
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic activation of a single C–F bond in trifluoromethyl arenes†
Hester
Dang
,
Aaron M.
Whittaker
and
Gojko
Lalic
*
Department of Chemistry, University of Washington, Seattle, Washington 98195, USA. E-mail: lalic@chem.washington.edu
First published on 23rd October 2015
Abstract
Synthetic methods for the direct transformation of ArCF3 to ArCF2R would enable efficient diversification of trifluoromethyl arenes and would be of great utility in medicinal chemistry. Unfortunately, the development of such methods has been hampered by the fundamental properties of C–F bonds, which are exceptionally strong and become stronger with increased fluorination of the carbon atom. Here, we describe a method for the catalytic reduction of ArCF3 to ArCF2H through a highly selective activation of a single C–F bond. Mechanistic studies reveal separate reaction pathways for the formation of ArCF2H and ArCH3 products and point to the formation of an unexpected intermediate as the source of the unusual selectivity for the mono-reduction.
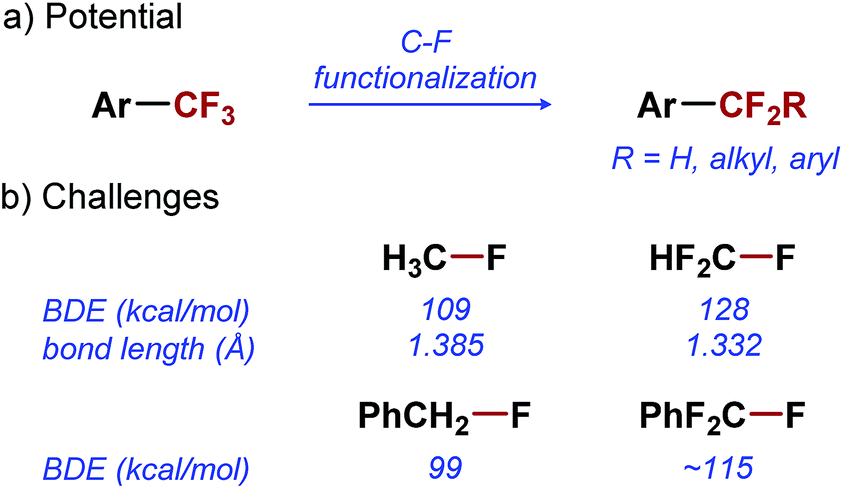
Fluorinated organic molecules play an important role in the pharmaceutical1,2 and agrochemical industries.3 Fluorine atoms are commonly introduced into organic molecules in an effort to manipulate the physical and chemical properties of these compounds, without making drastic changes to their overall structure.4,5 Trifluoromethylation of arenes is one of the most commonly used methods for fluorination of organic molecules, and can be accomplished using one of the numerous synthetic methods developed over the last two decades.6–9 This relatively small structural change results in increased bioavailability, lipophilicity, and metabolic stability of aromatic compounds,10,11 and often leads to an improved pharmacological profile. As a result of the favorable pharmacological properties imparted by trifluoromethylation, trifluoromethyl arenes are extensively used in medicinal chemistry and are present in a number of prescription drugs, including Prozac, Celebrex, and Januvia.
