
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand-controlled insertion regioselectivity accelerates copolymerisation of ethylene with methyl acrylate by cationic bisphosphine monoxide–palladium catalysts†
Yusuke
Mitsushige
 a,
Brad P.
Carrow
b,
Shingo
Ito
a and
Kyoko
Nozaki
*a
a,
Brad P.
Carrow
b,
Shingo
Ito
a and
Kyoko
Nozaki
*a
aDepartment of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: nozaki@chembio.t.u-tokyo.ac.jp
bDepartment of Chemistry, Princeton University, Princeton, New Jersey, USA
First published on 3rd November 2015
Abstract
A new series of palladium catalysts ligated by a chelating bisphosphine monoxide bearing diarylphosphino groups (aryl-BPMO) exhibits markedly higher reactivity for ethylene/methyl acrylate copolymerisation when compared to the first generation of alkyl-BPMO–palladium catalysts that contain a dialkylphosphino moiety. Mechanistic studies suggest that the origin of this disparate catalyst behavior is a change in regioselectivity of migratory insertion of the acrylate comonomer as a function of the phosphine substituents. The best aryl-BPMO–palladium catalysts for these copolymerisations were shown to undergo exclusively 2,1-insertion, and this high regioselectivity avoids formation of a poorly reactive palladacycle intermediate. Furthermore, the aryl-BPMO–palladium catalysts can copolymerise ethylene with other industrially important polar monomers.
Introduction
Migratory insertion of alkenes is a fundamental organometallic reaction involved in a number of industrial processes such as hydroformylation, hydrocyanation, hydrogenation, the Heck reaction in fine chemical synthesis, and olefin polymerisation. The regioselectivity of migratory insertion affects isomer distributions and thus product yields of catalytic processes that generate small molecules. When the alkene insertion is involved in polymerisation, regioselectivity during migratory insertion of substituted alkenes can influence the resulting material properties by affecting crystallinity,1 microstructure,1c,2 and molecular weight.3 Thus, appropriate control of alkene-insertion regioselectivity is an important consideration towards the development of efficient catalytic transformation of alkenes. In general, regioselectivity of alkene insertion into a palladium–carbon bond is affected by the electronic nature of alkenes. In palladium-catalysed reactions, for instance, mono-substituted alkenes bearing electron-withdrawing groups, such as acrylates or acrylonitrile, tend to undergo 2,1-insertion in their migratory insertion into a palladium–carbon bond, because the migratory group generally adds to the terminal sp2 carbon bearing a larger LUMO coefficient.4,5 The 2,1-insertion forms a 4-membered metallacycle or the corresponding multimer generated by intermolecular coordination (Scheme 1a). It is notable that a preferential 1,2-insertion of methyl acrylate (MA) into a palladium–carbon bond, forming a 5-membered metallacycle (Scheme 1b), was recently accomplished in a stoichiometric reaction using a palladium species ligated by an extremely bulky ligand.6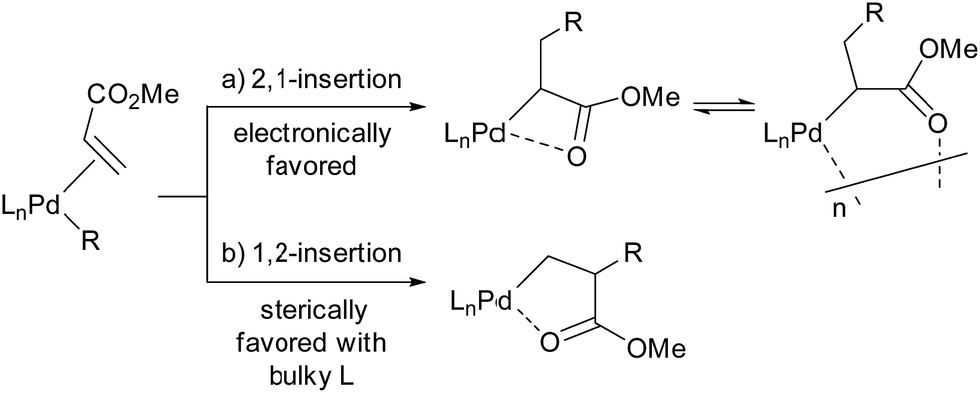 | ||
| Scheme 1 Schematic view of 2,1-insertion and 1,2-insertion of acrylates. | ||
In the last two decades, intensive studies have been devoted to catalyst development for coordination–insertion copolymerisation of olefins with polar vinyl monomers, aimed at the production of functionalised polyolefins.5 Acrylates are some of the most common polar vinyl monomers used for the copolymerisation with ethylene. Despite the importance, a limited number of papers have discussed the regioselectivity of acrylate insertion in the polymerisation processes. In the case of palladium/α-diimine catalysts, 2,1-insertion of MA selectively proceeds,7,8 although the formed 4-membered chelate complex rapidly isomerises via chain walking to form the corresponding 6-membered palladacycle before the next chain propagation.7 The same preferential 2,1-insertion of MA has been observed in the case of palladium/phosphine-sulfonate catalysts.9,10 As an exception, selective 1,2-insertion of MA was reported with a [P–SO3]–type ligand bearing a bulky diazaphospholidine group,6 although the catalyst did not promote ethylene/MA copolymerisation.6b Recently, ethylene/MA cooligomerization by palladium/phosphine-phosphonate catalysts was reported, in which chain-end analysis suggested coexistence of 2,1- and 1,2-insertion of MA.11 In this regard, we previously reported that cationic palladium complexes possessing a BPMO ligand (an analog of 1a bearing SbF6− in place of BArF4− (ArF = 3,5-bis(trifluoromethyl)phenyl) as a counter anion, and 2a (Fig. 1))12 mediate the copolymerisation of ethylene with a number of polar monomers such as acrylonitrile, vinyl acetate, allyl acetate, and butyl vinyl ether.13 Rather surprisingly, however, the best of the original BPMO–palladium catalysts were not applicable to the copolymerisation of ethylene and MA; only trace amounts of copolymer were formed after 15 h at 80 °C (vide infra). This observation was puzzling considering that MA has been the most reactive comonomer for copolymerisation with ethylene,5,14,15 and we set out to understand this phenomenon with the expectation that knowledge of the mechanistic limitations of 1a and 2a might shed light on the unique behavior of catalysts ligated by a BPMO compared to established polymerisation catalyst classes.16
Here we report catalyst-controlled 1,2- and 2,1-insertion regioselectivity in the ethylene/MA copolymerisation by palladium complexes possessing a chelating bisphosphine monoxide (BPMO) ligand. Detailed mechanistic studies revealed that a 5-membered palladacycle intermediate formed via 1,2-insertion of acrylate retards the (co)polymerisation.17 Newly designed BPMO ligands possessing aryl groups on the phosphine moiety (Fig. 1) are reported here to preferentially promote 2,1-insertion of acrylate and achieve the copolymerisation of ethylene and various polar comonomers.
