frequencies of 2 (1450 cm−1) and 2-13C (1417 cm−1) are comparable with the experimental IR values of the thorium oxycarbene complex [(C5Me5)2Th{η2-OCCH2C(CH3)3}Cl] (1469 cm−1) and its 13CO analogue (1434 cm−1), which are lower than those of transition-metal acyl complexes (1523–1666 cm−1).11 Further molecular orbital analysis of 2 suggests significant Sc1–O1 and Sc1–C1 bonding interactions with a minor contribution from the B1-2p orbital (see HOMO−1 in Fig. 2). HOMO−4 indicates π–bonding between C1, B1 and two N atoms of the boryl moiety (Fig. 2). The analysis of the donor–acceptor interactions on the basis of second-order perturbation theory13 revealed that the donation of σ(B1–C1) to Sc1 (177.7 kcal mol−1) is significantly stronger than that of σ(O1–C1) (96.0 kcal mol−1), and the donation of lone pair electrons of C1 to a vacant 3d orbital of Sc1 (170.3 kcal mol−1) is higher than that of O1 to Sc1 (83.3 kcal mol−1). In addition, the donation of the lone pair electrons of N2 (88.8 kcal mol−1) and N3 (57.9 kcal mol−1) atoms to B1 was also found in the boryl segment. Therefore, The boryl group plays an important role in stabilizing the Sc–(boryl)carbene moiety.
 | (1) |
 | ||
| Fig. 2 Selected molecular orbitals for 2 (all H atoms are omitted for clarify). | ||
Reaction of carbene with CO
When being exposed to CO (1 atm) in a benzene–THF solution at room temperature for 3 h, 2 was completely consumed, and a phenylamido- and boryl-substituted enediolate complex 3 was obtained in 81% yield as yellow crystals after crystallization from a hexane–benzene solution (Scheme 1). An X-ray crystallographic study established that C–C bond formation between the carbene atom in 2 and CO occurred, accompanied by cleavage of the Si–NPh bond and formation of the O2–Si and C2–NPh bonds (Fig. 3). The resulting C1–C2 bond in 3 could be assigned as a double bond (1.364(5) Å), which shows some interactions with the Sc atom (Sc1–C1 2.519(3) Å, Sc1–C2 2.477(3) Å). The two oxygen atoms (O1 and O2) attached to the C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2 double bond are trans to each other, so are the boryl and PhN groups.
C2 double bond are trans to each other, so are the boryl and PhN groups.
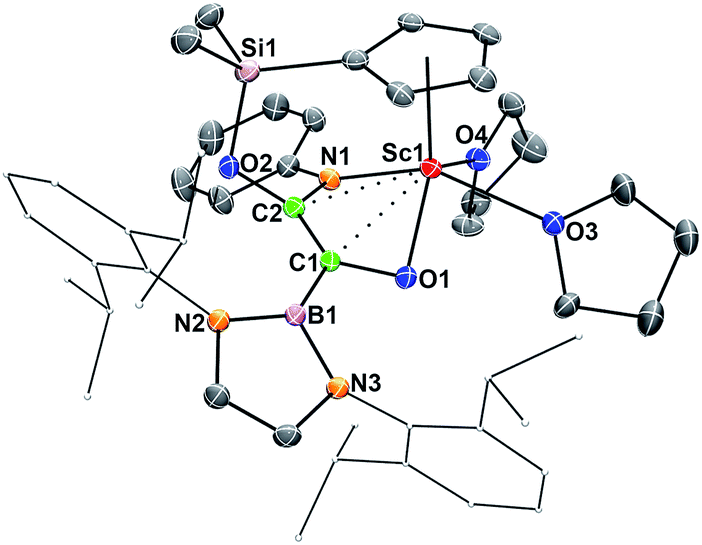 | ||
| Fig. 3 ORTEP drawing of 3 with thermal ellipsoids at the 30% probability except for the 2,6-(iPr)2C6H3 groups in the boryl unit. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å): Sc1–N1 2.086(3), Sc1–O1 2.005(2), Sc1–C1 2.519(3), Sc1–C2 2.477(3), Sc1–O3 2.335(3), Sc1–O4 2.291(2), C1–C2 1.364(5), C1–O1 1.383(4), C2–O2 1.390(4), C2–N1 1.405(4), B1–C1 1.567(5), Si1–O2 1.691(2). | ||
To further confirm the formation of 3, the 13C-enriched complexes 3-13C, 3-13C′ and 3-13C2 were synthesized analogously, as shown in eqn (2)–(4). The 13C NMR spectrum of 3-13C2 in benzene-d6 showed a broad doublet at δ 134.6 and a sharp doublet at δ 136.7 for the OCCO unit, whilst the 13C NMR spectra of 3-13C and 3-13C′ gave a singlet at δ 136.7 and δ 134.7, respectively. The 11B{H} NMR signal of 3 appeared at δ 23.8, which was 6.9 ppm downfield shifted compared to that of 2 (δ 16.9).