Difluoroalkyl arenes (ArCF2R) are analogues of ArCF3 that hold great potential for applications in medicinal chemistry. Difluoroalkyl substituents have a similar effect on the pharmacokinetic properties of arenes as CF3 substituent.12 Moreover, they offer a greater structural and functional diversity, which is often critical in the optimization of bio-active lead compounds.13 To fully exploit the potential of ArCF2R in medicinal chemistry, transformations that would enable their synthesis directly from ArCF3 precursors are needed (Scheme 1a).14–20 Direct mono-functionalization of ArCF3 would be the most efficient strategy for diversification of this important class of compounds, and would effectively leverage the reliable access to ArCF3 provided by synthetic methods developed in the last two decades.6–9
 | ||
| Scheme 1 Selective activation of a single C–F bond in ArCF3. | ||
Unfortunately, selective functionalization of a single C–F bond in ArCF3 remains one of the great unmet challenges in organic chemistry. The problems associated with the selective C–F functionalization become evident when we consider the fundamental properties of C–F bonds in ArCF3. The C–F bond is the strongest single bond to carbon, with a bond dissociation energy (BDE) of 109 kcal mol−1 in CH3–F.21 Even a relatively weak C–F bond in benzyl fluoride has a BDE of 99 kcal mol−1 (Scheme 1b).22 Not surprisingly, any transformation involving the activation of such strong bonds is difficult to accomplish, despite the significant progress made in the field of C–F bond activation.23–25 Furthermore, with increased fluorination of a carbon atom, C–F bonds become stronger and shorter, and thus less reactive (Scheme 1b).26–28 As a result, selective functionalization of a single C–F bond in ArCF3 poses an additional selectivity challenge: the product of the C–F functionalization (ArCF2R) is inherently more reactive than the starting material (ArCF3). Overcoming this selectivity problem has proven to be particularly difficult. Only reported examples of the selective monofunctionalization of ArCF3 are based on single electron transfer reactions achieved by electrochemistry29 or alkali earth metal reduction.30,31 So far, no catalytic methods for functionalization of a single C–F bond in a trifluoromethyl group have been reported in the literature.
In this report, we describe the discovery of a transition metal-catalyzed activation of a single C–F bond in ArCF3 that results in the selective formation of ArCF2H. We present the results of mechanistic studies that provide insight into the source of the unusual selectivity and the results of our exploration of the substrate scope.
We recently discovered that the reduction of 4-(4-MePh)C6H4CF3 (1) to 4-(4-MePh)C6H4CH3 (2) can be accomplished at room temperature using just 1 mol% of palladium(II) acetate as a catalyst, in the presence of triphenylsilane and potassium tert-butoxide (Table 1, entry 1). We also found that replacing potassium tert-butoxide with sodium tert-butoxide abolishes the reactivity. Even at a higher temperature reduction products (ArCH3 and ArCF2H) are obtained in low yields (Table 1, entry 2). However, the addition of a copper co-catalyst restores the reactivity, and leads to the formation of 4-(4-MePh)C6H4CF2H as the major product of the reaction (Table 1, entry 3). Considering that known methods for catalytic reduction of ArCF3 using Lewis acid catalysis, early transition metal catalysis or late transition metal catalysis, produce only the fully reduced ArCH3 products,32–38 we were intrigued by the unusual selectivity of our reduction reaction and decided to pursue the development of a synthetic method for the selective mono-reduction of ArCF3.
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Catalyst | Base | 2 | 3 | Conversion |
| a Reaction conditions: Ph3SiH (4 equiv.), base (5 equiv.), DMF, 25 °C, 1 h. b Reaction performed at 45 °C for 11 h then 60 °C for 17 h. c Reaction performed at 45 °C for 2 h, followed by 60 °C for 17 h. Ar = 4-(4-CH3Ph)C6H4; SIPr = N,N′-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene; DMF = N,N-dimethylformamide. All yields were determined by GC using an internal standard. | |||||
| 1 | Pd(OAc)2 1 mol% | KOt-Bu | 94% | 0% | 100% |
| 2b | Pd(OAc)2 1 mol% | NaOt-Bu | 8% | 7% | — |
| 3b | Pd(OAc)2 1 mol% & SIPrCuCl 1 mol% | NaOt-Bu | 12% | 52% | 90% |
| 4c | Pd(OAc)2 3 mol% & SIPrCuCl 20 mol% | NaOt-Bu | 15% | 51% | 100% |
| 5c | Pd(OAc)2 3 mol% & SIPrCuCl 20 mol% & 2-pyridone 5 mol% | NaOt-Bu | 3% | 56% | 100% |
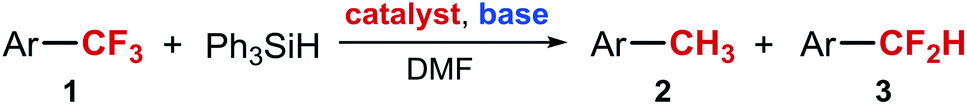
In the initial experiments we confirmed that both copper and palladium catalysts were necessary for the observed selectivity. Furthermore, we found that in the absence of both catalysts, starting material is fully recovered. Our initial attempts to optimize the reaction focused on improving the conversion of the starting material. We achieved complete conversion of 4-(4-MePh)C6H4CF3 by increasing the catalyst loading and modifying the ratio of the two catalysts (Table 1, entry 4). However, these changes did not improve the yield and instead decreased the selectivity. We also explored modifying the palladium catalyst with various classes of ligands and found that phosphine and N-heterocyclic carbene (NHC) ligands completely inhibited the reaction. The presence of amines, diamines, amides, and amino alcohols had a small, but consistently positive effect on selectivity. The greatest improvement was observed in the presence of 5 mol% of 2-pyridone (Table 1, entry 5). To facilitate further reaction optimization, we decided to focus on gaining a better understanding of the reaction mechanism and the source of the observed selectivity.
