
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-catalyzed trifluoromethylthiolation of Csp2–O bonds†
Alexander B.
Dürr‡
,
Guoyin
Yin‡
,
Indrek
Kalvet
,
François
Napoly
and
Franziska
Schoenebeck
*
RWTH Aachen University, Institute of Organic Chemistry, Landoltweg 1, 52074 Aachen, Germany. E-mail: franziska.schoenebeck@rwth-Aachen.de
First published on 30th October 2015
Abstract
While nickel catalysts have previously been shown to activate even the least reactive Csp2–O bonds, i.e. aryl ethers, in the context of C–C bond formation, little is known about the reactivity limits and molecular requirements for the introduction of valuable functional groups under homogeneous nickel catalysis. We identified that due to the high reactivity of Ni-catalysts, they are also prone to react with existing or installed functional groups, which ultimately causes catalyst deactivation. The scope of the Ni-catalyzed coupling protocol will therefore be dictated by the reactivity of the functional groups towards the catalyst. Herein, we showed that the application of computational tools allowed the identification of matching functional groups in terms of suitable leaving groups and tolerated functional groups. This allowed for the development of the first efficient protocol to trifluoromethylthiolate Csp2–O bonds, giving the mild and operationally simple C–SCF3 coupling of a range of aryl, vinyl triflates and nonaflates. The novel methodology was also applied to biologically active and pharmaceutical relevant targets, showcasing its robustness and wide applicability.
Introduction
Owing to nickel's non-precious nature and its higher reactivity in the first elementary step of cross coupling cycles, i.e. the oxidative addition, the field of homogeneous Ni-catalysis has long been considered promising, yet also challenging.1 This is because difficulties have frequently been encountered in taming nickel's reactive nature to achieve desired selectivities and scope.2 In spite of that, in recent years there has been impressive progress in the activation of the least reactive bonds, such as aromatic ethers or aryl fluorides.3 However, these milestones typically featured the conversion of C–OMe (or C–F4) to inert C–C or C–H bonds.5,6By contrast, less is known about the reactivity limits and molecular requirements for the installation of potentially reactive functional groups. We therefore envisioned that a computationally assisted development7 of an unprecedented Ni-catalyzed protocol for C-heteroatom bond formation presents an ideal challenge to (i) identify the general reactivity requirements for efficient Ni-catalysis and (ii) demonstrate the viability of applying computational tools to assess substrate scope.
As a suitable test case, we focused on the nickel-catalyzed trifluoromethylthiolation of Csp2–O bonds.8
The SCF3 group makes molecules more lipophilic, increasing their membrane permeability and bioavailability.9 These properties are of considerable interest in a pharmaceutical and agrochemical context. Consequently, numerous efforts have been undertaken to synthesize aryltrifluoromethyl sulfides.10,11 In particular the direct catalytic introduction of SCF3 is an attractive approach. While aryl halides12 or boronic acids13 have successfully been converted to C–SCF3via metal catalyzed cross-coupling strategies or oxidative protocols,14 to date, there is no report of a direct and catalytic trifluoromethylthiolation of Csp2–O bonds.
Results and discussion
Given the widespread abundance of phenols, the trifluoromethylthiolation of phenol derivatives would be highly attractive for synthetic diversity. In this context, the scope could in principle range from more activated derivatives (e.g. aryl triflates) to the least reactive derivatives, i.e. aryl ethers which are present in biomass feedstocks (such as lignin15).6 However, while Ni-catalysis has recently been successfully utilized to activate aromatic ethers,3 we hypothesized that there might be a fundamental reactivity conflict in introducing SCF3: the created SCF3-product would be expected to be inherently more reactive towards oxidative addition16 which may impede further transformation.To test this, we subjected Ni(cod)2/dppf to PhSCF31 (see Fig. 1). We recently showed that this system triggers the mild trifluoromethylthiolation of aryl chlorides, proceeding via Ni(0)/Ni(II) catalysis with [(dppf)Ni(cod)] formed as the active catalyst.12e In accordance with our hypothesis, the reaction of the [Ni(0)] catalyst with PhSCF3 is indeed seen, even under mild reaction conditions (45 °C), as judged by 31P-NMR spectroscopic analysis. A complete disappearance of the characteristic 31P-NMR singlet signal of [(dppf)Ni(0)(cod)] (33.8 ppm)12e occurred, and the formation of a new species was seen that appears as two triplets at 30.8 ppm (with J = 23.0 Hz) and at 22.1 ppm (with J = 37.6 Hz) by 31P-NMR spectroscopic analysis (see Fig. 1). While our efforts to structurally characterize the latter by X-ray crystallography have so far been unsuccessful, the formed species clearly constitutes a catalyst deactivation product. The subjection of this species as a catalyst (or also stoichiometrically) in the trifluoromethylthiolation of aryl chlorides did not yield ArSCF3. This indicates that oxidative addition by a [Ni(0)] catalyst to the product is facile and eventually leads to catalytically inactive species. To achieve productive catalysis and high overall conversion, it is therefore of utmost importance to avoid this deactivation process.
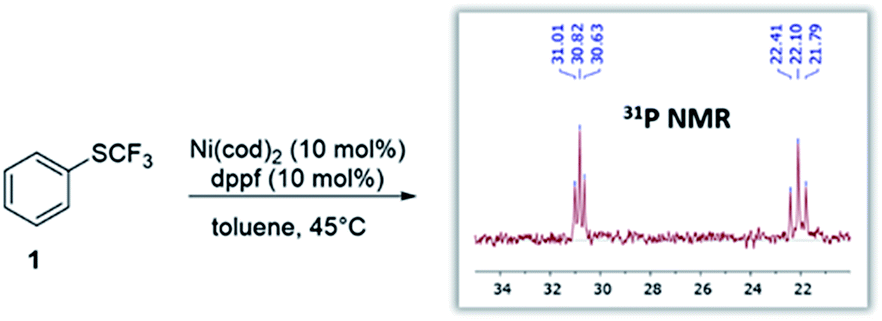 | ||
| Fig. 1 Reaction of catalyst [(dppf)Ni(cod)] with the desired product (ArSCF3) leads to catalyst deactivation. | ||
Our computational assessment17 of the oxidative addition of [(dppf)Ni(cod)] to Ph-SCF31 suggests an activation free energy barrier of ΔG‡ = 19.2 kcal mol−1, and it uses the M06L method with a CPCM solvation model to account for toluene and the mixed 6-311++G(d,p) and LANL2DZ (for Ni, Fe) basis set.17,18
This value now sets the bar for the possible reaction scope. The ‘to-be-transformed’ bond must show a barrier lower than 19.2 kcal mol−1 to avoid catalyst loss via an unproductive reaction with the product (ArSCF3).