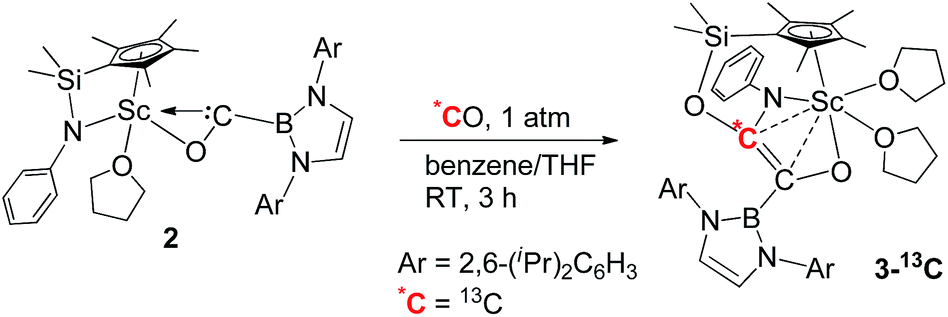 | (2) |
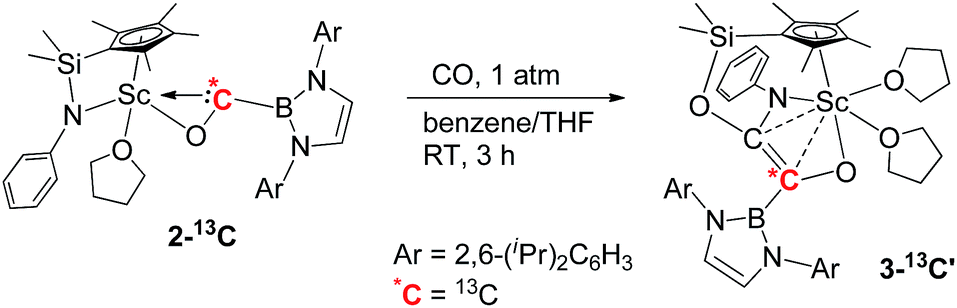 | (3) |
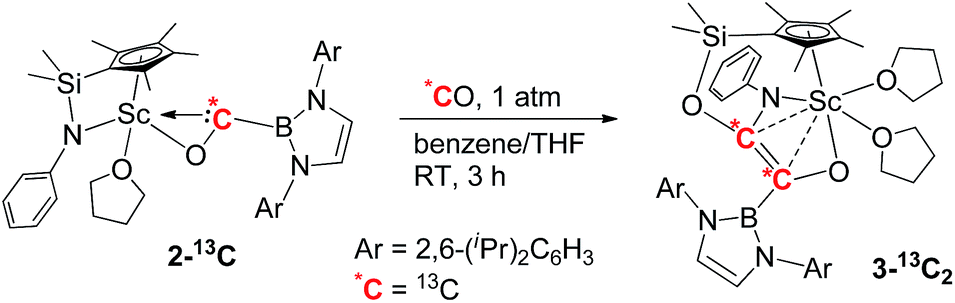 | (4) |
The formation of 3 may be achieved by insertion of CO into the Sc–carbene bond in 2 to give a ketene unit,14a–c followed by cleavage of the Si–N bond in the Cp–anilido ligand and formation of a Si–O bond and an N–C bond between the resulting silyl and PhN groups and the OCCO unit (cf.A and B in Scheme 1). Silylene-linked Cp–amido ligands have been used for the stabilization of various metal complexes, but examples of cleavage of the Si–N bond in these ligands are scarce.15 A possible driving force for the present Si–N cleavage could be the formation of stable Si–O and C–N bonds. A similar silyl migration reaction was also observed previously in the reaction of a bis(amidinate)-ligated scandium boryl complex with CO.9b The reaction of transition-metal acyl complexes [M–C(O)R] with CO were previously reported to give α-ketoacyl species such as [M–C(O)C(O)R].14d The reaction of a metallocene cerium hydride complex Cp′2CeH (Cp′ = 1,2,4-(tBu)3C5H2) with CO was reported to yield an enediolate complex [Cp′2CeOCHCHOCeCp′2] without observation of an isolable mono-CO insertion product.5l
Intra- and intermolecular insertion of carbene into C–H bonds
When complex 2 was heated at 100 °C in benzene for two days, intramolecular insertion of the carbene atom into a methine C–H bond in the boryl ligand took place to give the alkoxide complex 4 (Scheme 1). In this transformation, the Sc–carbene bond is broken, together with formation of a C54–H54 bond and a C54–C41 bond (Fig. 4). The Sc1–O1 bond distance in 4 (1.879(2) Å) is much shorter than that of the Sc–O1(oxycarbene) bond in 2 (2.114(2) Å), whilst the O1–C54 bond distance in 4 (1.424(4) Å) is much longer than that of the O1–C1(oxycarbene) bond in 2 (1.266(3) Å). The C–B bond distance in 4 (1.563(5) Å) is comparable with that in 2 (1.577(3) Å). The newly formed C54–C41 (1.561(5) Å) in 4 is best described as a single bond.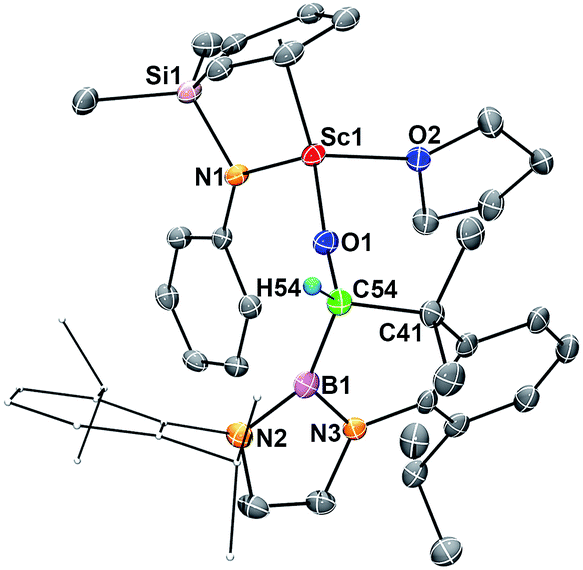 | ||