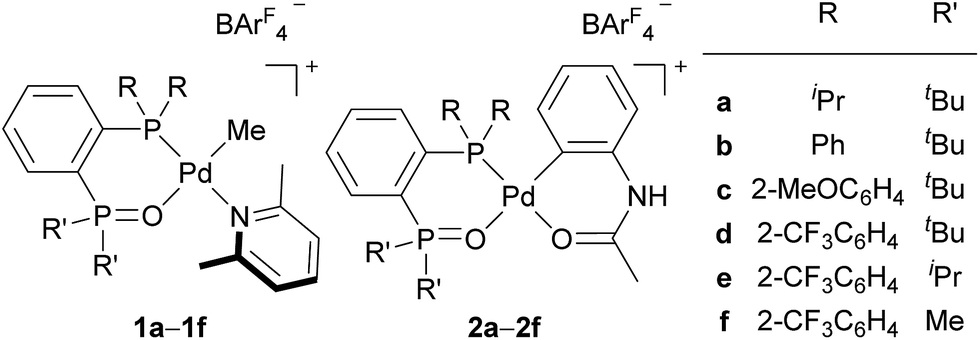 | ||
| Fig. 1 Examples of BPMO–palladium complexes. ArF = 3,5-bis(trifluoromethyl)phenyl. | ||
Results and discussion
Stoichiometric reactions of alkyl-BPMO–palladium complexes with methyl acrylate
We initially attempted to determine the organometallic product(s) from the reaction of an alkyl-BPMO–palladium complex with MA to gain insight into the structure of any potential deactivated catalyst states. Treatment of chloro(methyl)palladium complex 3a with silver hexafluorophosphate in the presence of MA in dichloromethane for 1 hour at room temperature (Scheme 2a) led to the formation of two distinct palladium products, as observed by 31P NMR spectroscopy. The two products, 4a and 5a, were formed in 29% and 71% yield, respectively, as estimated by integration of 1H coupled 31P NMR resonances against an external PPh3 standard. As the rapid decomposition of 4a during the evaporation of solvent made the isolation of 4a and 5a difficult, we repeated the same reaction in (trifluoromethyl)benzene, in which 4a is soluble but 5a is insoluble. This significant solubility difference of 4a and 5a in (trifluoromethyl)benzene facilitated their separation by fractional crystallization to give 16% and 52% isolated yields, respectively. Recrystallization of crude 4a from (trifluoromethyl)benzene/diethyl ether provided single crystals suitable for X-ray analysis (Scheme 2a). The solid state structure of the 5-membered palladacycle (4a) was consistent with NMR spectroscopic data in solution (see Fig. S37–46†). Complex 4a exhibited a pair of characteristic 31P NMR resonances at δP 63.2 and 55.8 ppm that correspond to the phosphine oxide and phosphine moieties of the BPMO (Fig. 2a). These characteristic resonances proved useful in determining the fate of the BPMO–palladium catalysts during ethylene/MA copolymerisations (vide infra). The formation of 4a must occur by initial 1,2-insertion of MA into the Pd–C bond of 3a, which is an uncommon regioselectivity for reactions of acrylates regardless of the nature of the transition metal complex.6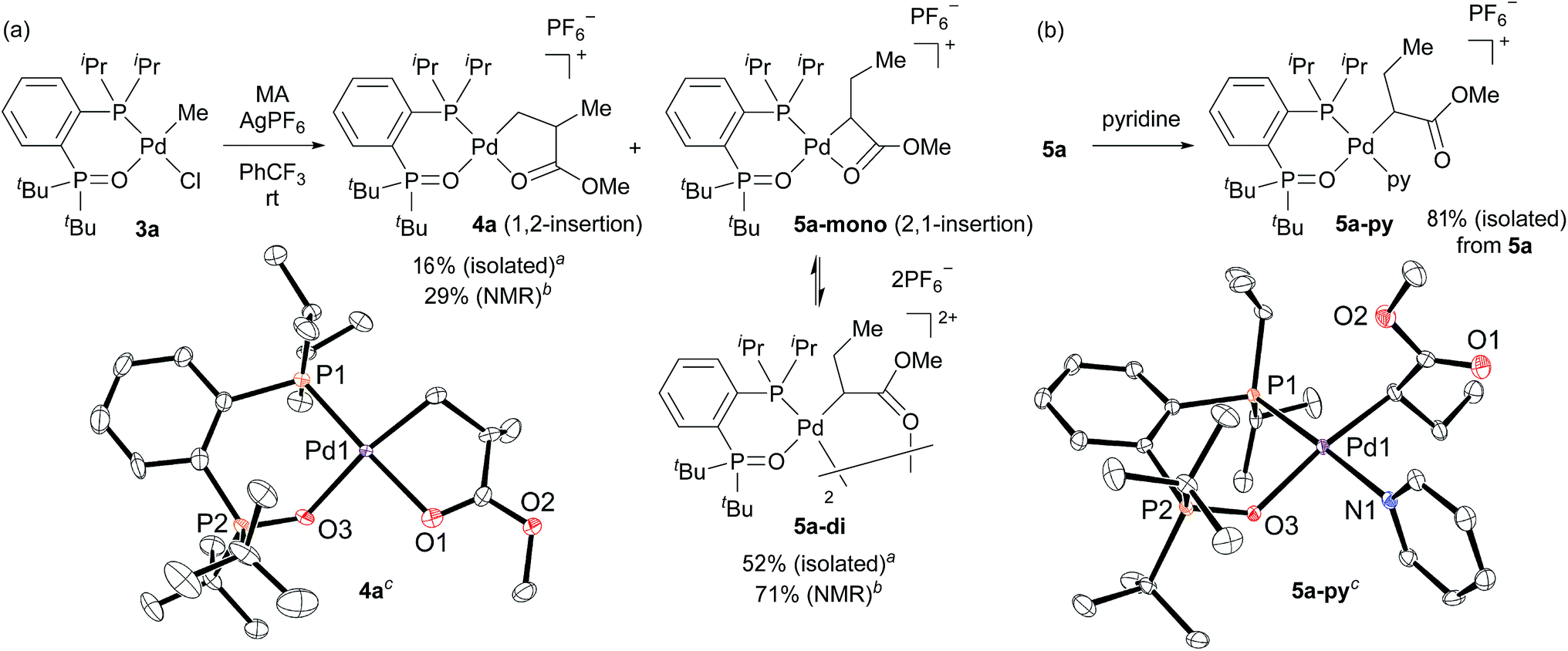 | ||