Our first hypothesis was that ArCF2H is an intermediate in the formation of ArCH3 and that high selectivity can be achieved only at a low conversion or with a low mass balance. However, the results of experiments in which we closely monitored the progress of the reaction shown in entry 3 of Table 1 contradicted this hypothesis. We found that the conversion of the starting material reached 90% and stopped after 11 hours at 45 °C. At this point, most of the fully reduced ArCH32 had already been formed (11%), while almost no ArCF2H 3 (4%) was present in the reaction mixture (see ESI for details†). At the same time point, the major component of the reaction mixture was intermediate X, which over the next 18 hours was converted into ArCF2H 3 (Scheme 2). Over the same period, essentially no more 2 was formed (12% vs. 11%). The same observations were made when the reaction was performed in the presence of 2-pyridone (Table 1, entry 5) (see ESI for details†). The only difference is that 2-pyridone inhibits the formation of 2. Overall, the results of these experiments suggest that the formation of ArCH3 and ArCF2H occurs through two independent reaction pathways, and that ArCF2H and intermediate X are not intermediates in the formation of ArCH3 (Scheme 2). Encouragingly, this finding suggested that, at least in principle, high yields of ArCF2H can be obtained with high selectivity.
 | ||
| Scheme 2 | ||
Our next goal was to determine the structure of intermediate X. The analysis of the reaction mixture by mass spectrometry suggested aldehyde 4 as the intermediate (Scheme 3). Furthermore, a reaction performed in DMF-d7 solvent resulted in the formation of the mono-deuterated intermediate (mass increased by 1 amu), indicating that the formyl group of ArCF2CHO is likely derived from DMF. From the reaction mixture containing intermediate X as the major species, we were also able to isolate the 2,4-dinitrophenyl hydrazone of 4-(4-MePh)C6H4CF2CHO (5) in 50% yield and characterize it by X-ray crystallography (Scheme 3). On the other hand, attempts to establish the presence of the aldehyde in the reaction mixture by in situ methods were not successful.
 | ||
| Scheme 3 Structure of intermediate X. | ||
For example, in situ analysis of the reaction mixture by 1H and 13C NMR indicated no presence of ArCF2CHO. Furthermore, a significant amount of the aldehyde could be identified in the mass spectrum only if the sample was exposed to air and silica gel. Overall, these results are consistent with the idea that intermediate X is a hemiacetal or a hemiaminal of ArCF2CHO 4, or a silylated version of the two, and that the aldehyde is revealed only in the presence of a protic acid (MeOH or moisture).