| Fig. 4 ORTEP drawing of 4 with thermal ellipsoids at the 30% level except for a 2,6-(iPr)2C6H3 group in the boryl unit. Hydrogen atoms (except H54) and the Me groups on the Cp ring have been omitted for clarity. Selected bond lengths (Å) and angles (°): Sc1–O1 1.879(2), Sc1–O2 2.184(2), C54–O1 1.424(4), C41–C54 1.561(5), O1–C54 1.424(4), B1–C54 1.563(5); Sc1–O1–C54 169.5(2), B1–C54–O1 111.3(3), C41–C54–O1 110.3(3). | ||
The hydrogen atom in the newly formed “HC(O)(B)C” unit in 4 gave a singlet at δ 3.91 in the 1H NMR spectrum in benzene-d6. The 11B{H} NMR signal of 4 is located at δ 21.8, which is comparable to that of 3 (δ 23.8) and is 4.9 ppm upfield shifted compared to that of 2 (δ 16.9). The transformation of 2 to 4 could be viewed as a typical reaction (C–H insertion) of a carbene species.16
The reaction of 2 with two equivalents of pyridine in benzene-d6 at room temperature yielded 5 as colourless crystals following recrystallization from hexane–benzene (Scheme 1; also see Fig. S1 in ESI†). In this reaction, the insertion of the carbene atom of 2 into an ortho-C–H bond of one molecule of pyridine took place, while another molecule of pyridine displaced the THF ligand of 2. The 1H NMR spectrum of the newly formed “HC(O)(Py)B” fragment in 5 showed a singlet at δ 5.75 in benzene-d6. The 11B{H} NMR spectrum of 5 showed a broad peak at δ 25.1 which is close to that of 3 (δ 23.8).
When 2 was allowed to react with 2-methylpyridine in benzene-d6 at room temperature for 26 h, the insertion of a sp3 C–H bond in the methyl group of 2-methylpyridine occurred to give complex 6 in 61% isolated yield (Scheme 1 and Fig. 5). The 1H NMR signals of the two protons on the resulting O–CH(B)–CH2C5H4N moiety appeared at δ 2.74 (dd, 10.6 Hz, 14.6 Hz) and δ 2.95 (d, 14.6 Hz), whilst the one of O–CH(B)–CH2C5H4N appeared at δ 4.50 (d, 10.6 Hz).