Identification of suitable leaving groups – computational assessment & experimental tests
We subsequently undertook computational studies to identify matching leaving groups ‘OR’ (Fig. 2) that would show the desired greater reactivity than the Csp2–SCF3 bond. For the cleavage of the C–O bonds, mechanistic support for Ni(0)/Ni(II)5i,6 and also Ni(I)-catalysis19 has previously been reported. However, on the basis of our previous mechanistic study12e and the observation that (dppf)Ni(I)Cl is ineffective as a catalyst in C–SCF3 bond formation,12e,20 as a first approximation, we calculated the activation barrier of oxidative addition using [(dppf)Ni(0)(cod)] to a range of phenol derivatives (Ph–OR), with R = alkyl (ether), R′C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (pivalate), SO2R′′ (sulfonic esters). Fig. 2 presents the results. This computational assessment suggests that in the context of C–O to C–SCF3 conversion, the inherently high reactivity of C–SCF3 only allows for triflate precursors as suitable starting materials. Alternative C–O leaving groups that have previously been employed in the Ni-catalyzed construction of inert C–C bonds, such as aryl ethers (OMe), mesylates (OMs), tosylates (OTs) or pivalates (OPiv)3,6 are predicted to be incompatible with Ni(0)-catalyzed trifluoromethylthiolation, as they would generally be less reactive than Ar–SCF3, hence favoring catalyst deactivation via reaction with the product.21
O (pivalate), SO2R′′ (sulfonic esters). Fig. 2 presents the results. This computational assessment suggests that in the context of C–O to C–SCF3 conversion, the inherently high reactivity of C–SCF3 only allows for triflate precursors as suitable starting materials. Alternative C–O leaving groups that have previously been employed in the Ni-catalyzed construction of inert C–C bonds, such as aryl ethers (OMe), mesylates (OMs), tosylates (OTs) or pivalates (OPiv)3,6 are predicted to be incompatible with Ni(0)-catalyzed trifluoromethylthiolation, as they would generally be less reactive than Ar–SCF3, hence favoring catalyst deactivation via reaction with the product.21
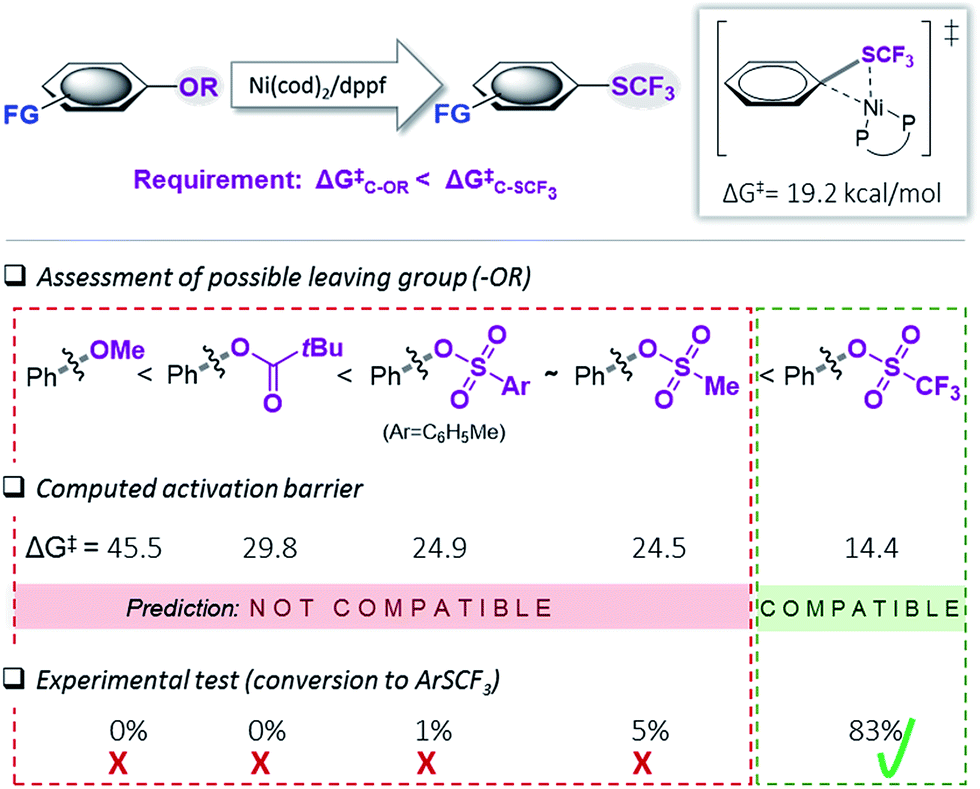 | ||
| Fig. 2 Calculated free energy barrier (ΔG‡) for the oxidative addition of [(dppf)Ni(0)(cod)] to various Ph–OR and the testing of the prediction. Free energies in kcal mol−1, calculated at CPCM (toluene) M06L/6-311++G(d,p) with LANL2DZ (for Ni, Fe).17 | ||
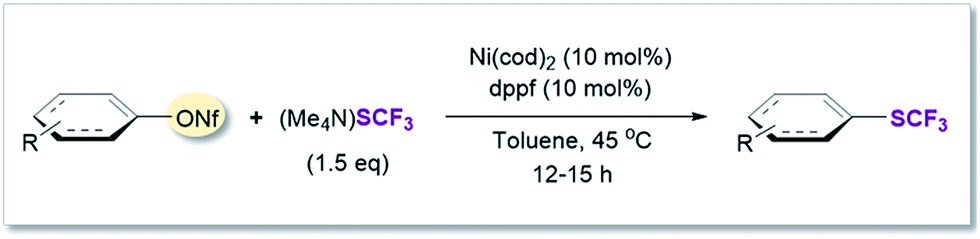
To experimentally test this computationally predicted trend, we subjected Ni(cod)2/dppf along with the easily accessible SCF3-source (Me4N)SCF3 to Ar–OR derivatives (in toluene at 45 °C), ranging from the predicted low (aryl ether) to high (aryl triflate) reactivity (Fig. 2). In accordance with expectations, at best, a low conversion was seen for phenyl mesylates (5%), tosylates (1%) or pivalates (0%). In stark contrast, phenyl triflate showed excellent conversion to PhSCF3 (83%).