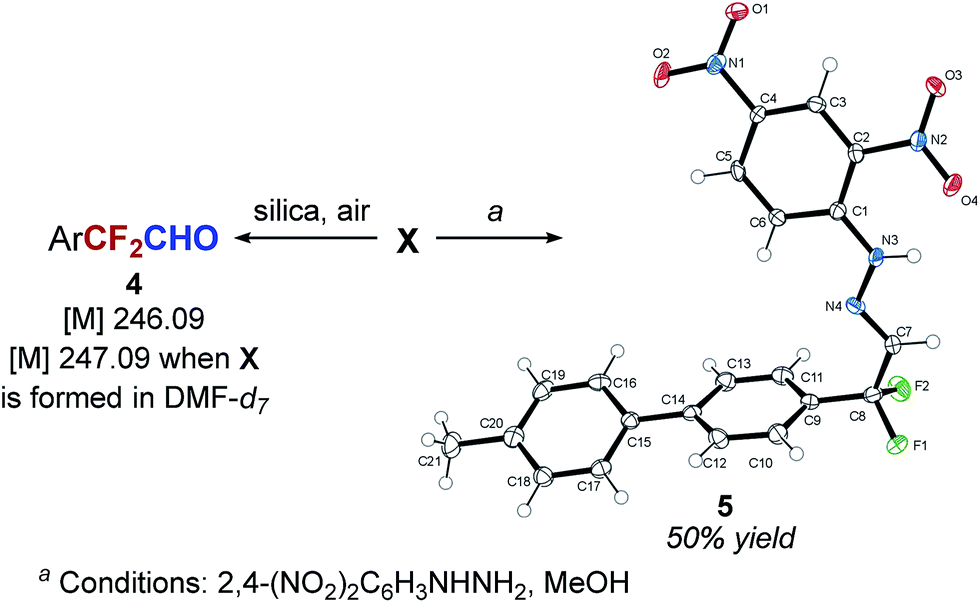
Based on the hypothesis that intermediate X is a silylated hemiacetal or a hemiaminal, we speculated that the relatively slow conversion of intermediate X to the aldehyde before decarbonylation to the difluoromethyl arene may be responsible for the low yield of the desired product. We reasoned that decomposition of the intermediate could be facilitated by the presence of a mild Brønsted acid. Indeed, we found that if 1 equivalent of tert-butanol was added to the reaction mixture after full conversion of the 4-(4-MePh)C6H4CF3 is achieved, there was a dramatic increase in both the rate of the reaction and in the yield of ArCF2H. The complete conversion of intermediate X was achieved in 2 hours at 60 °C, and ArCF2H 3 was obtained in 95% yield (Scheme 4). Similar results can be accomplished using phenol as an acid. Addition of an acid at the beginning of the reaction resulted in significantly lower yields.
 | ||
| Scheme 4 a) The effect of Brønsted acid b) Deuterium incorporation studies | ||
To gain more information about the mechanism of decarbonylation and the role of the acid we performed isotope labeling experiments. We found that the hydrogen atom in ArCF2H 3 is derived from tert-butanol, and not from the silane or from DMF (via intermediate X) (Scheme 4b) (see ESI for details†). Based on these results, we favor the mechanism of decarbonylation that involves a difluorobenzyl anion intermediate, akin to the haloform reaction. It is interesting to note that the presence of the two intermediates (intermediate X and difluorobenzyl anion) in this transformation provides a great opportunity for the development of other transformations of ArCF3.
Finally, we were interested in the fate of the fluoride removed from the ArCF3 and the mechanism of C–F activation. In a crude reaction mixture we were not able to identify any silyl fluoride, which we expected to be the major byproduct of the reaction. Instead, the only fluoride-containing byproduct was KF. In a control experiment, we found that Ph3SiF reacts with KOt-Bu under the reaction conditions to produce KF and Ph3SiOt-Bu, which further complicates interpretation of our findings in the context of the mechanism of the C–F activation.