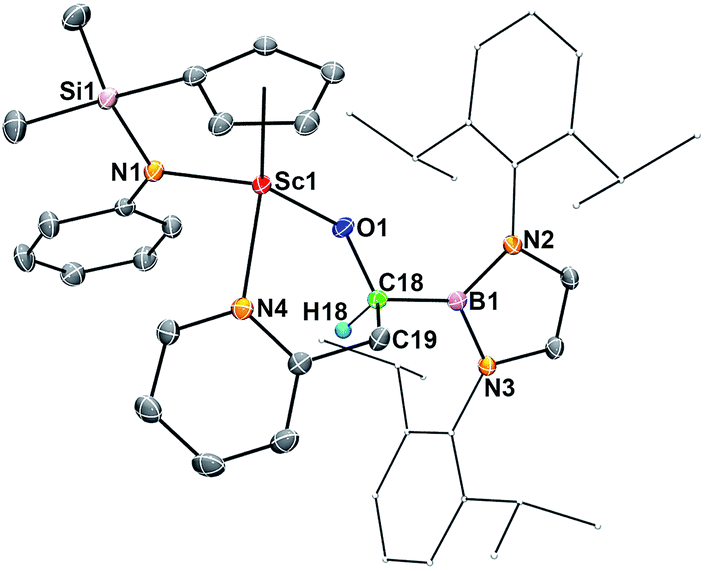 | ||
| Fig. 5 ORTEP drawing of 6 with thermal ellipsoids at the 30% level except for a 2,6-(iPr)2C6H3 group in the boryl unit. Hydrogen atoms (except H18) and the Me groups on the Cp ring have been omitted for clarity. Selected bond lengths (Å): Sc1–O1 1.9175(12), Sc1–N1 2.0947(15), Sc1–N4 2.2811(15), O1–C18 1.412(2), C18–C19 1.550(2), B1–C18 1.595(3). | ||
The molecular structures of 5 and 6 were also confirmed by X-ray crystallographic studies (Fig. S1† for 5 and Fig. 5 for 6), although there were disorder problems in the case of 5. The present C–H bond activation of pyridines by 2 is in contrast with what was observed previously in the reaction of conventional free carbene species with pyridines, in which a stable carbene–pyridine ylide complex was usually formed.17 The reason for the formation of the C–H activation products 5 and 6 is possibly because of facile coordination of the nitrogen atom of a pyridine unit to the electropositive Sc3+ centre, which could easily lead to activation of an ortho-C(sp2)–H or methyl C(sp3)–H bond by the highly active carbene species.18,5gortho-C–H activation of pyridine by a tantalum η2-acyl complex was reported previously.6a
Cyclopropanation of carbene with ethylene
The reaction of 2 with ethylene (1 atm) in benzene-d6 took place rapidly at room temperature, which was accompanied by a colour change from dark blue to colourless to give a borylcyclopropyloxy product 7via the cycloaddition of the carbene atom to ethylene (Scheme 1). The Sc–OCO bond distance in 7 (1.9083(14) Å) (Fig. 6) is comparable with that in 6 (1.9175(12) Å), as are the C–OCO bond distances (7: 1.402(2) Å; 6: 1.412(2) Å). The bond distances and angles of the triangular carbon skeleton in 6 are typical for a cyclopropyl unit (Fig. 6).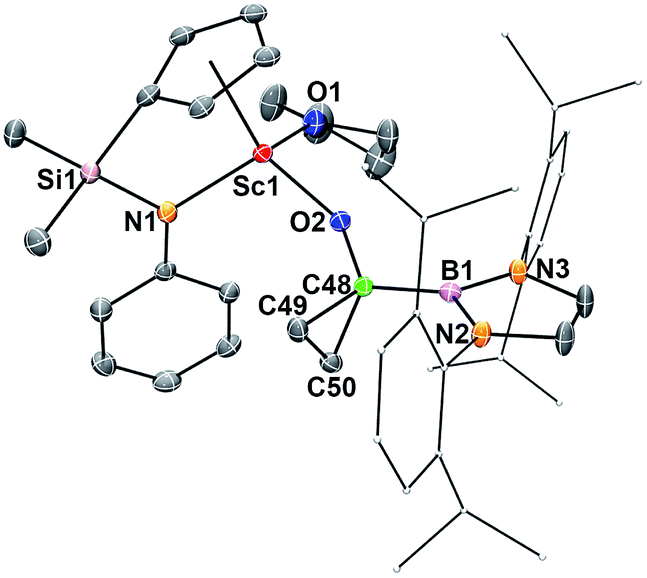 | ||
| Fig. 6 ORTEP drawing of 7 with thermal ellipsoids at the 30% level except for the 2,6-(iPr)2C6H3 groups in the boryl unit. Hydrogen atoms and the Me groups on the Cp ring have been omitted for clarity. Selected bond lengths (Å) and angles (°): Sc1–N1 2.1238(18), Sc1–O1 2.1577(15), Sc1–O2 1.9083(14), O2–C48 1.402(2), C48–C49 1.514(3), C48–C50 1.533(3), C49–C50 1.492(3), C48–B1 1.569(3); C49–C48–C50 58.64(14), C50–C49–C48 61.33(14), C49–C50–C48 60.02(14). | ||
The present formation of 7 represents a rare example of cyclopropanation of ethylene with a carbene species.19 It was known that carbenes could undergo cyclopropanation reactions with alkenes bearing polar substituents (either electron withdrawing or donating) but are usually inert towards simple alkenes such as ethylene.19 The cyclopropanation of ethylene with 2 may be promoted by coordination of ethylene to the electropositive Sc3+ center.20 The cyclopropanation of a cationic iron carbene complex [{Cp(CO)2FeCHC6H5}+{PF6}−] with ethylene was reported previously.19a The reaction of a classical acyl species M–C(O)R with ethylene usually gave a straightforward insertion product formulated as [M–CH2CH2C(O)R].21