We additionally followed the conversion ArOTf → ArSCF3 with ReactIR®. This analysis showed that the transformation was rapid, being essentially complete in 1.5 h with only little increase in conversion over the subsequent hours (see ESI, Fig. S1†). We also analyzed the reactions of those substrates that showed little conversion (≤5%), i.e. ArOMs and ArOTs, by 31P-NMR spectroscopic analyses. We observed that essentially all of the [Ni(0)] catalyst had transformed to the catalytically inactive species described in Fig. 1 within 3 h reaction time. This clearly highlights that while [Ni(0)] is in fact capable of reacting with Ph–OMs or –OTs, the catalyst is rapidly consumed as soon as some of the more reactive PhSCF3 molecules are generated. This corroborates with the strict requirement of suitably matching functionality and tailored reactivity progression from a “more” to “less reactive” functionality.
Computational assessment of functional group tolerance
We subsequently set out to test for the generality of the identified Ni-catalyzed trifluoromethylthiolation of activated C–O bonds and computationally assess the functional group (FG) tolerance (see Fig. 3). As we determined a barrier of ΔG‡ = 14.4 kcal mol−1 for the oxidative addition of [(dppf)Ni(0)(cod)] to Ph–OTf, all additional functional groups (FG) in the substrates will only be compatible if the reactivity of the C–FG bond is lower than that of Ph–OTf.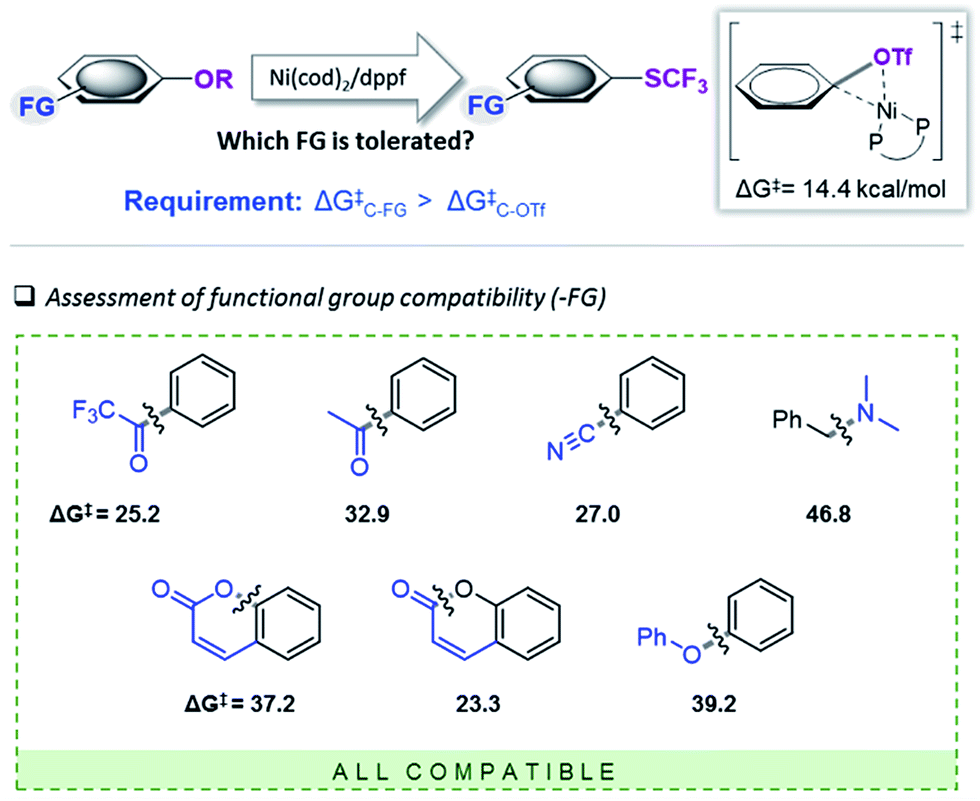 | ||
| Fig. 3 Computational scoping. Activation free energies (in kcal mol−1) calculated at CPCM (toluene) M06L/6-311++G(d,p) & LANL2DZ (for Ni, Fe)17 for the addition of [(dppf)Ni(cod)]. | ||
The computational results depicted in Fig. 3 suggest a tolerance of the protocol to ketone functional groups, C–C or benzylic C–O bonds. In all cases, the requirement of ΔG‡C–FG > 14.4 kcal mol−1 is fulfilled. Even aromatic C–CN bonds that were previously shown to be reactive under Ni-catalysis conditions22 are predicted to be compatible.