| Scheme 2 (a) Stoichiometric reaction of methyl acrylate with complex 3a and (b) reaction of 5a mixture with pyridine. a The reaction to isolate 4a and 5a was performed in (trifluoromethyl)benzene. b The reaction to determine the NMR yields was performed in dichloromethane. c For X-ray structures of 4a and 5a-py, thermal ellipsoids are shown at 50% probability. Hydrogen atoms, counter anions, and disordered fragments are omitted for clarity. | ||
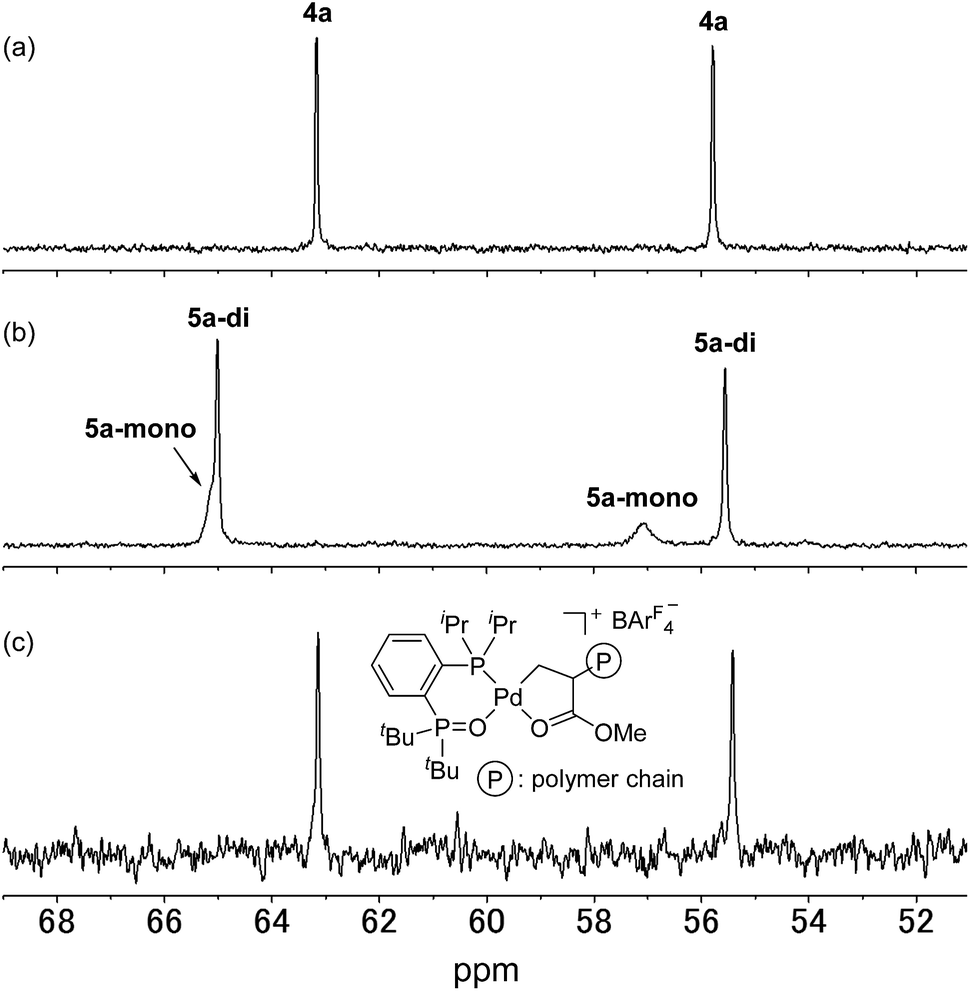 | ||
| Fig. 2 31P NMR spectra of (a) 4a, (b) 5a (3.0 × 10−2 M) and (c) the reaction mixture after copolymerisation of ethylene with MA catalysed by 1a (202 MHz, 1,1,2,2-tetrachloroethane-d2). | ||
Unfortunately, high quality crystals of 5a could not be obtained after repeated attempts, but a dimeric structure (5a-di) was suggested by low resolution X-ray diffraction data.18 NMR analysis of the isolated material in a 3.0 × 10−2 M solution in 1,1,2,2-tetrachloroethane-d2 at 25 °C exhibited two pairs of resonances at δP 65.1 and 57.1 ppm or 65.0 and 55.5 ppm in a 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :65 ratio indicative of a mixture of two BPMO–palladium complexes (Fig. 2b). These compounds were assigned as monomer 5a-mono and dimer 5a-di, respectively, in dynamic equilibrium at room temperature based on the correlation between the relative populations and solution concentration: the ratios of 5a-mono:5a-di were 50:50 and 58:42 in 1.5 × 10−2 and 1.0 × 10−2 M solutions, respectively.19 Reaction of pyridine and 5a, however, did converge to a single new species (5a-py) whose structure was successfully determined by X-ray analysis (Scheme 2b). This derivative complex corresponds to reaction of 3a and MA with 2,1-insertion regioselectivity. Thus, both organometallic products 4a and 5a arise from migratory insertion of MA, but occur with opposite regioselectivity.
