
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNegative ion photoelectron spectroscopy confirms the prediction that D3h carbon trioxide (CO3) has a singlet ground state†
David A.
Hrovat
 a,
Gao-Lei
Hou
b,
Bo
Chen
a,
Xue-Bin
Wang
*b and
Weston Thatcher
Borden
*a
a,
Gao-Lei
Hou
b,
Bo
Chen
a,
Xue-Bin
Wang
*b and
Weston Thatcher
Borden
*a
aDepartment of Chemistry and the Center for Advanced Scientific Computing and Modeling, University of North Texas, 1155 Union Circle, #305070, Denton, Texas 76203-5017, USA. E-mail: borden@unt.edu
bPhysical Sciences Division, Pacific Northwest National Laboratory, P. O. Box 999, MS K8-88, Richland, WA 99352, USA. E-mail: xuebin.wang@pnnl.gov
First published on 13th November 2015
Abstract
The CO3 radical anion (CO3˙−) has been formed by electrospraying carbonate dianion (CO32−) into the gas phase. The negative ion photoelectron (NIPE) spectrum of CO3˙− shows that, unlike the isoelectronic trimethylenemethane [C(CH2)3], D3h carbon trioxide (CO3) has a singlet ground state. From the NIPE spectrum, the electron affinity of D3h singlet CO3 was, for the first time, directly determined to be EA = 4.06 ± 0.03 eV, and the energy difference between the D3h singlet and the lowest triplet was measured as ΔEST = − 17.8 ± 0.9 kcal mol−1. B3LYP, CCSD(T), and CASPT2 calculations all find that the two lowest triplet states of CO3 are very close in energy, a prediction that is confirmed by the relative intensities of the bands in the NIPE spectrum of CO3˙−. The 560 cm−1 vibrational progression, seen in the low energy region of the triplet band, enables the identification of the lowest, Jahn–Teller-distorted, triplet state as 3A1, in which both unpaired electrons reside in σ MOs, rather than 3A2, in which one unpaired electron occupies the b2 σ MO, and the other occupies the b1 π MO.
Introduction
Carbon trioxide, CO3, is an unusual molecule with a long history. In 1962 CO3 was proposed by Katakis and Taube to be an intermediate in photoreaction of O3 with CO2.1 Four years later, CO3 was again postulated as a reactive intermediate, this time in the photoreaction of CO2 with itself.2Experimental confirmation of the existence of CO3 was obtained by IR spectroscopy on the matrix-isolated molecule, first by Moll, Clutter and Thompson in 1966,3 and subsequently by Weissberger, Breckenridge and Taube in 1967 (ref. 4) and by Jacox and Milligan in 1971.5 These experiments favored a C2v structure for CO3, containing a three-membered O–C–O ring and a carbonyl group. Nevertheless, a higher energy, D3h isomer was detected by Kaiser and coworkers in 2006.6 Very recently, the C2v and D3h isomers were reported by Sivaraman and coworkers to coexist in ion-irradiated CO2 ice.7
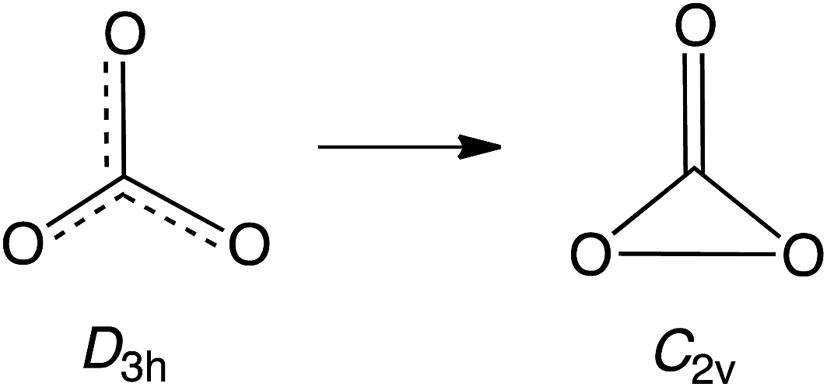
A number of theoretical studies from the 1960s to 1980s investigated the structure of CO3, mainly focusing on relative stabilities of the cyclic C2v structure, the acyclic Cs structure, and the linear C∞v structure.8 These INDO, EH, SCF, and MP2 calculations all found that the C2v isomer is the lowest in energy.9
Nevertheless, in 1987 CISD calculations by Mulder and coworkers found the D3h structure to be lower in energy than the C2v structure.10 However, subsequent calculations at higher levels of theory agree that the ground state of CO3 possesses a C2v structure, which is computed to be 1.8–6.4 kcal mol−1 lower in energy than the D3h isomer.11 A small barrier of 4.0–8.6 kcal mol−1 is calculated for the isomerization from the C2v to D3h structure.11b,e,12 The computational finding of separate C2v and D3h minima, with the former lower in energy than the latter, is, of course, consistent with the results of the experiments on matrix-isolated CO3.3–7