Conclusions
We have demonstrated that the reaction of a half-sandwich scandium boryl complex such as 1 with CO (1 atm) can afford a structurally characterizable oxycarbene complex including 2, which represents the first example of a well-defined boryl-substituted oxycarbene species. The scandium boryl oxycarbene complex 2 showed diverse reactivity, such as coupling with CO to form an enediolate complex 3, intramolecular C–H bond activation to give 4, insertion of the carbene atom into an ortho-C–H bond of pyridine or into a methyl C–H bond of 2-methylpyridine, and cyclopropanation with ethylene. The structure and reactivity of the carbene species in 2 are clearly affected by the scandium ion as well as the boryl substituent. Studies on the synthesis and reactions of other rare earth metal boryl complexes are in progress.Acknowledgements
We appreciate financial support through a Grant-in-Aid for Scientific Research (B) (24350030) and a Grant-in-Aid for Scientific Research (S) (26220802) from JSPS and grants from the National Natural Science Foundation of China (21174023, 21137001 and 21429201). We thank Mrs Akiko Karube for micro-elemental analyses.Notes and references
- (a) C. K. Rofer-Depoorter, Chem. Rev., 1981, 81, 447 CrossRef CAS; (b) A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692 CrossRef CAS PubMed.
- (a) K. Tatsumi, A. Nakamura, P. Hofmann, P. Stauffert and R. Hoffmann, J. Am. Chem. Soc., 1985, 107, 4440 CrossRef CAS; (b) W. J. Evans, J. W. Grate and R. J. Doedens, J. Am. Chem. Soc., 1985, 107, 1671 CrossRef CAS; (c) M. D. Curtis, K. B. Shiu and W. M. Butler, J. Am. Chem. Soc., 1986, 108, 1550 CrossRef CAS; (d) J. B. Sheridan, J. R. Johnson, B. M. Handwerker, G. L. Geoffroy and A. L. Rheingold, Organometallics, 1988, 7, 2404 CrossRef; (e) I. M. Arafa, K. Shin and H. M. Goff, J. Am. Chem. Soc., 1988, 110, 5228 CrossRef CAS; (f) J. W. Bruno, M. C. Fermin, S. E. Halfon and G. K. Schulte, J. Am. Chem. Soc., 1989, 111, 8738 CrossRef CAS; (g) J. D. Debad, P. Legzdins, R. J. Batchelor and F. W. B. Einstein, Organometallics, 1993, 12, 2094 CrossRef CAS; (h) M. Gomez, P. Gomez-Sal, G. Jimenez, A. Martin, P. Royo and J. Sanchez-Nieves, Organometallics, 1996, 15, 3579 CrossRef CAS; (i) H. Shen and R. F. Jordan, Organometallics, 2003, 22, 2080 CrossRef CAS; (j) W. J. Evans, S. A. Kozimor, G. W. Nyce and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 13831 CrossRef CAS PubMed; (k) M. L. Scheuermann, A. L. Rheingold and B. S. Williams, Organometallics, 2009, 28, 1613 CrossRef CAS; (l) O. Benaud, J. C. Berthet, P. Thuery and M. Ephritikhine, Inorg. Chem., 2010, 49, 8117 CrossRef CAS PubMed; (m) J. L. Smeltz, P. D. Boyle and E. A. Ison, J. Am. Chem. Soc., 2011, 133, 13288 CrossRef CAS PubMed.
- (a) P. T. Wolczanski and J. E. Bercaw, Acc. Chem. Res., 1980, 13, 121 CrossRef CAS; (b) T. J. Marks, Science, 1982, 217, 989 CAS; (c) L. D. Durfee and I. P. Rothwell, Chem. Rev., 1988, 88, 1059 CrossRef CAS; (d) B. E. Kahn and R. D. Rieke, Chem. Rev., 1988, 88, 733 CrossRef CAS.