The general understanding of the catalytic C–F activation by transition metals suggest that ArCF2H would be more reactive than ArCF3.21,39 However, in our reaction, we found that the opposite is true. With 4-(4-MePh)C6H4CF2H as the substrate, only ∼36% conversion is achieved at the time necessary for the full conversion of ArCF31 (2 h at 45 °C) (Scheme 5). One mechanistic hypothesis that accounts for this unusual order of reactivity (ArCF3 > ArCF2H) is C–F activation by a single electron transfer: ArCF3 are better electron acceptors than ArCF2H,29 although the difference in energy of σ*C–F orbitals is often too small to allow good selectivity.40,41 This mechanism would also explain why intermediate X is resistant to further reduction. Furthermore, mixtures of silanes and alkoxides have previously been implicated in single electron chemistry.42,43 However, it is important to note that the presence of transition metal catalysts is essential for C–F activation. In the absence of transition metals, we can fully recover the starting material, in agreement with the observations made by Grubbs and Stoltz.43
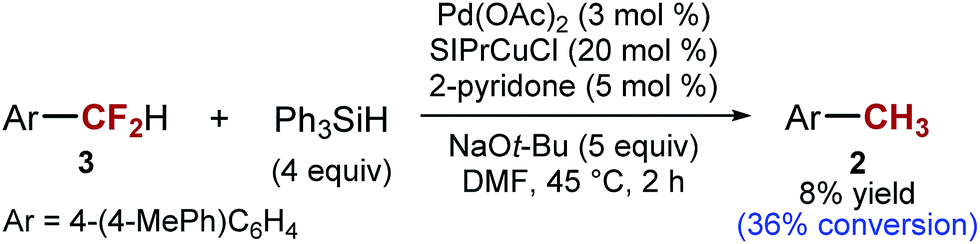 | ||
| Scheme 5 C–F activation of ArCF2H. | ||
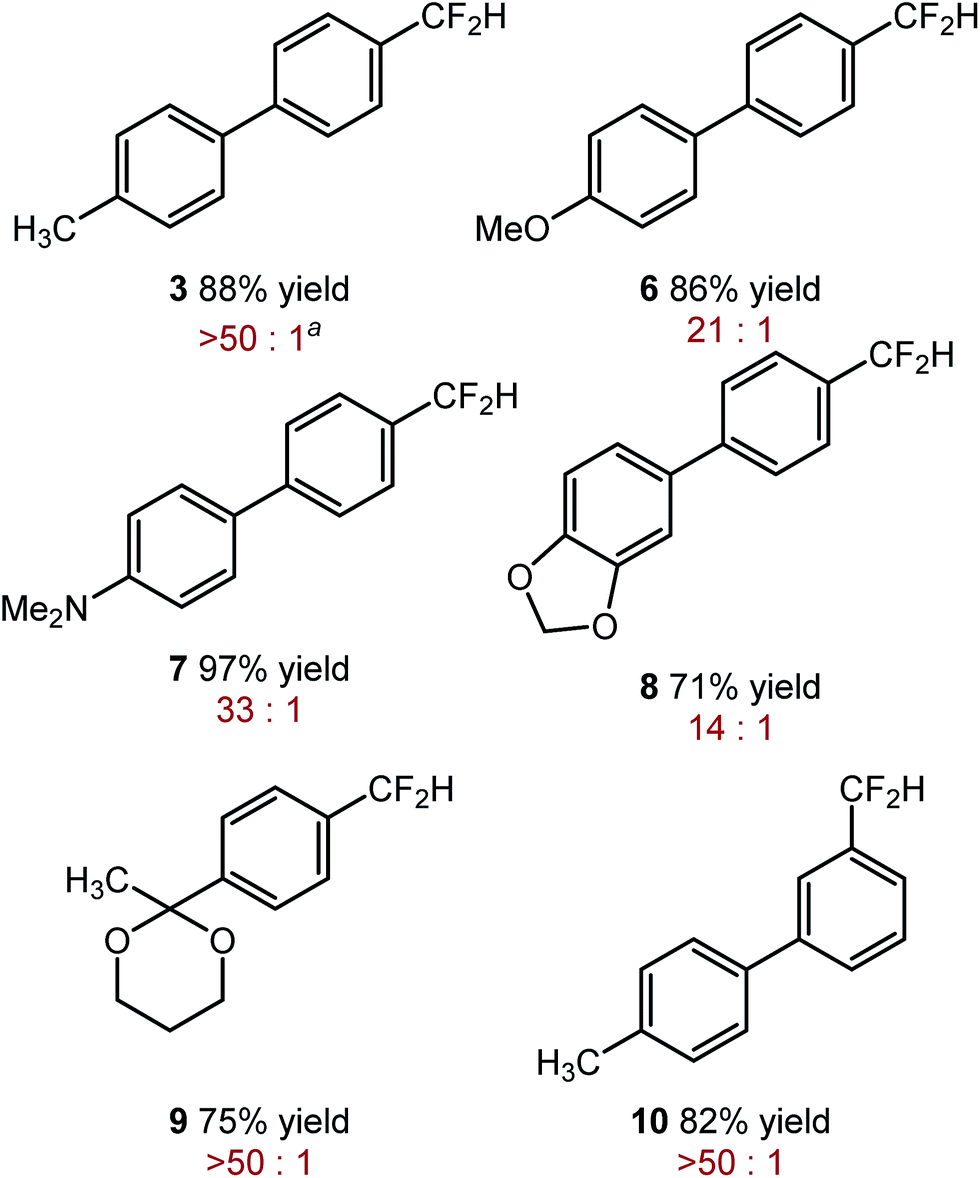
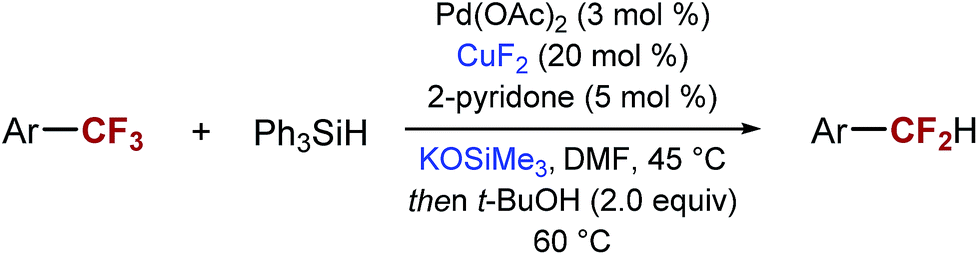
After exploring the mechanism of the mono-defluorination reaction, we turned our attention to further the reaction optimization and the exploration of the substrate scope. In the process, we found that SIPrCuCl could be replaced by significantly less expensive and commercially available CuF2 or CuBr, with CuF2 generally providing superior selectivity. Other copper(I) and copper(II) salts gave significantly lower yields of the desired product and/or significantly lower selectivity. We also found that consistently better selectivity could be obtained using KOSiMe3 instead of NaOt-Bu.
