
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structure and reactivity of a terminal magnesium fluoride compound, [TpBut,Me]MgF: hydrogen bonding, halogen bonding and C–F bond formation†
Michael
Rauch
,
Serge
Ruccolo
,
John Paul
Mester
,
Yi
Rong
and
Gerard
Parkin
*
Department of Chemistry, Columbia University, New York, New York 10027, USA. E-mail: parkin@columbia.edu
First published on 17th November 2015
Abstract
The bulky tris(3-tert-butyl-5-pyrazolyl)hydroborato ligand, [TpBut,Me], has been employed to obtain the first structurally characterized example of a molecular magnesium compound that features a terminal fluoride ligand, namely [TpBut,Me]MgF, via the reaction of [TpBut,Me]MgMe with Me3SnF. The chloride, bromide and iodide complexes, [TpBut,Me]MgX (X = Cl, Br, I), can also be obtained by an analogous method using Me3SnX. The molecular structures of the complete series of halide derivatives, [TpBut,Me]MgX (X = F, Cl, Br, I) have been determined by X-ray diffraction. In each case, the Mg–X bond lengths are shorter than the sum of the covalent radii, thereby indicating that there is a significant ionic component to the bonding, in agreement with density functional theory calculations. The fluoride ligand of [TpBut,Me]MgF undergoes halide exchange with Me3SiX (X = Cl, Br, I) to afford [TpBut,Me]MgX and Me3SiF. The other halide derivatives [TpBut,Me]MgX undergo similar exchange reactions, but the thermodynamic driving forces are much smaller than those involving fluoride transfer, a manifestation of the often discussed silaphilicity of fluorine. In accord with the highly polarized Mg–F bond, the fluoride ligand of [TpBut,Me]MgF is capable of serving as a hydrogen bond and halogen bond acceptor, such that it forms adducts with indole and C6F5I. [TpBut,Me]MgF also reacts with Ph3CCl to afford Ph3CF, thereby demonstrating that [TpBut,Me]MgF may be used to form C–F bonds.
Introduction
As a consequence of its small size, high electronegativity and low polarisability, the chemistry of fluorine is often distinctly different from that of the other halogens.1–4 For example, metal fluoride compounds often exhibit novel structures5 and reactivity,1b,2,6,7,8,9 but are generally more difficult to obtain than the other halide derivatives. As an illustration, while Grignard reagents (RMgX) are readily synthesized upon treatment of magnesium with RCl, RBr, or RI, the corresponding fluoro Grignard reagents are notoriously difficult to obtain2–4 and have been investigated to a negligible extent by comparison to the other halogen derivatives. The paucity of magnesium fluoride compounds is not, however, restricted to Grignard reagents, as illustrated by the fact that fluoride derivatives comprise only 2.4% of all structurally characterized magnesium halide compounds listed in the Cambridge Structural Database (CSD).10 Even more striking, in none of these compounds does fluorine serve the role of a terminal ligand. Magnesium fluoride compounds are, nevertheless, of considerable importance in view of the role that they have played in biological systems. For example, the use of in situ generated [MgF3]− to provide transition state analogues of the [PO3]− moiety has generated information concerned with the mechanism of phosphoryl transfer as catalyzed by enzymes.11–13 Here, we report the synthesis and structural characterization of a terminal magnesium fluoride complex, together with its ability to participate in (i) hydrogen bonding and halogen bonding interactions (both of which are important with respect to crystal engineering),14 and also (ii) C–F bond formation, which is of note because of the significance of introducing fluorine into organic molecules.1a,15Results and discussion
We have previously employed tris(pyrazolyl)hydroborato ligands, [TpR,R′],16 that feature bulky tert-butyl substituents, namely [TpBut], [TpBut,Me] and [ ], to provide a sterically demanding pocket about a metal center that enables the isolation of a variety of novel compounds. For example, [TpBut,R′] ligands provided the first structurally characterized examples of (i) a monomeric zinc hydride compound, [TpBut]ZnH,17 (ii) a monomeric terminal zinc hydroxide compound, [TpBut,Me]ZnOH,18 and (iii) a monomeric monovalent gallium compound, [
], to provide a sterically demanding pocket about a metal center that enables the isolation of a variety of novel compounds. For example, [TpBut,R′] ligands provided the first structurally characterized examples of (i) a monomeric zinc hydride compound, [TpBut]ZnH,17 (ii) a monomeric terminal zinc hydroxide compound, [TpBut,Me]ZnOH,18 and (iii) a monomeric monovalent gallium compound, [ ]Ga.19 This class of ligand was also used to synthesize monomeric magnesium chloride, bromide and iodide compounds,20,21 thereby suggesting the possibility that it could also afford a terminal magnesium fluoride compound.
]Ga.19 This class of ligand was also used to synthesize monomeric magnesium chloride, bromide and iodide compounds,20,21 thereby suggesting the possibility that it could also afford a terminal magnesium fluoride compound.
Significantly, we have discovered that the fluoride compound [TpBut,Me]MgF can indeed be obtained readily upon treatment of [TpBut,Me]MgMe22 in benzene with the tin reagent, Me3SnF,23 as illustrated in Scheme 1. The chloride, bromide and iodide complexes, [TpBut,Me]MgX can also be obtained by the analogous method using Me3SnX (X = Cl, Br, I; Scheme 1).