:65 ratio indicative of a mixture of two BPMO–palladium complexes (Fig. 2b). These compounds were assigned as monomer 5a-mono and dimer 5a-di, respectively, in dynamic equilibrium at room temperature based on the correlation between the relative populations and solution concentration: the ratios of 5a-mono:5a-di were 50:50 and 58:42 in 1.5 × 10−2 and 1.0 × 10−2 M solutions, respectively.19 Reaction of pyridine and 5a, however, did converge to a single new species (5a-py) whose structure was successfully determined by X-ray analysis (Scheme 2b). This derivative complex corresponds to reaction of 3a and MA with 2,1-insertion regioselectivity. Thus, both organometallic products 4a and 5a arise from migratory insertion of MA, but occur with opposite regioselectivity.
Analysis of catalyst residue after ethylene/methyl acrylate copolymerisation
We next analysed the palladium products formed during the copolymerisation of ethylene and MA using 1a to determine whether an analogue to palladacycles 4a or 5a formed by 1,2- or 2,1-insertion of MA, respectively, were also generated during catalysis.20 Following the reaction of ethylene and MA in the presence of 1a in toluene for 15 h at 80 °C, the resulting mixture was analysed by electrospray ionization (ESI) mass spectrometry. Two series of ion signals corresponding to [BPMO–Pd + (ethylene)n + MA + CH3] and [BPMO–Pd + (ethylene)m + MA + H] were observed, suggesting the insertion of one MA after several consecutive insertions of ethylene. After evaporation of solvent, the non-volatile residue was analysed by 1H NMR spectroscopy. A characteristic resonance at δH 2.82 ppm (dddd, J = 6,6,6,6 Hz) was assigned as a methine proton alpha to a coordinating ester group (Fig. S152†), similar to the methine resonance in the isolated 4a (Fig. S37†). This palladium product was the major species formed during the copolymerisation (>82% based on 1a). The 31P NMR chemical shifts at δP 63.1 and 55.4 ppm (Fig. 2c) are also similar to those of 4a (Fig. 2a).21 These data are consistent with 1,2-insertion of MA during attempted copolymerisation with 1a, and we suspected that this palladacyclic intermediate that lacks an open coordination site for monomer was functioning as a kinetic trap during catalysis.Ethylene polymerisation initiated and catalysed by alkyl-BPMO–palladium complexes
To probe the role of palladacycles as potential kinetic traps during catalysis, the activities of isolated metallacycles 4a and 5a-mono/5a-di towards ethylene polymerisation were compared to that of typical precatalyst 3a in the presence of silver hexafluorophosphate as a halogen scavenger (Table 1). In these experiments, the first insertion of ethylene to initiate the polymerisation occurs with a distinct palladium species in each case, but later propagation steps should occur through an identical catalytic species. At short reaction times the average reaction rate should be weighted towards the initiation phase of the polymerisation and thus qualitatively reflect how the first migratory insertion is affected by the stability of a metallacyclic complex. The results of these experiments (Table 1) clearly indicate that both palladacycle complexes react more slowly than methylpalladium precatalyst 3a. Notably, 5-membered palladacycle 4a is also significantly less reactive towards ethylene insertion compared to the dynamic mixture of 4-membered palladacycle 5a-mono and dimer 5a-di. These data suggest that any new BPMO–palladium catalyst that is active for polymerisations of acrylates likely would need to enforce high 2,1-insertion regioselectivity to avoid catalyst inhibition through formation of a stable 5-membered palladacycle.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Yield (g) | Activity (kg mol−1 h−1) | M n (103) | M w/Mnb |