The singlet-triplet energy difference (ΔEST) in CO3 has also been computed. ΔEST between the 1A1 singlet ground state and the 3A1 triplet state at their C2v equilibrium geometries was calculated by fourth-order MBPT calculations to be −20.5 kcal mol−1.11a (The negative sign indicates that the singlet is lower in energy than the triplet). Similar values were obtained by QCISD(T) calculations.11b,g The ΔEST of CO3 between the 1A1 singlet state and a different triplet state (3B2) was computed to be −22.5 kcal mol−1 at the MRCI+Q(16,13)/6-311+G(3df)//CASSCF(16,13)/6-311G(d) level of theory.12
Also of interest have been the roles of CO3 in the quenching of the singlet excited state of oxygen atom (1D) by CO2 and in the 18O enrichment in CO2 in the atmospheres of Earth and Mars.13 Singlet and triplet potential energy surfaces for the reaction of O with CO2 have both been calculated.9,11b,14
Our own interest in CO3 comes from the fact that it is the n = 3 member of the isoelectronic series of C(CH2)3−nOn diradicals, for which n = 0 is trimethylenemethane (TMM) and n = 1 is oxyallyl (OXA). Negative ion photoelectron spectroscopy (NIPES) has shown that the substitution of the oxygen in OXA for one CH2 group in TMM changes ΔEST by 17.5 kcal mol−1, from ΔEST = 16.2 kcal mol−1 for the triplet ground state of TMM15 to ΔEST = −1.3 kcal mol−1 for the singlet ground state of OXA.16
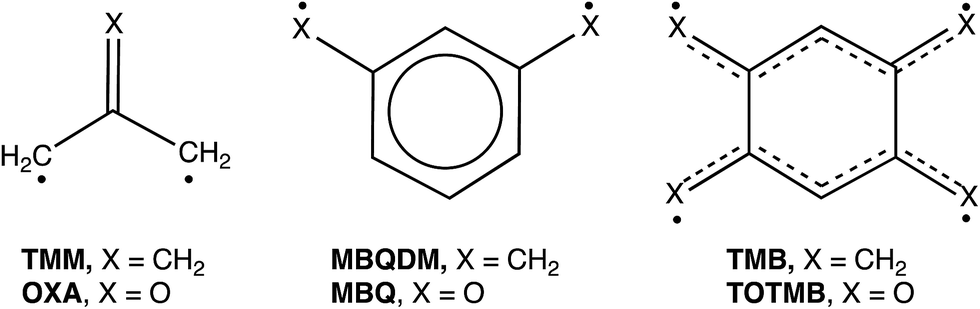
However, the substitution of oxygen for CH2 does not always have such a large effect on ΔEST in diradicals. For example, NIPES has shown that substitution of the oxygens in meta-benzoquinone (MBQ) for both CH2 groups in meta-benzoquino-dimethane (MBQDM) changes ΔEST by only 0.6 kcal mol−1, from ΔEST = 9.6 kcal mol−1 in MBQDM17 to ΔEST = 9.0 kcal mol−1 in MBQ.18 Substituting the oxygens in 1,2,4,5-tetraoxatetramethylenebenzene (TOTMB) for the four methylene groups in tetramethylenebenzene (TMB) has been predicted actually to destabilize the singlet, relative to the triplet, decreasing ΔEST by 2.7 kcal mol, from a calculated value of ΔEST = −6.2 kcal mol−1 in TMB to a value of ΔEST = −3.5 kcal mol−1, both calculated for and subsequently found by NIPES in TOTMB.19
As mentioned above, calculations have predicted a singlet ground state with C2v geometry for CO3, with ΔEST values ranging from −18.3 kcal mol−1 (ref. 11b) to −22.5 kcal mol−1.12 However, an experimental measurement of ΔEST in CO3 has not been published.
Similarly, the electron affinity (EA) of CO3 has been computed, with the best values ranging from EA = 3.84 eV to EA = 4.08 eV.20 However, the EA of CO3 has not been directly measured. With one exception,21 the experimental estimates are in the range EA = 1.8–3.5 eV,22 far below the best calculated values.20
In order to obtain accurate experimental values for both EA and ΔEST in CO3, we sought to obtain the NIPE spectrum of CO3˙−. Herein we report this spectrum and assign the peaks in it with the help of DFT and ab initio calculations. The NIPE spectrum and our analysis of it lead to values of EA = 4.06 ± 0.03 eV, and ΔEST = −17.8 ± 0.9 kcal mol−1 between the D3h1A′1 state and the Jahn–Teller distorted 3E′ state.