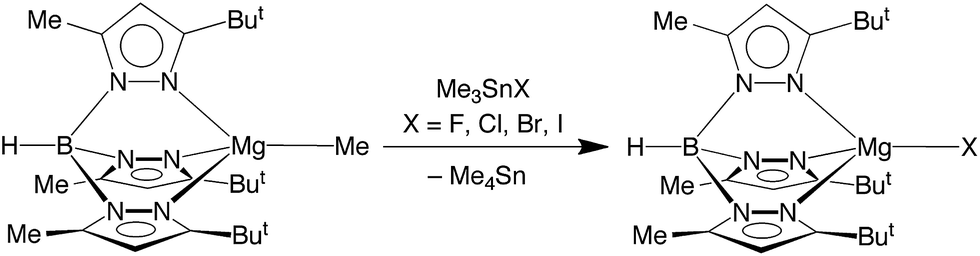 | ||
| Scheme 1 Synthesis of [TpBut,Me]MgX. | ||
The molecular structure of [TpBut,Me]MgF has been determined by X-ray diffraction (Fig. 1), thereby demonstrating that the compound is mononuclear and possesses a terminal fluoride ligand. As noted above, there are no similar compounds listed in the CSD, with other magnesium fluoride derivatives exhibiting various types of bridging interactions, which include μ2-,24–26 μ3-,27 and μ4-modes.28,29 Terminal Mg–F moieties have, nevertheless, been structurally characterized in protein structures.11a,d,e,h,i
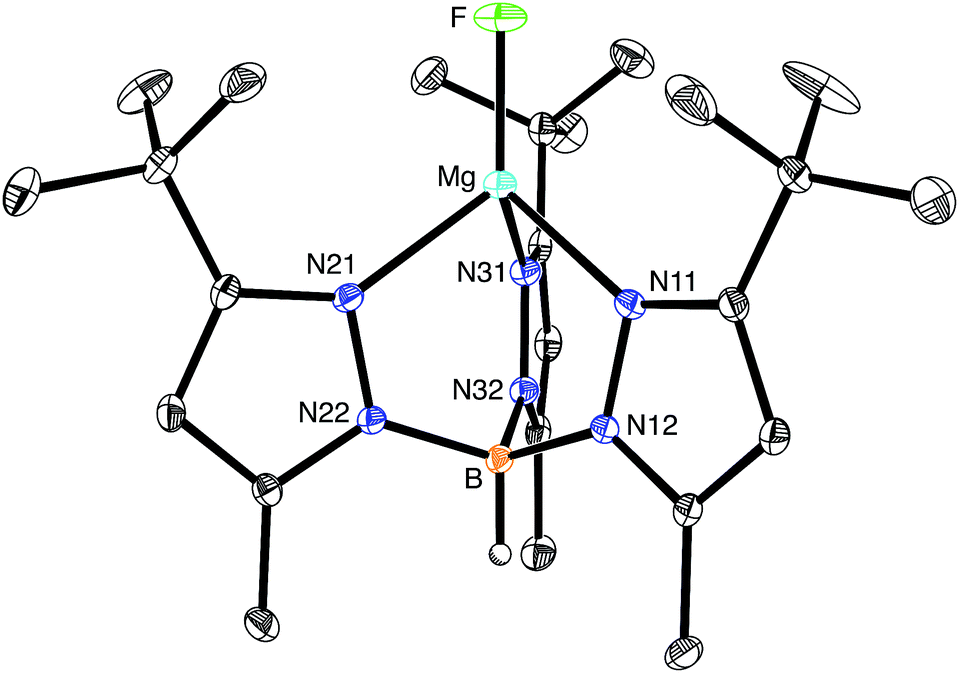 | ||
| Fig. 1 Molecular structure of [TpBut,Me]MgF. | ||
As would be expected, the Mg–F bond of [TpBut,Me]MgF [1.7977(11) Å] is considerably shorter than those with bridging fluoride ligands.30 For example, the Mg–F bond lengths of dinuclear {[BDIAr]Mg(μ-F)(THF)}2 with μ2-bridges are 1.951(2) Å,24,31 while those of trinuclear [Mg3(μ3-F)(μ2-TFA)6(OMe)2(py)]3− with a μ3-bridge range from 2.012(5) Å to 2.047(4) Å.27 Correspondingly, magnesium fluoride compounds that feature μ4-bridges exhibit even longer Mg–F bonds that range from 2.12 Å to 2.21 Å.28,32,33 In addition to the Mg–F bond of [TpBut,Me]MgF being shorter than other Mg–F bonds, it is also amongst the shortest Mg–X (X ≠ H) bonds listed in the CSD, as illustrated by the magnesium oxide and alkoxide complexes, [{(THF)[BDIAr]Mg}2(μ-O)] [1.8080(5) Å],34 [MesC{(C4N)Mes}2]Mg(OBut)(THF) [1.804(2) Å],35 and [(ArO)Mg(μ-OAr)2]2Mg (Ar = C6H3Pri2) [1.785(2) Å and 1.790(2) Å].36
Spectroscopically, [TpBut,Me]MgF is characterized by a 19F NMR signal at −169.3 ppm, which is within the range exhibited by the related beryllium and zinc complexes, namely [Tp]BeF (−149 ppm),37 [TpBut,Me]ZnF (−207 ppm),21a and [Tpp-Tol,Me]ZnF (−219 ppm),21a but is very different from the values observed for the dinuclear compounds, {[BDIAr]Mg(μ-F) (THF)}2 (−25 ppm) and {[BDIAr]Mg(μ-F)}2 (−26 ppm).24 While this large difference could be taken as an indication that 19F NMR spectroscopy could be used as a probe of fluoride coordination mode,38 we note that the chemical shift for [TpBut,Me]MgF (−169.3 ppm) is also comparable to the solid state value for Mg6F2(OMe)10(MeOH)14 (−174.5 ppm), which contains μ4-F atoms.28a As such, it is evident that 19F NMR chemical shift data do not provide a definitive probe for the fluoride coordination mode in these systems. Nevertheless, 19F NMR data in a comparable region to that of [TpBut,Me]MgF have been reported in protein systems;11c–h for example, PGM-MgF3-G6P-TSA in 100% H2O buffer exhibits 19F NMR chemical shifts of −147.0, −151.8, and −159.0 ppm.11d
The molecular structures of [TpBut,Me]MgX (X = Cl, Br, I) have also been determined by X-ray diffraction.39 In each case, the molecules possess approximately C3v symmetry, with a magnesium coordination geometry that is distorted considerably from tetrahedral. Specifically, the τ4 four-coordinate geometry indices40 range from 0.79 to 0.82 (Table 1) and deviate considerably from the value of 1.00 for that of an idealized tetrahedron.