SCF3-coupling of aryl triflates
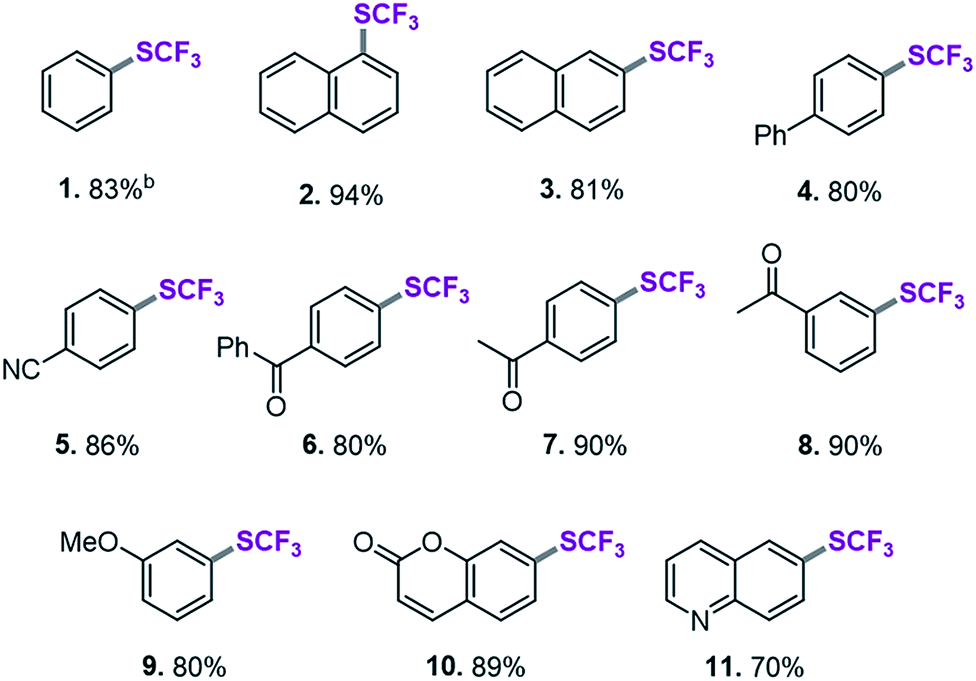
On the basis of this computationally guided substrate scope, we subjected a range of aryl triflates to standard catalysis conditions. Table 1 presents the results. A number of aryl- and heteroaryl triflates were coupled in good to excellent yields. The transformation was compatible with ketone (6, 7 and 8, Table 1), ether (9) and cyano (5) functional groups. Two heterocyclic examples (10, 11) were also trifluoromethylthiolated in good yields (see Table 1).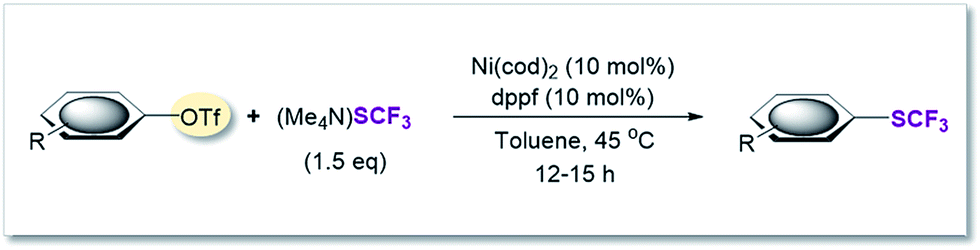
We next searched for bioactive molecules of greater complexity that would fulfil our reactivity requirements and show compatibility with the computationally predicted scope. Estrone (an estrogenic hormone), 6-hydroxy flavanone (a plant secondary metabolite used inter alia as an antioxidant) and δ-tocopherol (vitamin E) show an excellent functional group match, containing predominantly ketone and benzylic C–O bonds that are predicted to be less reactive than C–OTf and C–SCF3. Trifluoromethylthiolation was successfully accomplished in 62–96% yield, highlighting the potential of this method for pharmaceutical applications (see Scheme 1).
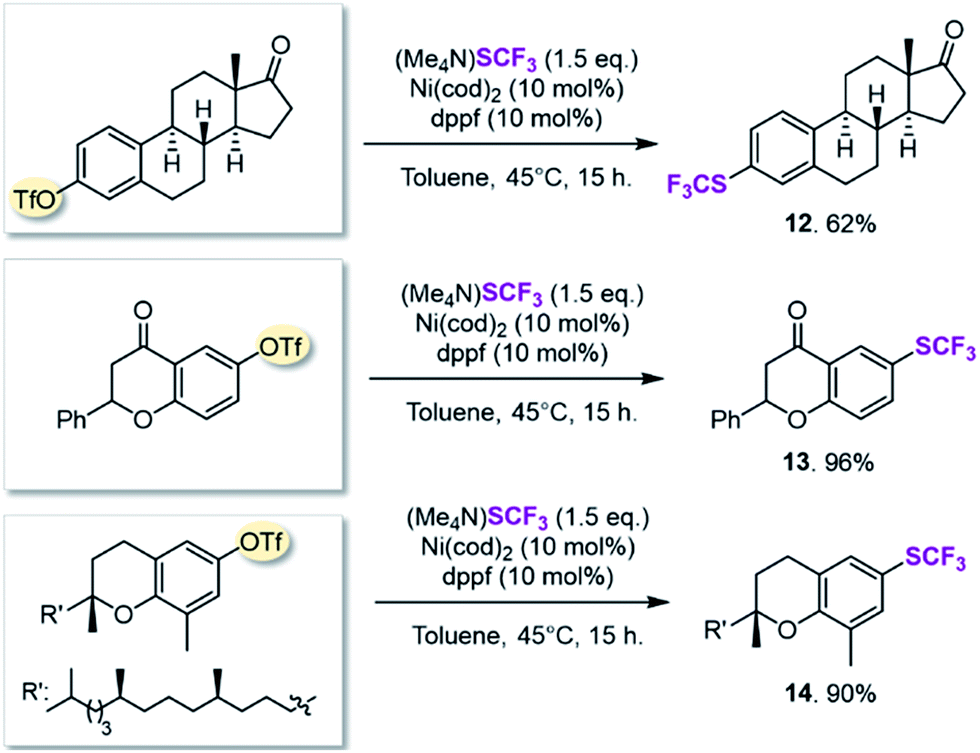 | ||
| Scheme 1 Synthesis of bioactive molecules. | ||
SCF3-coupling of vinyl triflates
Vinyl SCF3-compounds are also of significance, finding applications as herbicides for example.23 However, the current methodological repertoire to access these compounds relies predominantly on indirect strategies24 or requiring stoichiometric amounts of metal.13b,25 The direct construction of Cvinyl–SCF3 in a catalytic manner would be a highly attractive approach. It has been accomplished via the Cu-catalyzed trifluoromethylthiolation of vinyl boronic acids with electrophilic SCF3-sources.13c–e In a nucleophilic context, the catalytic installation of Cvinyl–SCF3 is limited to vinyl iodides and requires harsh reaction conditions (110 °C).26A mild Ni-catalyzed conversion of readily accessible Cvinyl–OR derivatives to Cvinyl–SCF3 would thus substantially widen the synthetic repertoire.