- (a) P. J. Fagan, J. M. Manriquez, T. J. Marks, V. W. Day, S. H. Vollmer and C. S. Day, J. Am. Chem. Soc., 1980, 102, 5393 CrossRef CAS; (b) E. A. Maatta and T. J. Marks, J. Am. Chem. Soc., 1981, 103, 3576 CrossRef CAS; (c) P. J. Fagan, K. G. Moloy and T. J. Marks, J. Am. Chem. Soc., 1981, 103, 6959 CrossRef CAS; (d) P. J. Fagan, J. M. Manriquez, S. H. Vollmer, C. S. Day, V. W. Day and T. J. Marks, J. Am. Chem. Soc., 1981, 103, 2206 CrossRef CAS; (e) K. G. Moloy, T. J. Marks and V. W. Day, J. Am. Chem. Soc., 1983, 105, 5696 CrossRef CAS; (f) K. G. Moloy and T. J. Marks, J. Am. Chem. Soc., 1984, 106, 7051 CrossRef CAS; (g) K. G. Moloy, P. J. Fagan, J. M. Manriquez and T. J. Marks, J. Am. Chem. Soc., 1986, 108, 56 CrossRef CAS.
- (a) W. J. Evans, A. L. Wayda, W. E. Hunter and J. L. Atwood, J. Chem. Soc., Chem. Commun., 1981, 706 RSC; (b) K. Tatsumi, A. Nakamura, P. Hofmann, R. Hoffmann, K. G. Moloy and T. J. Marks, J. Am. Chem. Soc., 1986, 108, 4467 CrossRef CAS; (c) W. J. Evans and D. K. Drummond, J. Am. Chem. Soc., 1988, 110, 2772 CrossRef CAS; (d) W. J. Evans, K. J. Forrestal and J. W. Ziller, J. Am. Chem. Soc., 1995, 117, 12635 CrossRef CAS; (e) C. J. Schaverien, N. Meijboom and A. G. Orpen, J. Chem. Soc., Chem. Commun., 1992, 124 RSC; (f) B. K. Campion, R. H. Heyn and T. D. Tilley, Organometallics, 1993, 12, 2584 CrossRef CAS; (g) B. J. Deelman, W. M. Stevels, J. H. Teuben, M. T. Lakin and A. L. Spek, Organometallics, 1994, 13, 3881 CrossRef CAS; (h) L. Lee, D. J. Berg, F. W. Einstein and R. J. Batchelor, Organometallics, 1997, 16, 1819 CrossRef CAS; (i) G. M. Ferrence, R. McDonald and J. Takats, Angew. Chem., Int. Ed., 1999, 38, 2233 CrossRef CAS; (j) T. M. Cameron, J. C. Gordon, B. L. Scott and W. Tumas, Chem. Commun., 2004, 1398 RSC; (k) T. Shima and Z. Hou, J. Am. Chem. Soc., 2006, 128, 8124 CrossRef CAS PubMed; (l) E. L. Werkema, L. Maron, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2007, 129, 6662 CrossRef CAS.
- (a) J. Arnold and T. D. Tilley, J. Am. Chem. Soc., 1985, 107, 6409 CrossRef CAS; (b) T. D. Tilley, J. Am. Chem. Soc., 1985, 107, 4084 CrossRef CAS; (c) J. Arnold, T. D. Tilley, A. L. Rheingold, S. J. Geib and A. M. Arif, J. Am. Chem. Soc., 1989, 111, 149 CrossRef CAS; (d) P. J. Fagan, J. M. Manriquez, S. H. Vollmer, C. S. Day, V. W. Day and T. J. Marks, J. Am. Chem. Soc., 1981, 103, 2206 CrossRef CAS; (e) P. L. Arnold, Z. R. Turner, R. M. Bellabarba and R. P. Tooze, Chem. Sci., 2011, 2, 77 RSC; (f) M. Porchia, N. Brianese, F. Ossola, G. Rossetto and P. Zanella, J. Chem. Soc., Dalton Trans., 1987, 691 RSC.
- (a) G. J. Irvine, M. J. Lesley, T. B. Marder, N. C. Norman, C. R. Rice, E. G. Robins, W. R. Roper, G. R. Whittell and L. J. Wright, Chem. Rev., 1998, 98, 2685 CrossRef CAS PubMed; (b) S. Aldridge and D. L. Coombs, Coord. Chem. Rev., 2004, 248, 535 CrossRef CAS; (c) L. Dang, Z. Lin and T. B. Marder, Chem. Commun., 2009, 3987 RSC; (d) H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924 CrossRef CAS PubMed; (e) I. A. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; (f) J. Cid, H. Gulyas, J. J. Carbo and E. Fernandez, Chem. Soc. Rev., 2012, 41, 3558 RSC; (g) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864 CrossRef CAS PubMed; (h) J. Cid, J. J. Carbo and E. Fernandez, Chem.–Eur. J., 2012, 18, 12794 CrossRef CAS PubMed.