| d(M–X)/Å | τ 4 | B⋯M–X/° | |
|---|---|---|---|
| [TpBut,Me]MgF | 1.7977(11) | 0.79 | 177.8 |
| [TpBut,Me]MgCl | 2.2701(15) | 0.81 | 179.2 |
| 2.2677(15) | 0.81 | 179.1 | |
| [TpBut,Me]MgBr | 2.425(2) | 0.81 | 178.9 |
| 2.425(2) | 0.82 | 179.0 | |
| [TpBut,Me]MgI | 2.6696(9) | 0.80 | 177.8 |
The availability of a complete series of structurally characterized halide compounds provides an opportunity to evaluate the bonding as a function of the halogen. The variation of the Mg–X bond lengths is illustrated in Table 1 and Fig. 2, which include, for comparison, the values predicted on the basis of the single bond covalent radii of the elements.
 | ||
| Fig. 2 Comparison of experimental and calculated Mg–X (X = F, Cl, Br, I) bond lengths, together with the sum of Pyykkö and Alvarez covalent radii. | ||
In this regard, it is pertinent to note that two sets of covalent radii have been recently proposed by Alvarez41 and Pyykkö,42,43 and that in each case the experimental Mg–X bond lengths are consistently smaller than those predicted by the sum of the covalent radii. With the exception of the fluoride derivative, the Pyykkö estimates are closer to the experimental bond lengths than are the Alvarez values. Specifically, the experimental Mg–X bond lengths are 0.13–0.19 Å shorter than the Alvarez values, and 0.05–0.23 Å shorter than the Pyykkö values. The magnesium–methyl bond length of [TpBut,Me]MgMe [2.119(3) Å]22 is also shorter than the predicted values, although the difference (0.05 Å, Alvarez; 0.02 Å, Pyykkö) is much smaller than those for the halide derivatives. In addition to being smaller than the sum of the covalent radii, the experimental bond lengths are also shorter than the sum of the respective ionic radii.44
In principle, M–X bond lengths that are shorter than the sum of single-bond covalent radii can be a consequence of either (i) an ionic contribution to the bonding or (ii) π-bonding.45 To investigate this issue, we have examined the series of compounds, [TpBut,Me]MgX (X = F, Cl, Br, I), computationally. Firstly, density functional theory (DFT) geometry optimization calculations reproduce the experimental structures very well, as indicated by the close correspondence between the experimental and calculated Mg–X bond lengths (Fig. 2). Secondly, the calculations indicate that the bonds have a significant ionic component, as illustrated by the atomic charges on the halogen, be they derived from Mulliken, electrostatic potential, or Natural population analysis. Thirdly, the bonds have no M–X π-interactions,46 such that it is the ionic component which provides a mechanism to shorten the Mg–X bond from that predicted by the sum of the covalent radii.47 Thus, both the experimental observations and the theoretical calculations are consistent with the Mg–X bonds having a significant ionic component; furthermore, the calculations indicate that this is greatest for the fluoride derivative (Table 2).
| NPA | Mulliken | ESP | ||||
|---|---|---|---|---|---|---|
| q Mg/e | q X/e | q Mg/e | q X/e | q Mg/e | q X/e | |
| F | 1.733 | −0.828 | 0.658 | −0.496 | 0.334 | −0.516 |
| Cl | 1.660 | −0.809 | 0.526 | −0.408 | 0.358 | −0.431 |
| Br | 1.624 | −0.767 | 0.516 | −0.385 | 0.400 | −0.426 |
| I | 1.597 | −0.736 | 0.534 | −0.400 | 0.448 | −0.393 |
In terms of reactivity, the fluoride compound [TpBut,Me]MgF reacts with Me2Mg to regenerate the methyl derivative, [TpBut,Me]MgMe (Scheme 2). Furthermore, the well known silaphilicity of fluorine48,49 provides a means to convert the fluoride complex [TpBut,Me]MgF to the other halide derivatives via reaction with Me3SiX (X = Cl, Br, I),50 as illustrated in Scheme 2.