Our calculation of the barrier for the oxidative addition of [Ni(0)] to Cvinyl–SCF3 indicated ΔG‡ = 18.8 kcal mol−1. This barrier constitutes the upper limit for the reactivity of a potential leaving group (OR). Cvinyl–OPiv and Cvinyl–OMs show higher or similarly high barriers for oxidative addition (ΔG‡ = 22.1 and 17.7 kcal mol−1) and are hence ruled out. Cvinyl–OTf on the other hand is predicted to be highly reactive (ΔG‡ = 5.2 kcal mol−1) and should hence be a compatible match.
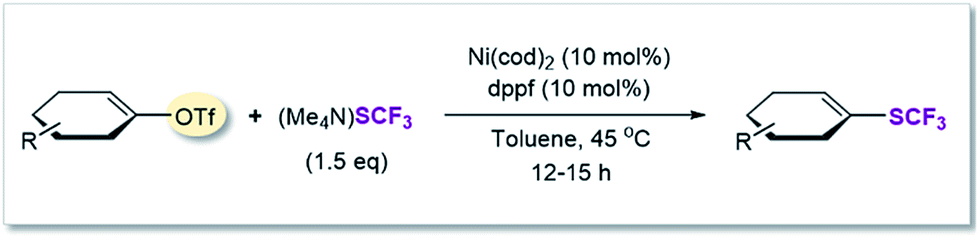
After applying standard catalysis conditions,27 we successfully transformed a number of vinyl triflates to the corresponding trifluoromethylthiolated counterparts (see Table 2).
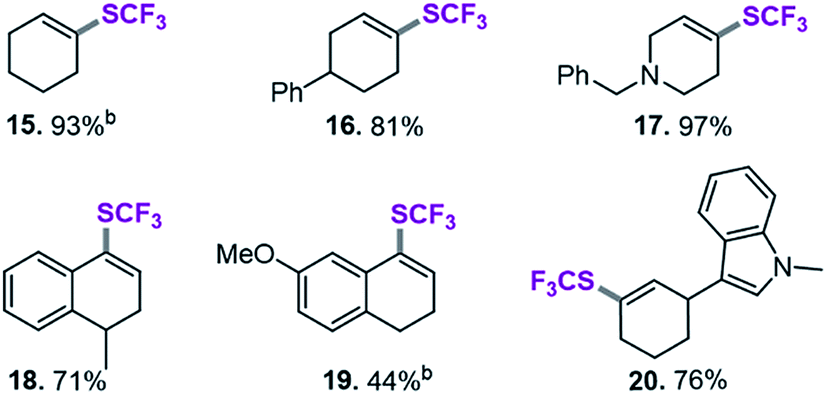
The protocol proved to be compatible with a heterocyclic moiety (20, Table 2), a benzyl protecting group (17), and was successful for fully aliphatic (15) as well as conjugated (18, 19) vinyl triflate derivatives. Compound 19 (Table 2) was afforded in a slightly lower yield (44%). However, upon closer inspection, it became clear that this was related to the inherent instability of the vinyl triflate starting material.
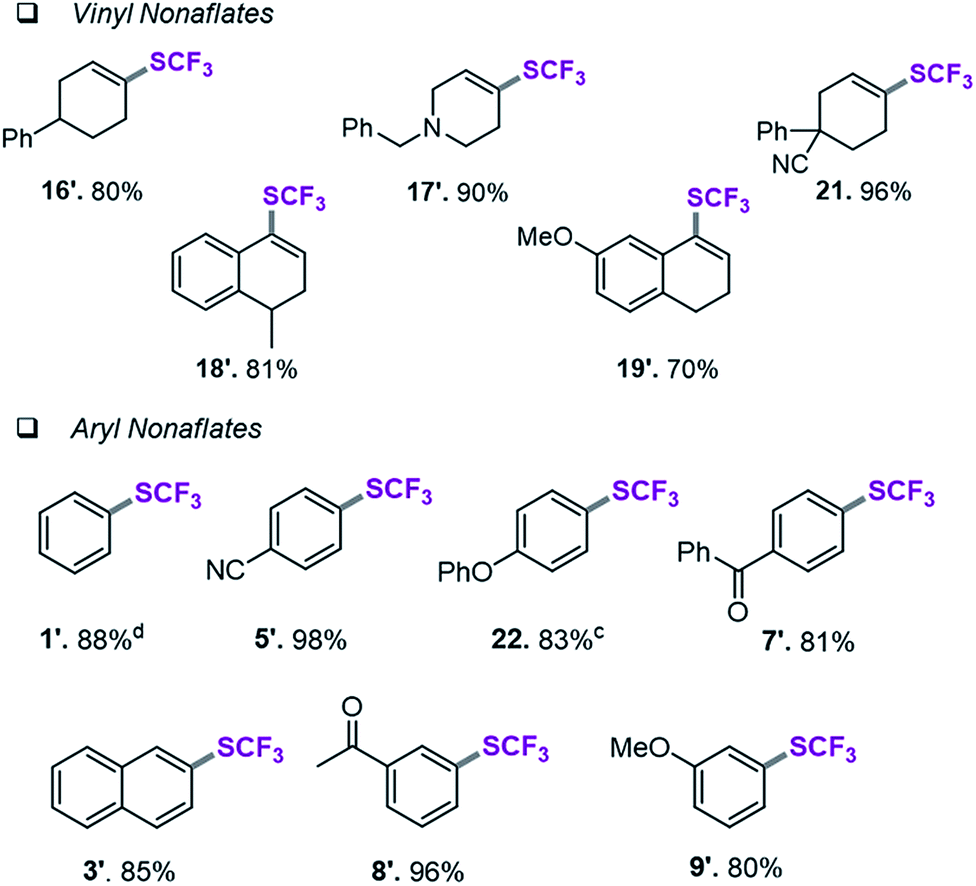
Assessment of aryl and vinyl nonaflates
We therefore shifted our attention to potentially more stable analogues and considered nonaflates.28 Both, aryl and vinyl nonaflates are computationally predicted to be compatible with Ni-catalyzed trifluoromethylthiolation, showing similarly low or even lower barriers for oxidative addition by [Ni(0)] than the corresponding triflates (ΔG‡ = 4.8 for addition to Cvinyl–ONf and ΔG‡ = 10.6 kcal mol−1 for addition to Ph–ONf). In accordance with these computational predictions, excellent conversions to aryl– and Cvinyl–SCF3 were observed (see Table 3). Particularly notable is the synthesis of 19′ (Table 3) which was now high-yielding (as opposed to its preparation in Table 2), reflecting the greater robustness of vinyl nonaflates over vinyl triflates.29|
|
|---|
| a Conditions for the coupling of vinyl nonaflates: Ni(cod)2 (5.5 mg, 0.02 mmol), dppf (11.1 mg, 0.02 mmol), vinyl nonaflate (0.2 mmol), (Me4N)SCF3 (52 mg, 0.3 mmol), PhCN (20.6 mg, 0.2 mmol),27 toluene (1 mL), under inert atmosphere, isolated yield. b Conditions for the coupling of aryl nonaflates Ni(cod)2 (11.0 mg, 0.04 mmol), dppf (22.2 mg, 0.04 mmol), aryl nonaflate (0.4 mmol), (Me4N)SCF3 (104 mg, 0.6 mmol), toluene (2 mL), under inert atmosphere, isolated yield. c Reaction performed with MeCN (16.4 mg, 0.4 mmol). d Yield determined by 19F-NMR analysis using PhCF3 as the internal standard. |
|
Conclusions
The inherently high reactivities of Ni-catalysts may be fundamentally at conflict with introducing a wide range of functional groups, as shown here for the introduction of the pharmaceutically and agrochemically valuable SCF3 group. We identified that the reaction of the Ni-catalyst with the desired product, ArSCF3, triggers undesirable catalyst deactivation reactions that ultimately inhibit catalysis. The overall substrate scope is therefore dictated by the reactivity of