| a Conditions: toluene (15 mL), ethylene (3.0 MPa), and palladium catalyst (6 μmol) were stirred in a 50 mL stainless autoclave for 1 h at 80 °C. b Determined by SEC analysis using polystyrene as an internal standard and calibrated by universal calibration. c 6 μmol of AgPF6 was added. | |||||
| 1 | 3a + AgPF6c | 2.51 | 420 | 23 | 3.3 |
| 2 | 4a | 0.05 | 8 | 30 | 2.3 |
| 3 | 5a-mono/5a-di | 0.32 | 54 | 23 | 3.0 |

Ethylene polymerisation initiated and catalysed by aryl-BPMO–palladium complexes
Because triarylphosphines are generally air stable and can be prepared in a modular fashion from haloarene starting materials, we also developed in parallel to our mechanistic study a new series of BPMO ligands and corresponding BPMO–palladium catalysts that incorporated this structural motif. The easily derivatised BPMO framework with a diarylphosphino moiety ultimately allowed us to empirically identify several new BPMO–palladium catalysts that were similar to 1a and 2a in activity for ethylene polymerisation (Table 2), but several of these were also substantially more active during copolymerisations with acrylate monomers (vide infra).| Entry | Catalyst | Yield (g) | Activity (kg mol−1 h−1) | M n /103 | M w/Mnb | Me br.c (/103 C) |
|---|---|---|---|---|---|---|
| a Conditions: toluene (15 mL), ethylene (3.0 MPa), and palladium catalyst (0.75 μmol) were stirred in a 50 mL stainless autoclave for 1 h at 100 °C. b Determined by SEC analysis using polystyrene as an internal standard and calibrated by universal calibration. c Determined by quantitative 13C NMR analysis.. | ||||||
| 1 | 1a | 2.00 | 2700 | 31 | 3.1 | 5 |
| 2 | 1b | 0.10 | 130 | 17 | 2.4 | 11 |
| 3 | 1c | 1.74 | 2300 | 12 | 4.2 | 14 |
| 4 | 1d | 0.79 | 1100 | 21 | 2.8 | 17 |
| 5 | 2a | 2.11 | 2800 | 29 | 2.1 | 5 |
| 6 | 2c | 1.40 | 1900 | 10 | 5.5 | 11 |
| 7 | 2d | 1.64 | 2200 | 14 | 2.4 | 22 |
| 8 | 2e | 1.75 | 2300 | 10 | 4.5 | 14 |
| 9 | 2f | 1.17 | 1600 | 9.3 | 3.1 | 2 |
As we previously reported,13 complex 1b with a simple diphenylphosphino group displayed modest ethylene polymerisation activity (Table 2, entry 2). In sharp contrast, BPMO–palladium complexes with ortho-substituted aryl groups on the phosphine exhibited markedly improved activity. Complexes 1c and 1d with bis(2-methoxyphenyl)phosphino and bis[2-(trifluoromethyl)phenyl]phosphino groups, respectively, promoted ethylene polymerisation with activity comparable to the best first generation catalyst 2a (entries 3 and 4). Palladacycle analogues 2c and 2d performed similarly (entries 6 and 7) to the methylpalladium-type precatalysts. Notably, an increase in the methyl branching ratio was detected in polyethylenes formed by these diarylphosphino BPMO–palladium complexes (compare entries 2–4 with 1; entries 6 and 7 with 5) relative to 1a or 2a. Palladium complexes with the same bis[2-(trifluoromethyl)phenyl]phosphino group but differing alkyl substituents at the phosphine oxide position of the BPMO (2d–2f) were also evaluated (entries 7–9). Decreasing substituent size from tert-butyl (2d) to isopropyl (2e) to methyl (2f) had a small effect on the molecular weight of the resulting polyethylene, but a significant change in methyl branching ratio from 22 to 14 to 2 per 1000 carbons, respectively, was observed. This data emphasises that alteration of the substituents at the phosphine oxide ligand in a BPMO–palladium catalyst offers an additional and useful site of perturbation to tune catalyst function and the structure of the resulting polymers, which is not possible with existing catalyst families such as Drent-type palladium/phosphine-sulfonate complexes.
Copolymerisation of ethylene and methyl acrylate