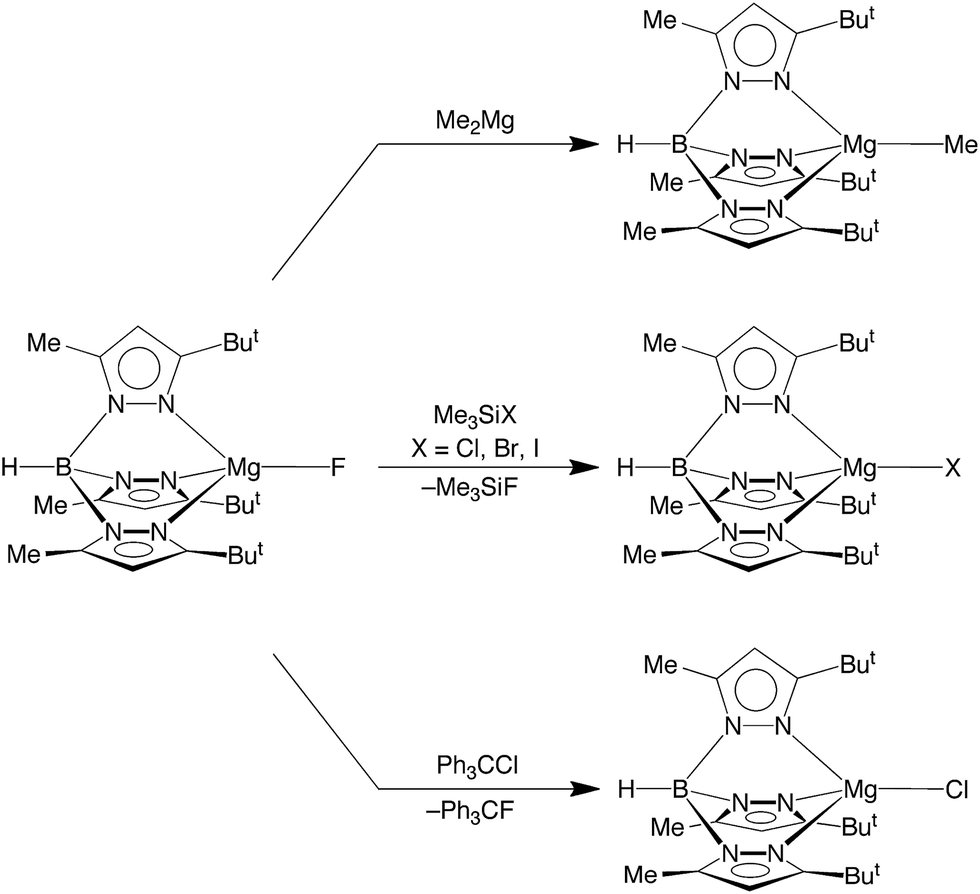 | ||
| Scheme 2 Reactivity of [TpBut,Me]MgF. | ||
More interesting than its reactivity towards Me3SiX, [TpBut,Me]MgF also reacts with Ph3CCl to afford [TpBut,Me]MgCl and Ph3CF (Scheme 2). The ability of [TpBut,Me]MgF to fluorinate Ph3CCl is of note because of the current significance of introducing fluorine into organic molecules,1a,15 which is of interest due to their role in pharmaceuticals and agrochemicals. The incorporation of fluorine into such molecules is, however, nontrivial, due to the facts that (i) fluoride has a large hydration energy and (ii) bonds to fluorine are strong.1 Therefore, considerable attention has been directed towards using metal-mediated transformations for introducing fluorine. The majority of studies, however, have focused on the use of transition metals.15 For example, [RuF(dppp)2]+ has also been used to convert Ph3CCl to Ph3CF.51 Thus, the corresponding reaction of [TpBut,Me]MgF provides a novel example of C–F bond formation mediated by a covalent main group metal compound.
In addition to [TpBut,Me]MgF undergoing halogen exchange with Me3SiX (X = Cl, Br, I), the chloride and bromide complexes, [TpBut,Me]MgCl and [TpBut,Me]MgBr, also undergo halogen exchange with the heavier Me3SiX derivatives (Scheme 3). The magnitude of the equilibrium constants are such that they may be determined by NMR spectroscopy (Table 3), thereby indicating that the thermodynamics for the exchange between congeneric pairs of halogens, i.e. [TpBut,Me]MgY (Y = F, Cl, Br) and Me3SiX (X = Cl, Br, I), becomes less exoergic upon descending the periodic table. The derived equilibrium constants for the reactions of [TpBut,Me]MgY (Y = F, Cl, Br, I) with Me3SiI are also listed in Table 3, which indicates that the reaction which involves formation of the Si–F bond is more exoergic than that which involves formation of the Si–I bond. As such, the data provide quantitative evidence that the phenomenological silaphilicity of the halogens increases in the sequence I ≈ Br < Cl ≪ F. While this trend is in accord with the Si–F bond being stronger than the Si–I bond,52 it is important to emphasize that the thermodynamics are actually dictated by the relative values of Mg–X and Si–X bond energies.