the desired functionality towards the catalyst (here: C–SCF3). The application of computational tools allowed for the identification of matching functional groups in terms of suitable leaving groups and tolerated functional groups. As a result, the first Ni-catalyzed C–SCF3 coupling of aryl and vinyl C–O bonds has been developed. Given the highly reactive nature of C–SCF3, only those C–OR derivatives of even greater reactivity, i.e. triflates and nonaflates, allow for efficient C–SCF3 coupling. The protocol is mild, general and operationally simple.Given that computational methods, software and hardware have evolved to a level, at which calculations can nowadays frequently be done faster than experiments,30 we anticipate that the herein applied approach will find applications in the development of, but not limited to, homogeneous Ni-catalysis.
Acknowledgements
We thank the MIWF NRW and the RWTH Aachen University for financial support. The authors gratefully acknowledge the computing time granted on the RWTH Bull Cluster in Aachen (grant number JARA0091).Notes and references
- For reviews, see: (a) Y. Tamaru, in Modern Organonickel Chemistry, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed; (c) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270 RSC; (d) J. Montgomery, Organonickel Chemistry, in Organo-metallics in Synthesis: Fourth Manual, ed. B. H. Lipshutz, Wiley, Hoboken, N.J., 2013, pp. 319–428 Search PubMed.
- C. C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef PubMed.
- For an overview, see: S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed.
- Examples of Ni-catalyzed C–F activation, see: (a) T. Braun, S. P. Foxon, R. N. Perutz and P. H. Walton, Angew. Chem., Int. Ed., 1999, 38, 3326 CrossRef CAS; (b) L. Ackermann, R. Born, J. H. Spatz and D. Meyer, Angew. Chem., Int. Ed., 2005, 44, 7216 CrossRef CAS PubMed; (c) S. A. Johnson, C. W. Huff, F. Ferheen Mustafa and M. Saliba, J. Am. Chem. Soc., 2008, 130, 17278 CrossRef CAS PubMed; (d) M. Tobisu, T. Xu, T. Shimasaki and N. Chatani, J. Am. Chem. Soc., 2011, 133, 19505 CrossRef CAS PubMed.
- For examples, see: (a) J. W. Dankwardt, Angew. Chem., Int. Ed., 2004, 43, 2428 CrossRef CAS PubMed; (b) M. Tobisu, T. Shimasaki and N. Chatani, Angew. Chem., Int. Ed., 2008, 47, 4866 CrossRef CAS PubMed; (c) B.-T. Guan, S.-K. Xiang, T. Wu, Z.-P. Sun, B.-Q. Wang, K.-Q. Zhao and Z.-J. Shi, Chem. Commun., 2008, 1437 RSC; (d) P. Alvarez-Bercedo and R. Martin, J. Am. Chem. Soc., 2010, 132, 17352 CrossRef CAS PubMed; (e) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439 CrossRef CAS PubMed; (f) M. Tobisu, K. Yamakawa, T. Shimasaki and N. Chatani, Chem. Commun., 2011, 47, 2946 RSC; (g) A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226 CrossRef CAS PubMed; (h) A. R. Ehle, Q. Zhou and M. P. Watson, Org. Lett., 2012, 14, 1202 CrossRef CAS PubMed; (i) K. Muto, J. Yamaguchi, A. Lei and K. Itami, J. Am. Chem. Soc., 2013, 135, 16384 CrossRef CAS PubMed; (j) Y. Hoshimoto, H. J. Yabuki, R. Kumar, H. Suzuki, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2014, 136, 16752 CrossRef CAS PubMed.