The optimized reaction conditions shown in Table 2, were used in mono-reduction of several trifluoromethyl arenes. We found that the reaction can be accomplished in the presence of amines, ethers, and acetals with several biphenyl and aryl substrates. Reducible groups, such as aryl halides, nitro arenes, or nitriles are reduced under the reaction conditions. ortho-Substituted trifluoromethyl arenes provided low yields of the desired products.
|
|
|---|
| a Ratio of ArCF2H and ArCH3. See ESI for a detailed description of experimental procedure. |
|
Conclusions
We have discovered a combination of palladium and copper catalysts that allows selective activation of a single C–F bond in trifluoromethyl arenes under relatively mild conditions. This discovery allowed the development of a method for selective reduction of ArCF3 to ArCHF2. More importantly, the unique mechanism of the reaction and the unusual source of the selectivity provide new opportunities for the development of useful transformations based on selective C–F activation.Acknowledgements
Financial support by NSF (NSF CAREER award #1254636) is gratefully acknowledged. We gratefully acknowledge Sarah E. Flowers for assistance with X-ray crystallography.Notes and references
- K. L. Kirk, J. Fluorine Chem., 2006, 127, 1013–1029 CrossRef CAS PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed.
- B. E. Smart, J. Fluorine Chem., 2001, 109, 3–11 CrossRef CAS.
- K. L. Kirk, Curr. Top. Med. Chem., 2006, 6, 1447–1456 CrossRef CAS.
- C. Alonso, E. Martínez de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847–1935 CrossRef CAS PubMed.
- Z. Jin, G. B. Hammond and B. Xu, Aldrichimica Acta, 2012, 45, 67–83 CAS.
- G. Shi, C. Shao, S. Pan, J. Yu and Y. Zhang, Org. Lett., 2015, 17, 38–41 CrossRef CAS PubMed.
- M. Shang, S.-Z. Sun, H.-L. Wang, B. N. Laforteza, H.-X. Dai and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 10439–10442 CrossRef CAS PubMed.