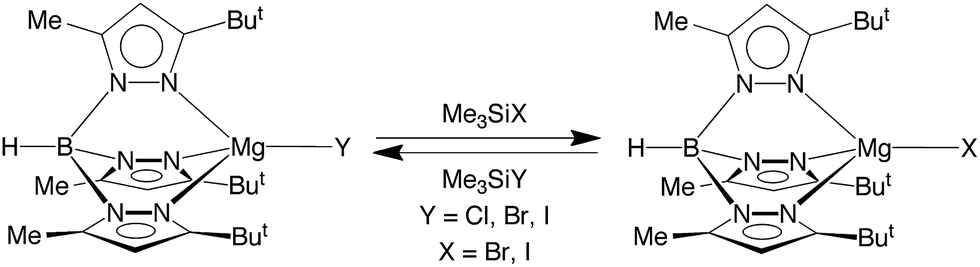 | ||
| Scheme 3 Halide exchange reactions. | ||
| Reactantsa | Productsa | K |
|---|---|---|
| a [Mg] = [TpBut,Me]Mg. b Experimental value. c Derived from experimentally measured K values. d Defined value. | ||
| [Mg]F + Me3SiCl | [Mg]Cl + Me3SiF | >1000b |
| [Mg]Cl + Me3SiBr | [Mg]Br + Me3SiCl | 13.4 ± 1.2b |
| [Mg]Br + Me3SiI | [Mg]I + Me3SiBr | 0.93 ± 0.15b |
| [Mg]F + Me3SiI | [Mg]I + Me3SiF | >12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500c 500c |
| [Mg]Cl + Me3SiI | [Mg]I + Me3SiCl | 12.5c |
| [Mg]Br + Me3SiI | [Mg]I + Me3SiBr | 0.93b |
| [Mg]I + Me3SiI | [Mg]I + Me3SiI | 1d |
Another interesting aspect of the reactivity of [TpBut,Me]MgF pertains to its ability to participate in intermolecular interactions. In this regard, while fluorine is well recognized as an important structure-directing element by virtue of its ability to bridge two or more metal centers (vide supra),53 it may also serve a structural role by participating in hydrogen bonding54 and halogen bonding55–57 interactions. The latter is a directional attractive noncovalent interaction between a covalently bound halogen atom (X), e.g. R–X or X–X, and a Lewis base, and results from the electron density distribution about X being anisotropic, such that it creates a belt of high electron density perpendicular to the covalent bond, but a region of low electron density (a so-called σ-hole) in the direction of the bond.55 Albeit much less heavily investigated than hydrogen bonding, halogen bonding has been shown to be an important tool in crystal engineering,55 with geometrical preferences that are similar to hydrogen bonding interactions, i.e. linear A⋯X–D motifs, where A is the acceptor for the halogen bond and D is the donor. However, despite many structural investigations pertaining to intermolecular interactions involving metal fluoride ligands,54b,g there are few reports that detail the thermodynamics associated with either hydrogen bonding,50b,58–60 or halogen bonding interactions.50b,58,61,62 Therefore, we have examined the ability of the fluoride ligand of [TpBut,Me]MgF to serve as a hydrogen bond and halogen bond acceptor.
Hydrogen bonding interactions involving magnesium fluoride species are of relevance to the use of in situ generated [MgF3]− to provide transition state analogues of phosphoryl transfer.11,12,63 In this regard, indole is a useful probe for quantitative studies because, although it is a good hydrogen bond donor, it is neither a good hydrogen bond acceptor nor a good nitrogen donor ligand,58,64 both of which would otherwise complicate the analysis. In this regard, Job plots65 based on 1H and 19F NMR spectroscopic data demonstrate that the interaction between [TpBut,Me]MgF and indole involves formation of a 1:1 adduct in benzene (Scheme 4 and Fig. 3).66 Analysis of the variation of the 19F NMR chemical shift as a function of indole concentration provides a binding constant of K = 39 ± 6 M−1 at 300 K for formation of the 1:1 adduct, [TpBut,Me]MgF·indole.67 For comparison, there are few reports pertaining to the thermodynamics of hydrogen bonding of indole to a terminal fluoride ligand, namely [κ4-Tptm]ZnF (85 M−1),50b (Et3P)2Ni(C5NF4)F (57.9 M−1),58,68 and Cp*2MF2 (M = Ti, 5.4 M−1; M = Zr, 1.4 M−1; M = Hf, 1.4 M−1),69 from which it is evident that [TpBut,Me]MgF must be considered a significant hydrogen bond acceptor.
 | ||
| Scheme 4 Hydrogen and halogen bonding interactions of [TpBut,Me]MgF. | ||
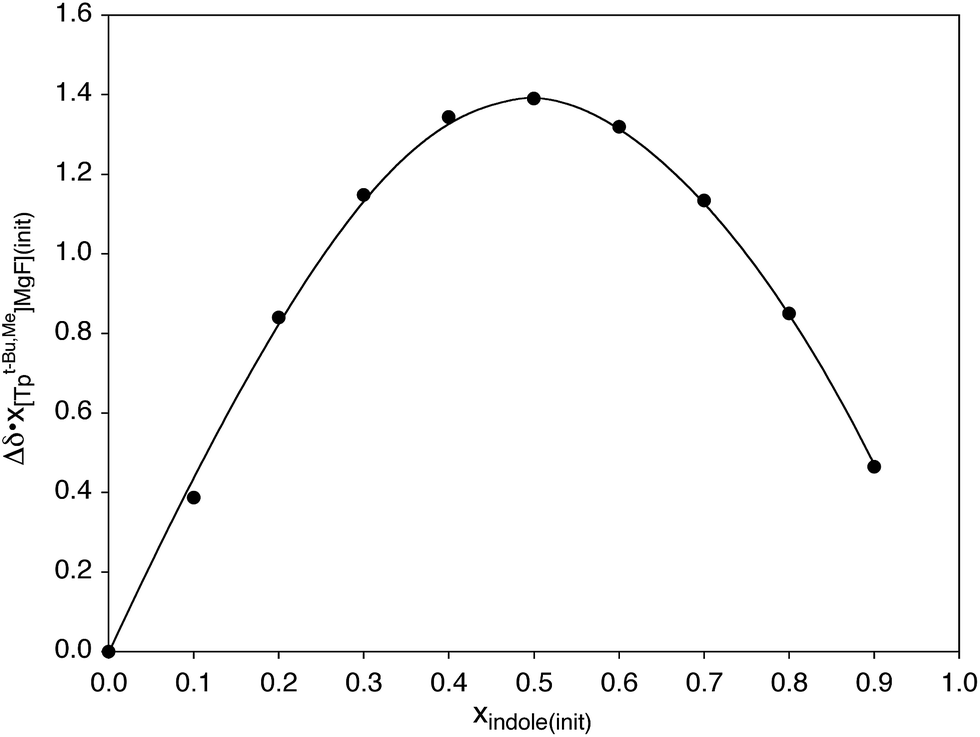 | ||