Most importantly, a significant improvement in the activity for copolymerisation of ethylene and MA was observed using BPMO–palladium catalysts that possess an ortho-substituted diarylphosphino moiety (Table 3). Complex 1b with a simple diphenylphosphino fragment were inert for copolymerisation (entry 2), but dramatically improved activity was observed using 1c, 1d, 2c, or 2d (entries 3, 4, 6, and 7). The low activity of phenyl-substituted ligands could be attributed to fast chain transfer reactions, as is generally observed in related catalysts.5,11,15e Ligands bearing electron-donating MeO groups, 1c and 2c, gave higher copolymer molecular weights and incorporation ratios of MA than those bearing electron-withdrawing CF3 groups, 1d and 2d.22 It is worth noting that the molecular weight and the incorporation ratio of MA were comparable to copolymers formed using state-of-the-art Drent-type phosphine-sulfonate palladium complexes.15b–e All copolymers were linear and random as determined by 1H NMR and quantitative 13C NMR spectroscopic analysis. The identity of the phosphine oxide substituents also had an important influence on reaction rate; drastically improved activity occurred as the size of substituent decreased from tert-butyl to isopropyl to methyl (entries 7–10). The exceptionally high catalytic activity using complex 2f (540 kg mol−1 h−1; entry 10),23,24 again highlights the power of exploiting steric perturbation near the oxygen atom of this chelating ligand, which is not possible with many established catalysts with [P–O]-type ancillary ligands such as a phosphine-sulfonate.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalystb | Time (h) | Yield (g) | Activity (kg mol−1 h−1) | M n /103 | M w/Mnc | Incorp.d (mol%) |
| a Conditions: ethylene, palladium catalyst, and comonomer were stirred in a 50 mL stainless autoclave at 80 °C. b Numbers in parenthesis are the amount of catalyst (μmol). c Determined by SEC analysis using polystyrene as an internal standard and calibrated by universal calibration. d Incorporation of MA determined by quantitative 13C NMR analysis. e Incorporation of MA determined by 1H NMR analysis. | |||||||
| 1 | 1a (10) | 15 | 0.03 | 0.2 | 1.6 | 1.7 | 2.5 |
| 2 | 1b (10) | 15 | 0 | — | — | — | — |
| 3 | 1c (10) | 15 | 0.61 | 4.1 | 33 | 2.3 | 2.3 |
| 4 | 1d (10) | 15 | 0.89 | 5.9 | 14 | 2.1 | 0.9 |
| 5 | 2a (10) | 15 | 0.01 | 0.1 | — | — | 3.3e |
| 6 | 2c (10) | 15 | 0.40 | 2.7 | 24 | 2.8 | 3.4 |
| 7 | 2d (10) | 15 | 1.37 | 9.1 | 17 | 2.6 | 1.0 |
| 8 | 2e (0.75) | 15 | 0.37 | 33 | 17 | 3.6 | 0.9 |
| 9 | 2e (0.75) | 1 | 0.03 | 41 | 18 | 2.3 | 1.2e |
| 10 | 2f (0.75) | 1 | 0.41 | 540 | 6.9 | 2.9 | 0.5 |
Copolymerisation of ethylene and other polar monomers by aryl-BPMO–palladium complexes
We performed copolymerisation of ethylene with various polar monomers using complexes bearing 2-methoxyphenyl group (1c) and 2-(trifluoromethyl)phenyl group (2d, 2f) (Table 4). Both catalysts 1c and 2d catalysed copolymerisation of ethylene and allyl acetate with comparable performance to common phosphine-sulfonate catalysts25 and the original alkyl-BPMO catalyst 2a13 in terms of activity, molecular weight, and incorporation ratio (entries 1 and 2). In contrast, catalyst 2f showed about 6-fold higher activity than 2d, while maintaining similar molecular weight and incorporation ratio (entry 3). Thus, exceptionally high activity of 2f was not limited to the copolymerization of acrylates. In the case of butyl vinyl ether, the copolymerisation using catalyst 1c proceeded (entry 4), while no or little incorporation of vinyl ether into polyethylene was observed, along with the formation of poly(butyl vinyl ether), when 2d or 2f was used (entries 5 and 6). This significant difference of catalyst behaviour is probably due to the decreased electrophilicity of 1c by electron-donating methoxy group that suppress the cationic polymerisation of butyl vinyl ether observed in entry 5 and 6. The same trend was observed in the copolymerisation of