- (a) Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113 CrossRef CAS PubMed; (b) T. B. Marder, Science, 2006, 314, 69 CrossRef CAS PubMed; (c) H. Braunschweig, Angew. Chem., Int. Ed., 2007, 46, 1946 CrossRef CAS PubMed; (d) M. Yamashita, Y. Suzuki, Y. Segawa and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 9570 CrossRef CAS PubMed; (e) Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069 CrossRef CAS PubMed; (f) L. Weber, Eur. J. Inorg. Chem., 2012, 5595 CrossRef CAS; (g) A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500 CrossRef CAS PubMed; (h) N. Dettenrieder, H. M. Dietrich, C. Schadle, C. M. Mossmer, K. W. Tornroos and R. Anwander, Angew. Chem., Int. Ed., 2012, 51, 4461 CrossRef CAS PubMed; (i) N. Dettenrieder, C. Schadle, C. Mossmer, P. Sirsch and R. Anwander, J. Am. Chem. Soc., 2014, 136, 886 CrossRef CAS PubMed; (j) R. T. Baker, D. W. Ovenall, J. C. Calabrese, S. A. Westcott, N. J. Taylor, I. D. Williams and T. B. Marder, J. Am. Chem. Soc., 1990, 112, 9399 CrossRef CAS; (k) K. M. Waltz and J. F. Hartwig, Science, 1997, 277, 211 CrossRef CAS; (l) Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2007, 46, 6710 CrossRef CAS PubMed; (m) T. Kajiwara, T. Terabayashi, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2008, 47, 6606 CrossRef CAS PubMed; (n) Y. Segawa, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 9201 CrossRef CAS PubMed; (o) T. Terabayashi, T. Kajiwara, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14162 CrossRef CAS PubMed; (p) Y. Okuno, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2011, 50, 920 CrossRef CAS PubMed.
- (a) L. M. Saleh, K. H. Birjkumar, A. V. Protchenko, A. D. Schwarz, S. Aldridge, C. Jones, N. Kaltsoyannis and P. Mountford, J. Am. Chem. Soc., 2011, 133, 3836 CrossRef CAS PubMed; (b) S. Li, J. Cheng, Y. Chen, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 6360 CrossRef CAS PubMed; (c) B. Wang, M. Nishiura, J. Cheng and Z. Hou, Dalton Trans., 2014, 14215 RSC.
- (a) R. Frank, J. Howell, R. Tirfoin, D. Dange, C. Jones, D. M. P. Mingos and S. Aldridge, J. Am. Chem. Soc., 2014, 136, 15730 CrossRef CAS PubMed; (b) R. Frank, J. Howell, J. Campos, R. Tirfoin, N. Phillips, S. Zahn, D. M. P. Mingos and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 9586 CrossRef CAS PubMed; (c) H. Braunschweig, M. Koster and K. W. Klinkhammer, Angew. Chem., Int. Ed., 1999, 38, 2229 CrossRef CAS; (d) H. Braunschweig, C. Kollann, M. Koster, U. Englert and M. Muller, Eur. J. Inorg. Chem., 1999, 2277 CrossRef CAS; (e) H. Braunschweig, K. W. Klinkhammer, M. Koster and K. Radacki, Chem.–Eur. J., 2003, 9, 1303 CrossRef CAS PubMed.
- (a) E. J. M. Deboer, L. C. T. Cate, A. G. J. Staring and J. H. Teuben, J. Organomet. Chem., 1979, 181, 61 CrossRef CAS; (b) J. Jeffery, M. F. Lappert, N. T. Luongthi, M. Webb, J. L. Atwood and W. E. Hunter, J. Chem. Soc., Dalton Trans., 1981, 1593 RSC; (c) Z. Y. Guo, D. C. Swenson, A. S. Guram and R. F. Jordan, Organometallics, 1994, 13, 766 CrossRef CAS; (d) J. Vicente, M. T. Chicote, A. J. Martinez-Martinez, A. Abellan-Lopez and D. Bautista, Organometallics, 2010, 29, 5693 CrossRef CAS; (e) D. F. Chen, R. Scopelliti and X. L. Hu, J. Am. Chem. Soc., 2010, 132, 928 CrossRef CAS PubMed.