| Fig. 3 Job plot for coordination of indole to [TpBut,Me]MgF as measured by 1H NMR spectroscopy. | ||
The ability of [TpBut,Me]MgF to participate in halogen bonding interactions has been investigated by a related study using C6F5I. Thus, 19F NMR spectroscopy demonstrates that the fluoride ligand of [TpBut,Me]MgF serves as a halogen bond acceptor (Scheme 4) with the 19F NMR chemical signal shifting downfield upon addition of C6F5I.70 The derived binding constant (1.6 ± 0.3 M−1) is approximately an order of magnitude smaller than the hydrogen bonding interaction involving indole, but is comparable to the few reports of halogen bonding interactions involving fluoride ligands, namely [κ4-Tptm]ZnF (9.0 M−1) and trans-(R3P)2M(Ar)F (M = Ni, Pd, Pt; 2.4 to 5.2 M−1).58,61a
Conclusions
In summary, the first structurally characterized example of a molecular magnesium compound that features a terminal fluoride ligand, namely [TpBut,Me]MgF, has been obtained by the reaction of [TpBut,Me]MgMe with Me3SnF. The chloride, bromide and iodide complexes, [TpBut,Me]MgX, can also be obtained by analogous methods using Me3SnX (X = Cl, Br, I). Structural characterization by X-ray diffraction demonstrates that, in each case, the Mg–X bond lengths are shorter than the sum of the covalent radii, thereby indicating that there is a significant ionic component to the bonding, which is in accord with density functional theory calculations.The fluoride ligand of [TpBut,Me]MgF undergoes halide exchange with Me3SiX (X = Cl, Br, I) to afford [TpBut,Me]MgX. The other halide derivatives [TpBut,Me]MgX undergo similar exchange reactions, but the thermodynamic driving forces are much smaller than those involving fluoride transfer, a manifestation of the often discussed silaphilicity of fluorine. [TpBut,Me]MgF also undergoes metathesis with Ph3CCl to afford Ph3CF, thereby demonstrating that [TpBut,Me]MgF has applications in the formation of C–F bonds.
In accord with the highly polarized nature of the Mg–F bond, the fluoride ligand of [TpBut,Me]MgF is capable of serving as a hydrogen bond and halogen bond acceptor to indole and C6F5I, respectively. The ability of [TpBut,Me]MgF to participate in hydrogen bonding interactions mimics the involvement of magnesium fluoride species in biological systems.
Acknowledgements
We thank the National Science Foundation (CHE-1465095) for support of this research.Notes and references
-
(a) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC
; (b) K. Fagnou and M. Lautens, Angew. Chem., Int. Ed., 2002, 41, 26–47 CrossRef CAS
- J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Rev., 1994, 94, 373–431 CrossRef CAS
- B. R. Jagirdar, E. F. Murphy and H. W. Roesky, Prog. Inorg. Chem., 1999, 48, 351–455 CrossRef CAS
-
(a) E. C. Ashby, S. H. Yu and R. G. Beach, J. Am. Chem. Soc., 1970, 92, 433–435 CrossRef CAS
- H. W. Roesky and I. Haiduc, J. Chem. Soc., Dalton Trans., 1999, 2249–2264 RSC
-
(a) N. M. Doherty and N. W. Hoffman, Chem. Rev., 1991, 91, 553–573 CrossRef CAS
- T. Hascall, D. Rabinovich, V. J. Murphy, M. D. Beachy, R. A. Friesner and G. Parkin, J. Am. Chem. Soc., 1999, 121, 11402–11417 CrossRef CAS
-
(a)
R. N. Perutz and T. Braun, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, 2007, vol. 1, ch. 1.26 Search PubMed
-
(a) B. L. Pagenkopf and E. M. Carreira, Chem.–Eur. J., 1999, 5, 3437–3442 CrossRef CAS
- F. H. Allen and O. Kennard, Cambridge Structural Database (version 5.36). 3D Search and Research Using the Cambridge Structural Database, Chemical Design Automation News, 1993, 8(1), pp. 1 & 31–37.