- For recent reviews on Ni-catalyzed C–O bond cleavages, see: (a) B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886 CrossRef CAS PubMed; (b) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081 RSC; (c) T. Mesganaw and N. K. Garg, Org. Process Res. Dev., 2013, 17, 29 CrossRef CAS; (d) J. Yamaguchi, K. Muto and K. Itami, Eur. J. Org. Chem., 2013, 19 CrossRef CAS; (e) A. Correa, J. Cornella and R. Martin, Angew. Chem., Int. Ed., 2013, 52, 1878 CrossRef CAS PubMed; (f) B.-J. Li, D.-G. Yu, C.-L. Sun and Z.-J. Shi, Chem.–Eur. J., 2011, 17, 1728 CrossRef CAS PubMed; (g) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346 CrossRef CAS PubMed; (h) D.-G. Yu, B.-J. Li and Z.-J. Shi, Acc. Chem. Res., 2010, 43, 1486 CrossRef CAS PubMed.
- For recent reviews on combining experiment and computation, see: (a) K. J. Bonney and F. Schoenebeck, Chem. Soc. Rev., 2014, 43, 6609 RSC; (b) A. S.-K. Tsang, I. A. Sanhueza and F. Schoenebeck, Chem.–Eur. J., 2014, 20, 16432 CrossRef CAS PubMed; (c) G.-J. Cheng, X. Zhang, L. W. Chung, L. Xu and Y.-D. Wu, J. Am. Chem. Soc., 2015, 137, 1706 CrossRef CAS PubMed.
- For examples of our activities to introduce fluorine containing groups, see: (a) P. Anstaett and F. Schoenebeck, Chem.–Eur. J., 2011, 17, 12340 CrossRef CAS PubMed; (b) I. A. Sanhueza, M. C. Nielsen, M. Ottiger and F. Schoenebeck, Helv. Chim. Acta, 2012, 95, 2231 CrossRef CAS; (c) I. A. Sanhueza, K. J. Bonney, M. C. Nielsen and F. Schoenebeck, J. Org. Chem., 2013, 78, 7749 CrossRef CAS PubMed; (d) M. C. Nielsen, K. J. Bonney and F. Schoenebeck, Angew. Chem., Int. Ed., 2014, 53, 5903 CrossRef CAS PubMed; (e) M. Aufiero, T. Sperger, A. S.-K. Tsang and F. Schoenebeck, Angew. Chem., Int. Ed., 2015, 54, 10322 CrossRef CAS PubMed.
- (a) A. Leo, P. Y. C. Jow, C. Silipo and C. Hansch, J. Med. Chem., 1975, 18, 865 CrossRef CAS PubMed; (b) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- For non-catalytic or indirect synthetic methods to generate ArSCF3 from aryl sulfides or disulfides with trifluoromethylation reagents, see: (a) V. N. Boiko, G. M. Shchupak and L. M. Yagupolskii, Journal of Organic Chemistry of the USSR, 1977, 13, 972 Search PubMed; (b) C. Wakselman and M. Tordeux, Chem. Commun., 1984, 793 RSC; (c) T. Umemoto and S. Ishihara, Tetrahedron Lett., 1990, 31, 3579 CrossRef CAS; (d) C. Wakselman, M. Tordeux, J. L. Clavel and B. R. Langlois, Chem. Commun., 1991, 993 RSC; (e) N. Roques, J. Fluorine Chem., 2001, 107, 311 CrossRef CAS; (f) G. Blond, T. Billard and B. R. Langlois, Tetrahedron Lett., 2001, 42, 2473 CrossRef CAS; (g) C. Pooput, M. Medebielle and W. R. Dolbier, Org. Lett., 2004, 6, 301 CrossRef CAS PubMed; (h) F. Leroux, P. Jeschke and M. Schlosser, Chem. Rev., 2005, 105, 827 CrossRef CAS PubMed. For syntheses in which ‘SCF3’ functions as nucleophile or electrophile, see: (i) T. Billard and B. R. Langlois, Tetrahedron Lett., 1996, 37, 6865 CrossRef CAS; (j) D. J. Adams, A. Goddard, J. H. Clark and D. J. Macquarrie, Chem. Commun., 2000, 987 RSC; (k) D. J. Adams and J. H. Clark, J. Org. Chem., 2000, 65, 1456 CrossRef CAS PubMed; (l) I. Kieltsch, P. Eisenberger and A. Togni, Angew. Chem., Int. Ed., 2007, 46, 754 CrossRef CAS PubMed; (m) S. Capone, I. Kieltsch, O. Flögel, G. Lelais, A. Togni and D. Seebach, Helv. Chim. Acta, 2008, 91, 2035 CrossRef CAS; (n) K. Stanek, R. Koller and A. Togni, J. Org. Chem., 2008, 73, 7678 CrossRef CAS PubMed; (o) B. Manteau, S. Pazenok, J.-P. Vors and F. R. Leroux, J. Fluorine Chem., 2010, 131, 140 CrossRef CAS; (p) F. Baert, J. Colomb and T. Billard, Angew. Chem., Int. Ed., 2012, 51, 10382 CrossRef CAS PubMed.
- For recent reviews, see: (a) G. Landelle, A. Panossian, S. Pazenok, J.-P. Vors and F. R. Leroux, Beilstein J. Org. Chem., 2013, 9, 2476 CrossRef PubMed; (b) P. Chen and G. Liu, Synthesis, 2013, 45, 2919 CrossRef CAS; (c) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (d) F. Toulgoat, S. Alazet and T. Billard, Eur. J. Org. Chem., 2014, 2415 CrossRef CAS.
- (a) G. Teverovskiy, D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 7312 CrossRef CAS PubMed; (b) C.-P. Zhang and D. A. Vicic, J. Am. Chem. Soc., 2012, 134, 183 CrossRef CAS PubMed; (c) Z. Weng, W. He, C. Chen, R. Lee, D. Tan, Z. Lai, D. Kong, Y. Yuan and K.-W. Huang, Angew. Chem., Int. Ed., 2013, 52, 1548 CrossRef CAS PubMed; (d) J. Xu, X. Mu, P. Chen, J. Ye and G. Liu, Org. Lett., 2014, 16, 3942 CrossRef CAS PubMed; (e) G. Yin, I. Kalvet, U. Englert and F. Schoenebeck, J. Am. Chem. Soc., 2015, 137, 4164 CrossRef CAS PubMed; (f) G. Yin, I. Kalvet and F. Schoenebeck, Angew. Chem., Int. Ed., 2015, 54, 6809 CrossRef CAS PubMed.