acrylonitrile (entries 7–9). Thus, catalyst 1c could afford ethylene/acrylonitrile copolymer, but 2d afforded only oligoethylene and 2f afforded even no solid product. In this case, strong σ-coordination of acrylonitrile to the electrophilic palladium centre of 2d and 2f would prevent the copolymerisation with ethylene. This higher electron-withdrawing ability of 2-(trifluoromethyl)phenyl than 2-methoxyphenyl group may partially explain the higher activity of 1d and 2d toward copolymerisation of ethylene with MA compared to 1c and 2c (compare entries 3 and 4 in Table 3, entries 6 and 7 in Table 3).26|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | FG | Comonomer (mL) | Toluene (mL) | Temperature (°C) | Time (h) | Yield (g) | Activity (kg mol−1 h−1) | M n (103) | M w/Mn | Incorp.c (mol%) |
| a Conditions: ethylene, palladium catalyst (10 μmol), and comonomer were stirred in a 50 mL stainless autoclave at an indicated temperature. b Determined by SEC analysis using polystyrene as an internal standard and calibrated by universal calibration. c Incorporation ratio of polar monomer determined by quantitative 13C NMR analysis. d Determined by 1H NMR spectrum. e Yield after washing with dichloromethane to remove the homopolymer of butyl vinyl ether formed as a side product. | |||||||||||
| 1 | 1c | CH2OAc | 3.0 | 12 | 80 | 12 | 0.35 | 2.9 | 5.1 | 2.0 | 0.9 |
| 2 | 2d | CH2OAc | 3.0 | 12 | 80 | 8 | 0.50 | 6.3 | 6.4 | 2.3 | 0.8d |
| 3 | 2f | CH2OAc | 3.0 | 12 | 80 | 8 | 3.02 | 38 | 5.2 | 4.0 | 0.6 |
| 4 | 1c | OBu | 5.0 | 10 | 80 | 26 | 0.20 | 0.8 | 5.7 | 2.2 | 0.7 |
| 5 | 2d | OBu | 5.0 | 10 | 80 | 20 | 2.91e | 15 | 18 | 2.6 | 0 |
| 6 | 2f | OBu | 5.0 | 10 | 80 | 20 | 1.96e | 9.8 | 11 | 3.8 | 0.1 |
| 7 | 1c | CN | 2.5 | 2.5 | 100 | 72 | 0.12 | 0.2 | 1.9 | 3.4 | 2.4 |
| 8 | 2d | CN | 2.5 | 2.5 | 100 | 72 | 0.09 | 0.1 | 0.4 | 1.6 | 0d |
| 9 | 2f | CN | 2.5 | 2.5 | 100 | 72 | 0 | — | — | — | — |
Origin of high copolymerisation activity
Finally, we conducted stoichiometric experiments to understand the origin of the much higher activity of this second generation of aryl-BPMO–palladium catalysts towards ethylene/MA copolymerisation. Reaction of bis(2-methoxyphenyl)phosphine complex 3c with silver hexafluorophosphate in the presence of MA at room temperature resulted in formation of 5c-di in 72% isolated yield (Scheme 3a). The dimeric structure of 5c-di was determined by single crystal X-ray analysis (Fig. 3), which verified the major palladium product from this reaction resulted from 2,1-insertion of MA. It is worth noting that the distance of Pd1–O5 is ca. 3.45 Å, which suggests no interaction between the methoxy group and the palladium centre.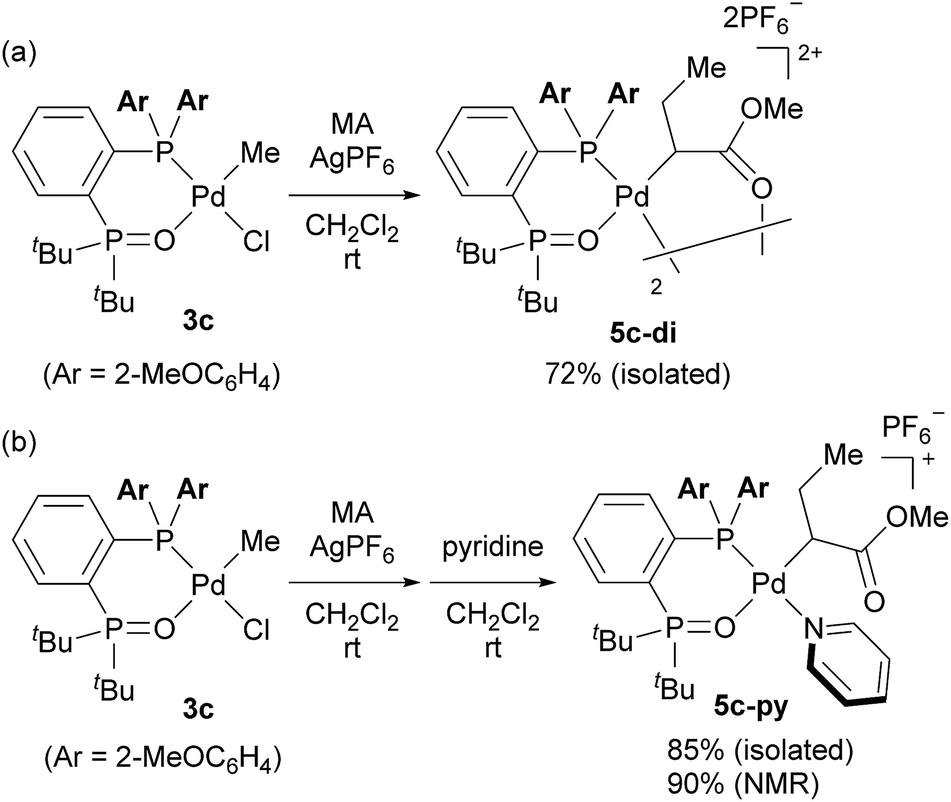 | ||
| Scheme 3 Insertion of methyl acrylate into aryl-BPMO–palladium complex 3c. | ||
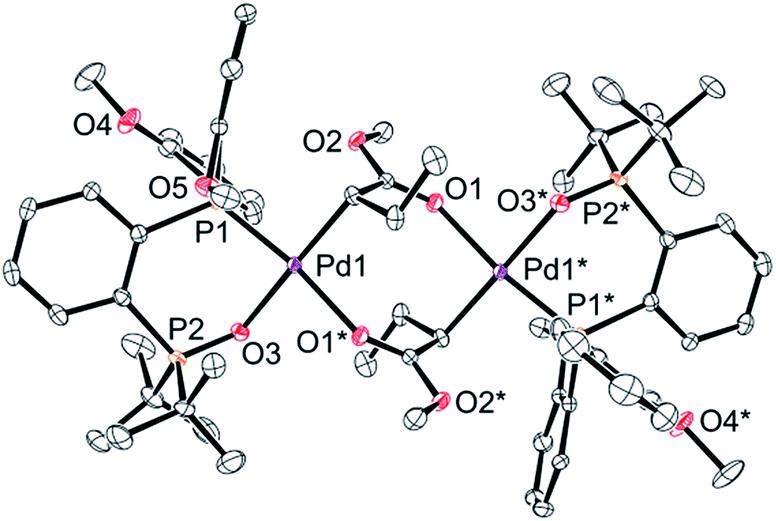 | ||
| Fig. 3 An X-ray structure of complex 5c-di. Thermal ellipsoids are shown at 50% probability. Hydrogen atoms and counter anions are omitted for clarity. | ||
Additionally, reaction of 3c with silver hexafluorophosphate in the presence of MA followed by trapping with pyridine afforded 5c-py in high overall yield (90%) as determined by 1H NMR yield (Scheme 3b). Importantly, a palladium complex corresponding to 1,2-insertion of MA was not detected in either case. The selective 2,1-insertion of MA was also suggested for the reaction of MA with an analogous aryl-BPMO–palladium complex with a bis[2-(trifluoromethyl)phenyl]phosphino group (3d; see ESI†). These experiments indicate a clear difference in insertion regioselectivity for BPMO–palladium catalysts that are, or are not, active for copolymerisation of ethylene and MA, favoring exclusive 2,1-insertion regioselectivity of MA for highly active BPMO–palladium complexes with an ortho-substituted diarylphosphino group.