-
(a) D. L. Graham, P. N. Lowe, G. W. Grime, M. Marsh, K. Rittinger, S. J. Smerdon, S. J. Gamblin and J. F. Eccleston, Chem. Biol., 2002, 9, 375–381 CrossRef CAS PubMed
- It has also been reported that magnesium fluoride can activate heterotrimeric G proteins and that the structures of some protein·nucleotide complexes that have previously been proposed to contain aluminum fluoride may, in fact, be magnesium derivatives. See: D. L. Graham, J. F. Eccleston, C. W. Chung and P. N. Lowe, Biochemistry, 1999, 38, 14981–14987 CrossRef CAS PubMed
-
In situ generated [MgF4]2− has also been employed. See, for example:
(a) M. Laursen, M. Bublitz, K. Moncoq, C. Olesen, J. V. Møller, H. S. Young, P. Nissen and J. P. Morth, J. Biol. Chem., 2009, 284, 13513–13518 CrossRef CAS PubMed
-
(a) F. Meyer and P. Dubois, CrystEngComm, 2013, 15, 3058–3071 RSC
-
(a) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed
-
(a)
S. Trofimenko, Scorpionates – The Coordination Chemistry of Polypyrazolylborate Ligands, Imperial College Press, London, 1999 Search PubMed
-
(a) R. Han, I. B. Gorrell, A. G. Looney and G. Parkin, J. Chem. Soc., Chem. Commun., 1991, 717–719 RSC
-
(a) A. Looney, R. Han, K. McNeill and G. Parkin, J. Am. Chem. Soc., 1993, 115, 4690–4697 CrossRef CAS
- M. C. Kuchta, J. B. Bonanno and G. Parkin, J. Am. Chem. Soc., 1996, 118, 10914–10915 CrossRef CAS
-
(a) R. Han and G. Parkin, J. Am. Chem. Soc., 1992, 114, 748–757 CrossRef CAS
- For [TpR,R′]MF compounds for metals other than magnesium, see:
(a) W. Kläui, U. Schilde and M. Schmidt, Inorg. Chem., 1997, 36, 1598–1601 CrossRef
- O. Michel, H. M. Dietrich, R. Litlabø, K. W. Törnroos, C. Maichle-Mössmer and R. Anwander, Organometallics, 2012, 31, 3119–3127 CrossRef CAS
- For the use of Me3SnF as a fluorinating agent, see: A. Herzog, F.-Q. Liu, H. W. Roesky, A. Demsar, K. Keller, M. Noltemeyer and F. Pauer, Organometallics, 1994, 13, 1251–1256 CrossRef CAS
- H. Hao, H. W. Roesky, Y. Ding, C. Cui, M. Schormann, H.-G. Schmidt, M. Noltemeyer and B. Žemva, J. Fluorine Chem., 2002, 115, 143–147 CrossRef CAS
- J. A. Rood, S. E. Hinman, B. C. Noll and K. W. Henderson, Eur. J. Inorg. Chem., 2008, 3935–3942 CrossRef CAS
- A. Distler, D. L. Lohse and S. C. Sevov, J. Chem. Soc., Dalton Trans., 1999, 1805–1812 RSC
- J. Noack, C. Fritz, C. Flugel, F. Hemmann, H.-J. Glasel, O. Kahle, C. Dreyer, M. Bauer and E. Kemnitz, Dalton Trans., 2013, 42, 5706–5710 RSC
-
(a) S. Wuttke, A. Lehmann, G. Scholz, M. Feist, A. Dimitrov, S. I. Troyanov and E. Kemnitz, Dalton Trans., 2009, 4729–4734 RSC
- For examples of compounds in which fluoride bridges magnesium and another metal, see:
(a) F.-Q. Liu, A. Kuhn, R. Herbst-Irmer, D. Stalke and H. W. Roesky, Angew. Chem., Int. Ed. Engl., 1994, 33, 555–556 CrossRef
- The Mg–F bond length is also comparable to values that have been observed in protein systems (ref. 11a, d, e, h and i). For example, PGM-MgF3-G6P-TSA exhibits Mg–F bond lengths of 1.8–1.9 Å (ref. 11d).
- [BDIAr] = HC{C(Me)NAr}2, Ar = 2,6-Pri2C6H3. This ligand is also referred to as [DipNacnac].
- For further comparison, the Mg–F bond lengths in MgF2 are 1.984(1) Å at 300 K and 1.979 Å at 52 K. See: G. Vidal-Valat, J.-P. Vidal, C. M. E. Zeyen and K. Kurki-Suonio, Acta Crystallogr., 1979, 35, 1584–1590 CrossRef
- Mg–F bond lengths of 1.759(28) Å and 1.902(27) Å have been reported for a bridging fluoride ligand in a compound that was obtained as a minor side product of a reaction, with no other characterization. However, a different crystalline form of the same compound is characterized by significantly a different pair of Mg–F bond lengths of 1.851(8) Å and 1.828(8) Å. The site occupied by the proposed fluoride ligand in these structures was also disordered with an unidentified organic ligand and so it was noted that the bond lengths associated with this bridging fluoride ligand were to be treated with caution. In particular, the short value of 1.759(28) Å is questionable. See ref. 25.
- R. Lalrempuia, A. Stasch and C. Jones, Chem. Sci., 2013, 4, 4383–4388 RSC
- M. H. Chisholm, K. Choojun, J. C. Gallucci and P. M. Wambua, Chem. Sci., 2012, 3, 3445–3457 RSC
- C. A. Zechmann, T. J. Boyle, M. A. Rodriguez and R. A. Kemp, Polyhedron, 2000, 19, 2557–2564 CrossRef CAS
- D. Naglav, D. Bläser, C. Wölper and S. Schulz, Inorg. Chem., 2014, 53, 1241–1249 CrossRef CAS PubMed
- Indeed, terminal fluoride ligands are also observed at higher field than μ-F bridging fluoride ligands in Cu, Ag and Au complexes. See: C. M. Wyss, B. K. Tate, J. Bacsa, M. Wieliczko and J. P. Sadighi, Polyhedron, 2014, 84, 87–95 CrossRef CAS
- See ESI†.
-
τ
4 = [360 − (α + β)]/141, where α + β is the sum of the two largest angles. See: L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC
-
(a) P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef PubMed
- The principal difference between these two sets of covalent radii are that (i) the Alvarez values are determined by analysis of more than 228000 experimental bond distances in the CSD that involve either carbon (0.762 Å), nitrogen (0.706 Å) or oxygen (0.661 Å), and using fixed values of these elements to determine the covalent radius of the partner, while (ii) the Pyykkö radii do not use fixed values for any element, but are derived self-consistently by a least-squares fit of an array of bond lengths that include both experimental and theoretical values (albeit a much smaller array of 410 data points than that used by Alvarez). However, the Pyykkö values exclude transition metal halide and chalcogenide compounds due to the possibility of the values being influenced by multiple bonding.