- (a) C. Chen, Y. Xie, L. Chu, R.-W. Wang, X. Zhang and F.-L. Qing, Angew. Chem., Int. Ed., 2012, 51, 2492 CrossRef CAS PubMed; (b) C.-P. Zhang and D. A. Vicic, Chem.–Asian J., 2012, 7, 1756 CrossRef CAS PubMed; (c) X. Shao, X. Wang, T. Yang, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2013, 52, 3457 CrossRef CAS PubMed; (d) R. Pluta, P. Nikolaienko and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 1650 CrossRef CAS PubMed; (e) Q. Glenadel, S. Alazet, A. Tlili and T. Billard, Chem.–Eur. J., 2015, 21, 14694 CrossRef CAS PubMed.
- For examples of metal-catalyzed C–H trifluoromethyl-thiolation of arenes, see: (a) L. D. Tran, I. Popov and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 18237 CrossRef CAS PubMed; (b) C. Xu and Q. Shen, Org. Lett., 2014, 16, 2046 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552 CrossRef CAS PubMed.
- For precedence of aryl sulfide cleavage under Ni-catalysis, see: (a) S. Kanemura, A. Kondoh, H. Yorimitsu and K. Oshima, Synthesis, 2008, 2659 CAS; (b) K. Lee, C. M. Counceller and J. P. Stambuli, Org. Lett., 2009, 11, 1457 CrossRef CAS PubMed; (c) L. Melzig, A. Metzger and P. Knochel, Chem.–Eur. J., 2011, 17, 2948 CrossRef CAS PubMed; (d) N. Barbero and R. Martin, Org. Lett., 2012, 14, 796 CrossRef CAS PubMed.
- Gaussian 09, Revision D.01, M. J. Frisch, et al. Search PubMed, [see ESI† for full reference], The geometries were optimized in the gas-phase at B3LYP/6-31G(d) with LANL2DZ (for Ni, Fe).
- For appropriateness of method, see: (a) T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532 CrossRef CAS PubMed; (b) M. T. Haynes, P. Liu, R. D. Baxter, A. J. Nett, K. N. Houk and J. Montgomery, J. Am. Chem. Soc., 2014, 136, 17495 CrossRef CAS PubMed; (c) T. Mesganaw, A. L. Silberstein, S. D. Ramgren, N. Fine Nathel, X. Hong, P. Liu and N. K. Garg, Chem. Sci., 2011, 2, 1766 RSC; (d) See also: ref. 12e.
- For mechanistic support of Ni(I) as catalyst in C–O cleavage, see: J. Cornella, E. Gomez-Bengoa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1997 CrossRef CAS PubMed.
- Using 10 mol% of [(dppf)Ni(I)(Cl)] complex as catalyst for the trifluoromethylthiolation of PhOTf with (Me4N)(SCF3) at 45 °C for 12 h in toluene, no reaction was seen.
- We did not consider the influence of other substituents or alternative mechanisms on this trend. It is likely, that specialized substrates with electronic bias may also allow for selected examples of these functional groups to be converted. The goal of this assessment was to identify compatible functionality that would allow for widest possible scope in the context of [Ni(0)] catalysis.
- (a) J. J. Garcia, N. M. Brunkan and W. D. Jones, J. Am. Chem. Soc., 2002, 124, 9547 CrossRef CAS PubMed; (b) D.-G. Yu, M. Yu, B.-T. Guan, B.-J. Li, Y. Zheng, Z.-H. Wu and Z.-J. Shi, Org. Lett., 2009, 11, 3374 CrossRef CAS PubMed; (c) J.-S. Zhang, T. Chen, J. Yang and L.-B. Han, Chem. Commun., 2015, 51, 7540 RSC.
- S. Scheiblich, T. Maier and H. Baltruschat, PCT Int. Appl., CODEN: PIXXD2, WO 01/36410A1, 2001, 41 pp.
- For a review, see: A. Y. Sizov, A. N. Kovregin and A. F. Ermolov, Russ. Chem. Rev., 2003, 72, 357 CrossRef CAS.
- Y. Huang, J. Ding, C. Wu, H. Zheng and Z. Weng, J. Org. Chem., 2015, 80, 2912 CrossRef CAS PubMed.
- M. Rueping, N. Tolstoluzhsky and P. Nikolaienko, Chem.–Eur. J., 2013, 19, 14043 CrossRef CAS PubMed.
- PhCN was also added. For the beneficial effect of nitrile, see: ref. 12e.
- J. Högermeier and H.-U. Reissig, Adv. Synth. Catal., 2009, 351, 2747 CrossRef.
- For examples of the superiority of nonaflates: (a) S. Bräse and A. de Meijere, Angew. Chem., Int. Ed., 1995, 34, 2545 CrossRef; (b) K. Voigt, P. von Zezschwitz, K. Rosauer, A. Lansky, A. Adams, O. Reiser and A. de Meijere, Eur. J. Org. Chem., 1998, 152 Search PubMed; (c) A. E. Jensen, W. Dohle and P. Knochel, Tetrahedron, 2000, 56, 4197 CrossRef CAS; (d) G. Dunet and P. Knochel, Synlett, 2006, 407 CAS.
- The total time necessary for optimization, frequency and energy calculation of the oxidative addition TSs was on average 11.5 h (using 6 cores and 10GB memory on a MPI-S node of the RWTH Bull cluster).
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, ReactIR studies, computational information and Cartesian coordinates of calculated species as well as full ref. 17 are given. See DOI: 10.1039/c5sc03359d |
| ‡ These authors contributed equally. The authors declare no competing financial interests. |
| This journal is © The Royal Society of Chemistry 2016 |