Conclusions
In conclusion, a new generation of cationic bisphosphine monoxide–palladium catalysts with a diarylphosphino moiety was shown to exhibit markedly improved performance for the copolymerisation of ethylene with methyl acrylate. Mechanistic studies revealed that the contrasting reactivity between these aryl-BPMO and previously reported inactive alkyl-BPMO catalysts was a shift to higher 2,1-insertion regioselectivity of methyl acrylate that avoids generation of a stable palladacycle intermediate that is poorly reactive towards additional monomer enchainment. Newly developed aryl-BPMO catalysts can also copolymerise ethylene with other industrially important polar monomers. Future studies will be directed toward revealing the reason why the higher 2,1-selectivity was observed for reaction of methyl acrylate with 3c or 3d as compared to 3a.27Acknowledgements
This work was supported by JST and CREST. Y. M. is grateful to Program for Leading Graduate Schools (MERIT) from JSPS.Notes and references
- Crystalinity of polypropylene:
(a) J. C. Chadwick, F. P. T. J. van der Burgt, S. Rastogi, V. Busico, R. Cipullo, G. Talarico and J. J. R. Heere, Macromolecules, 2004, 37, 9722 CrossRef CAS
; (b) C. de Rosa, F. Auriemma, M. Paolillo, L. Resconi and I. Camurati, Macromolecules, 2005, 38, 9143 CrossRef CAS
- α-Diimine/metal catalysts afford various microstructures in polymerisations of α-olefins depending on regioselectivity. Review:
(a) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169 CrossRef CAS PubMed
- Regioselectivity effect on β-elimination during copolymerisation catalysed by late-transition metals:
(a) R. A. Stockland Jr and R. F. Jordan, J. Am. Chem. Soc., 2000, 122, 6315 CrossRef
-
(a) R. F. Heck, Acc. Chem. Res., 1979, 12, 146 CrossRef CAS
- A. Nakamura, S. Ito and K. Nozaki, Chem. Rev., 2009, 109, 5215 CrossRef CAS PubMed
-
(a) P. Wucher, L. Caporaso, P. Roesle, F. Ragone, L. Cavallo, S. Mecking and I. Göttker-Schnetmann, Proc. Natl. Acad. Sci., U. S. A, 2011, 108, 8955 CrossRef CAS PubMed
- For experimental results, see: S. Mecking, L. K. Johnson, L. Wang and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 888 CrossRef CAS
- For calculation results, see:
(a) A. Michalak and T. Ziegler, J. Am. Chem. Soc., 2001, 123, 12266 CrossRef CAS PubMed
- For insertion into a palladium–alkyl bond, see:
(a) D. Guironnet, P. Roesle, T. Rünzi, I. Göttker-Schnetmann and S. Mecking, J. Am. Chem. Soc., 2009, 131, 422 CrossRef CAS PubMed
- For insertion into a palladium–acyl bond, see:
(a) A. Nakamura, K. Munakata, T. Kochi and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 8128 CrossRef CAS PubMed
- N. D. Contrella, J. R. Sampson and R. F. Jordan, Organometallics, 2014, 33, 3546 CrossRef CAS
- The BArF4 complexes generally showed higher catalytic activity for polymerisation than the corresponding SbF6 complexes. Thus, all the (co)polymerisations in this manuscript were performed using BArF4 complexes.
- B. P. Carrow and K. Nozaki, J. Am. Chem. Soc., 2012, 134, 8802 CrossRef CAS PubMed
-
(a) A. Berkefeld and S. Mecking, Angew. Chem., Int. Ed., 2008, 47, 2538 CrossRef CAS PubMed
-
(a) L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267 CrossRef CAS
- While the manuscript was under review, palladium catalysts ligated by phosphine-phosphonic amide, an analog of BPMO, was reported to promote ethylene/polar monomer copolymerisations. See: X. Sui, S. Dai and C. Chen, ACS Catal., 2015, 5, 5932 CrossRef CAS
- Mecking et al., pointed out that the 1,2-insertion products may retard ethylene/methyl acrylate copolymerisation by palladium/phosphine-sulfonate catalysts. See ref. 9c.
- Although we could not obtain high quality single crystals of 5a, low quality single crystals of the dimer of 5a were grown from dichloromethane/diethyl ether. A primitive X-ray analysis data of dimer 5a-di was presented in ESI†.
- See ESI† for the NMR spectra.
- Complex 4a in the solution state is stable at up to 100 °C. In contrast, complex 5a in tetrachloroethane-d2, was gradually decomposed upon heating at 80 °C to give methyl crotonate via β-hydride elimination. This observation suggests that 5a undergoes decomposition before the rearrangement to the 5-membered palladacycle as observed for α-diimine palladium catalysts under the assumption that stability of 4a and 5-membered palladacycle, which can be formed by β-hydride elimination and subsequent re-insertion, is similar.
- It is possible that rapid chain transfer after β-H elimination prevented the chain growth. If that is the case, a significant amount of oligomer should be formed during copolymerisation. However, oligomer was not detected by gas chromatography analysis of the reaction mixture after copolymerisation. See ESI†.
- The reason is not unclear at present.
- Since the copolymerisation with 10 μmol of 2f for 15 hours produced a saturated amount of copolymer in a stainless autoclave, the reaction needs to be performed with lower catalyst loading and shorter time. See ESI† for full data of polymerisation.
- Given that the incorporation ratio of MA in entry 10 was slightly lower than those in other entries, the higher catalytic activity may partly be attributed to 2f's preference of ethylene insertion to MA insertion.
- S. Ito, M. Kanazawa, K. Munakata, J. Kuroda, Y. Okumura and K. Nozaki, J. Am. Chem. Soc., 2011, 133, 1232 CrossRef CAS PubMed
- P. Wucher, V. Goldbach and S. Mecking, Organometallics, 2013, 32, 4516 CrossRef CAS
- We calculated % Vbur values of each BPMO ligand and found the BPMO ligand in 3a and 3c were sterically similar while the BPMO ligand in 3d was suggested to be bulkier. See ESI† for the discussion on regioselectivity of MA based on the steric bulkiness of BPMO ligands.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, NMR spectra of complexes and (co)polymers, and X-ray crystallographic data. CCDC (3c: 1408864, 4a: 14088655a-py: 1408866, 5c-di: 14088675d-py: 1408868). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03361f |
| This journal is © The Royal Society of Chemistry 2016 |