-
d(Mg–F) = 1.90 Å; d(Mg–Cl) = 2.38 Å; d(Mg–Br) = 2.53 Å; d(Mg–I) = 2.77 Å. See: R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
- For discussions pertaining to the ionic shortening of covalent bonds, see:
(a) R. Blom and A. Haaland, J. Mol. Struct., 1985, 128, 21–27 CrossRef CAS
- The absence of π-interactions is in accord with calculations on [Tp]GaE (E = O, S, Se, Te) which indicate that each chalcogen possesses three lone pairs so that the bonding is best represented as [Tp]Ga+–E−. See: J. C. Green and J. L. Suter, J. Chem. Soc., Dalton Trans., 1999, 4087–4092 RSC
- Despite the highly polar nature of the bonding, it is still appropriate to consider the molecules covalent. For a discussion, see: A. Haaland, T. U. Helgaker, K. Ruud and D. J. Shorokhov, J. Chem. Educ., 2000, 77, 1076–1080 CrossRef CAS
- S. K. Agbossou, C. Roger, A. Igau and J. A. Gladysz, Inorg. Chem., 1992, 31, 419–424 CrossRef CAS
-
M. A. Brook, Silicon in Organic, Organometallic and Polymer Chemistry, Wiley, New York, 2000, pp. 27–38 Search PubMed
- For other examples of M–F substitution employing Me3SiX reagents, see:
(a)
ref. 21a and b
;
(b) W. Sattler, S. Ruccolo and G. Parkin, J. Am. Chem. Soc., 2013, 135, 18714–18717 CrossRef CAS PubMed
-
(a) P. Barthazy, R. M. Stoop, M. Wörle, A. Togni and A. Mezzetti, Organometallics, 2000, 19, 2844–2852 CrossRef CAS
- For example, the Si–X bond dissociation energies of Me3SiX decrease from F to I (F, 159.9 kcal mol−1; Cl, 109.5 kcal mol−1; Br, 101.4 kcal mol−1; I, 82.1 kcal mol−1). See: Luo Y.-R., in Comprehensive Handbook of Chemical Bond Energies, CRC Press, 2007, ch. 9.
- H. W. Roesky and I. Haiduc, J. Chem. Soc., Dalton Trans., 1999, 2249–2264 RSC
- For examples of hydrogen bonding interactions involving halogens as hydrogen bond acceptors, see:
(a) L. Brammer, E. A. Bruton and P. Sherwood, Cryst. Growth Des., 2001, 1, 277–290 CrossRef CAS
-
(a) P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed
-
(a) L. Brammer, G. M. Espallargas and S. Libri, CrystEngComm, 2008, 10, 1712–1727 RSC
-
(a) T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC
- S. Libri, N. A. Jasim, R. N. Perutz and L. Brammer, J. Am. Chem. Soc., 2008, 130, 7842–7844 CrossRef CAS PubMed
- C. E. Osterberg, M. A. King, A. M. Arif and T. G. Richmond, Angew. Chem., Int. Ed. Engl., 1990, 29, 888–890 CrossRef
- D.-H. Lee, H. J. Kwon, B. P. Patel, L. M. Liable-Sands, A. L. Rheingold and R. H. Crabtree, Organometallics, 1999, 18, 1615–1621 CrossRef CAS
-
(a) T. Beweries, L. Brammer, N. A. Jasim, J. E. McGrady, R. N. Perutz and A. C. Whitwood, J. Am. Chem. Soc., 2011, 133, 14338–14348 CrossRef CAS PubMed
- For an example of halogen bonding involving a metal hydride derivative, see: D. A. Smith, L. Brammer, C. A. Hunter and R. N. Perutz, J. Am. Chem. Soc., 2014, 136, 1288–1291 CrossRef CAS PubMed
- It is worth noting that, in aqueous solution, the only magnesium fluoride species present in significant concentration is cationic, i.e. [MgF]+, although anionic variants have been evaluated theoretically. See:
(a) M. R. Montes, M. S. Ferreira-Gomes, M. Centeno and R. C. Rossi, Biochim. Biophys. Acta, 2015, 1848, 1514–1523 CrossRef CAS PubMed
-
(a) J. Wessel, J. C. Lee Jr, E. Peris, G. P. A. Yap, J. B. Fortin, J. S. Ricci, G. Sini, A. Albinati, T. F. Koetzle, O. Eisenstein, A. L. Rheingold and R. H. Crabtree, Angew. Chem., Int. Ed. Engl., 1995, 34, 2507–2509 CrossRef CAS
-
(a) J. S. Renny, L. L. Tomasevich, E. H. Tallmadge and D. B. Collum, Angew. Chem., Int. Ed. Engl., 2013, 52, 11998–12013 CrossRef CAS PubMed
- It should be noted that 19F NMR chemical shifts are also influenced by the hydrogen bonding environment of [MgF3]− in proteins. See, for example, ref. 11c–g and K. N. Leigh and C. E. Webster, Dalton Trans., 2014, 43, 3039–3043 RSC
- Binding constants were determined by using WinEQNMR2. See: M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC
- The activity based equilibrium constant reported was based on the assumption that the activity was equal to the molar concentration and so the M−1 units have been included here to facilitate direct comparison.
- D. A. Smith, T. Beweries, C. Blasius, N. Jasim, R. Nazir, S. Nazir, C. C. Robertson, A. C. Whitwood, C. A. Hunter, L. Brammer and R. N. Perutz, J. Am. Chem. Soc., 2015, 137, 11820–11831 CrossRef CAS PubMed
- There are relatively few studies pertaining to the use of 19F NMR spectroscopy to measure halogen bonding interactions but, for some examples, see ref. 58 and
(a) D. Hauchecorne, B. J. van der Veken, W. A. Herrebout and P. E. Hansen, Chem. Phys., 2011, 381, 5–10 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. CCDC 1424708, 1424709, 1424759 and 1424773. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03504j |
| This journal is © The Royal Society of Chemistry 2016 |