- G. Gerebtzoff, X. Li-Blatter, H. Fischer, A. Frentzel and A. Seelig, ChemBioChem, 2004, 5, 676–684 CrossRef CAS PubMed.
- B. K. Park and N. R. Kitteringham, Drug Metab. Rev., 1994, 26, 605–643 CrossRef CAS PubMed.
- G. D. Diana, P. Rudewicz, D. C. Pevear, T. J. Nitz, S. C. Aldous, D. J. Aldous, D. T. Robinson, T. Draper and F. J. Dutko, J. Med. Chem., 1995, 38, 1355–1371 CrossRef CAS.
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
- For examples of catalytic methods that can be used to prepare ArCF2R from other precursors, see ref. 12–17.
- Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494–1497 CrossRef CAS PubMed.
- P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5524–5527 CrossRef CAS PubMed.
- Q.-Q. Min, Z. Yin, Z. Feng, W.-H. Guo and X. Zhang, J. Am. Chem. Soc., 2014, 136, 1230–1233 CrossRef CAS PubMed.
- K. Fujikawa, Y. Fujioka, A. Kobayashi and H. Amii, Org. Lett., 2011, 13, 5560–5563 CrossRef CAS PubMed.
- J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494–17500 CrossRef CAS PubMed.
- P. Xu, S. Guo, L. Wang and P. Tang, Angew. Chem., Int. Ed., 2014, 53, 5955–5958 CrossRef CAS PubMed.
- J. Burdeniuc, B. Jedlicka and R. H. Crabtree, Chem. Ber., 1997, 130, 145–154 CrossRef CAS PubMed.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Taylor and Francis Group, LLC, Boca Raton, 2007 Search PubMed.
- M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328–3348 CrossRef PubMed.
- H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed.
- T. A. Unzner and T. Magauer, Tetrahedron Lett., 2015, 56, 877–883 CrossRef CAS PubMed.
- J. A. Pople, L. Radom and W. J. Hehre, J. Am. Chem. Soc., 1971, 93, 289–300 CrossRef CAS.
- K. B. Wiberg and P. R. Rablen, J. Am. Chem. Soc., 1993, 115, 614–625 CrossRef CAS.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- Y. Yamauchi, T. Fukuhara, S. Hara and H. Senboku, Synlett, 2008, 438–442 CAS.
- H. Amii, Y. Hatamoto, M. Seo and K. Uneyama, J. Org. Chem., 2001, 66, 7216–7218 CrossRef CAS.
- S. Utsumi, T. Katagiri and K. Uneyama, Tetrahedron, 2012, 68, 1085–1091 CrossRef CAS PubMed.
- C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188–1190 CrossRef CAS PubMed.
- R. Panisch, M. Bolte and T. Müller, J. Am. Chem. Soc., 2006, 128, 9676–9682 CrossRef CAS PubMed.
- T. Stahl, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 1248–1251 CrossRef CAS PubMed.
- K. Fuchibe and T. Akiyama, J. Am. Chem. Soc., 2006, 128, 1434–1435 CrossRef CAS PubMed.
- K. Fuchibe, Y. Ohshima, K. Mitomi and T. Akiyama, Org. Lett., 2007, 9, 1497–1499 CrossRef CAS PubMed.
- S. Sabater, J. A. Mata and E. Peris, Nat. Commun., 2013, 4, 2553 Search PubMed.
- W. Zhao, J. Wu and S. Cao, Adv. Synth. Catal., 2012, 354, 574–578 CrossRef CAS PubMed.
- T. L. Gianetti, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2013, 135, 8145–8148 CrossRef CAS PubMed.
- P. Clavel, G. Lessene, C. Biran, M. Bordeau, N. Roques, S. Trévin and D. D. Montauzon, J. Fluorine Chem., 2001, 107, 301–310 CrossRef CAS.
- J. H. Stocker and R. M. Jenevein, Chem. Commun., 1968, 934–935 RSC.
- A. Fedorov, A. A. Toutov, N. A. Swisher and R. H. Grubbs, Chem. Sci., 2013, 4, 1640–1645 RSC.
- A. A. Toutov, W.-B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80–84 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1423553. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03415a |
| This journal is © The Royal Society of Chemistry 2016 |