- DFT-M06: Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- F. Lavigne, E. Maerten, G. Alcaraz, N. Saffon-Merceron, C. Acosta-Silva, V. Branchadell and A. Baceiredo, J. Am. Chem. Soc., 2010, 132, 8864 CrossRef CAS PubMed.
- (a) W. A. Herrmann, J. Plank, M. L. Ziegler and K. Weidenhammer, J. Am. Chem. Soc., 1979, 101, 3133 CrossRef CAS; (b) W. Sander, G. Bucher and S. Wierlacher, Chem. Rev., 1993, 93, 1583 CrossRef CAS; (c) V. Lavallo, Y. Canac, B. Donnadieu, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed., 2006, 45, 3488 CrossRef CAS PubMed; (d) G. L. Geoffroy, J. B. Sheridan, S. L. Bassner and C. Kelley, Pure Appl. Chem., 1989, 61, 1723 CrossRef CAS.
- The Si–N bond cleavage of [Me2Si(C5Me4)(NPh)M(NMe2)3] (M = Nb, Ta) by photo-irradiation was reported: W. A. Herrmann and W. Baratta, J. Organomet. Chem., 1996, 506, 357 CrossRef CAS.
- (a) S. K. Zhao, C. Knors and P. Helquist, J. Am. Chem. Soc., 1989, 111, 8527 CrossRef CAS; (b) H. M. L. Davies and T. Hansen, J. Am. Chem. Soc., 1997, 119, 9075 CrossRef CAS; (c) J. L. Wood, B. M. Stoltz, H. J. Dietrich, D. A. Pflum and D. T. Petsch, J. Am. Chem. Soc., 1997, 119, 9641 CrossRef CAS; (d) R. D. Haszeldine and J. G. Speight, Chem. Commun., 1967, 995 RSC; (e) R. N. Haszeldine, R. Rowland, J. G. Speight and A. E. Tipping, J. Chem. Soc., Perkin Trans. 1, 1979, 1943 RSC.
- (a) J. E. Jackson, N. Soundararajan, M. S. Platz and M. T. H. Liu, J. Am. Chem. Soc., 1988, 110, 5595 CrossRef CAS; (b) C. S. Ge, E. G. Jang, E. A. Jefferson, W. G. Liu, R. A. Moss, J. Wlostowska and S. Xue, J. Chem. Soc., Chem. Commun., 1994, 1479 RSC; (c) A. Kuhn, C. Plug and C. Wentrup, J. Am. Chem. Soc., 2000, 122, 1945 CrossRef CAS.
- For examples of C–H activation of pyridines by rare-earth alkyl complexes, see: (a) B. Guan and Z. Hou, J. Am. Chem. Soc., 2011, 133, 18086 CrossRef CAS PubMed; (b) B. Guan, B. Wang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2013, 52, 4418 CrossRef CAS PubMed; (c) G. Song, W. O and Z. Hou, J. Am. Chem. Soc., 2014, 136, 12209 CrossRef CAS PubMed.
- (a) M. Brookhart, M. B. Humphrey, H. J. Kratzer and G. O. Nelson, J. Am. Chem. Soc., 1980, 102, 7802 CrossRef CAS; (b) M. Brookhart, D. Timmers, J. R. Tucker, G. D. Williams, G. R. Husk, H. Brunner and B. Hammer, J. Am. Chem. Soc., 1983, 105, 6721 CrossRef CAS; (c) M. Brookhart and W. B. Studabaker, Chem. Rev., 1987, 87, 411 CrossRef CAS; (d) D. F. Harvey and D. M. Sigano, Chem. Rev., 1996, 96, 271 CrossRef CAS PubMed; (e) C. K. Murray, D. C. Yang and W. D. Wulff, J. Am. Chem. Soc., 1990, 112, 5660 CrossRef CAS; (f) J. Pfeiffer and K. H. Dotz, Angew. Chem., Int. Ed., 1997, 36, 2828 CrossRef CAS; (g) J. Pfeiffer, M. Nieger and K. H. Dotz, Eur. J. Org. Chem., 1998, 1011 CrossRef CAS.
- For examples of reactions of alkenes with scandium alkyls, see: (a) M. Nishiura and Z. Hou, Nat. Chem., 2010, 2, 257 CrossRef CAS PubMed; (b) M. Nishiura, F. Guo and Z. Hou, Acc. Chem. Res., 2015, 48, 2209 CrossRef CAS PubMed.
- P. Braunstein, C. Frison and X. Morise, Angew. Chem., Int. Ed., 2000, 39, 2867 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization of compounds 2–7. CCDC 981561–981563, 1042268 and 1042269. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03138a |
| This journal is © The Royal Society of Chemistry 2016 |