Experimental methodology
The NIPES experiments were performed with an apparatus that consisted of an electrospray ionization source, a cryogenic ion trap, and a magnetic-bottle time-of-flight (TOF) photoelectron spectrometer.23 Electrospraying an aqueous methanolic solution of Na2CO3 into a vacuum afforded generation of a weak CO3˙− radical anion beam, although HCO3− was always the dominant anion formed.24 The anions generated were guided by quadrupole ion guides into an ion trap, where they were accumulated and cooled by collisions with cold buffer gas, before being transferred into the extraction zone of a TOF mass spectrometer.The CO3˙− radical anions were carefully mass selected, and decelerated before being photodetached with a laser beam of 193 nm (6.424 eV) from an ArF laser in the photodetachment zone. The laser was operated at a 20 Hz repetition rate with the ion beam off at alternating laser shots, to enable shot-to-shot background subtraction to be carried out. Photoelectrons were collected at ∼100% efficiency with the magnetic bottle and analyzed in a 5.2 m long electron flight tube.
The TOF photoelectron spectra were converted into electron kinetic energy spectra by calibration with the known NIPE spectra of I− and Cu(CN)2−. The electron binding energies, given in the spectrum in Fig. 1 were obtained by subtracting the electron kinetic energies from the detachment photon energy.
 | ||
| Fig. 1 The 20 K NIPE spectrum of CO3˙− at 193 nm (6.424 eV). The intensity of the low binding energy band X is ca. one sixth of the high binding energy band A. The inset in blue is the X band enlarged by a factor of 6. The spectrum yields values of EA = 4.06 ± 0.03 eV, and ΔEST = −0.77 ± 0.04 eV = −17.8 ± 0.9 kcal mol−1. | ||
The best instrumental resolution was 20 meV full width at half maximum for 1 eV kinetic energy electrons, as demonstrated in the I− spectrum after a maximum deceleration. However, due to the weak mass intensity and light mass of CO3˙−, the NIPE spectra of CO3˙− were obtained under compromised conditions with 4% energy resolution, i.e., 40 meV for 1 eV kinetic energy electrons.
Computational methodology
In order to help analyze the NIPE spectrum of CO3˙−, three different types of electronic structure calculations were performed – B3LYP DFT calculations,25 CCSD(T) coupled cluster calculations,26 and (16/13)CASPT2 calculations.27 In the CASPT2 calculations second-order perturbation theory was used to add the effects of dynamic electron correlation28 to a (16/13)CASSCF wavefunction. The (16/13)CASSCF active space consisted of all the configurations that can be generated by distributing four valence electrons from carbon and four from each of the three oxygens among 13 MOs. The MOs were those formed from the σ and π 2p lone-pair AOs on each oxygen in CO3, the three C–O bonding and three C–O antibonding σ orbitals, and the 2p–π AO on carbon.All of the calculations were performed using the aug-cc-pVTZ basis set.29 The B3LYP and CCSD(T) calculations and vibrational analyses at these two levels of theory were carried out using the Gaussian09 suite of programs.30 The CASSCF and CASPT2 calculations were performed with MOLCAS.31 The program ezSpectrum32 was used to compute the Franck–Condon factors33 that were necessary in order to simulate the vibrational progressions in the NIPE spectrum of CO3˙−.
Results and discussion
The NIPE spectrum of CO3˙−
Fig. 1 shows the 20 K NIPE spectrum of CO3˙− at 193 nm. A weak band, X, peaked at electron binding energy (EBE) of ∼4.1 eV, and a strong band A, peaked at EBE of ∼4.9 eV, are observed in the spectrum. The intensity of the X band is ca. one sixth of the A band.For statistical reasons, formation of a triplet state is a factor of three more probable than formation of a singlet state; so triplet states invariably give the most intense peaks in NIPE spectra.34 Thus, the NIPE spectrum of CO3˙− indicates that the ground state of CO3 is a singlet and that the lowest excited state is a triplet. However, the factor of about six difference between the intensities of the X and A bands in the NIPE spectrum in Fig. 1 suggests that CO3 has two triplet states with very similar energies and that both can be formed in the photodetachment of an electron from CO3˙−.
From the rising edge of the X band, we estimate the adiabatic detachment energy (ADE) of CO3˙− (or, equivalently, the electron affinity, EA of CO3) to be 4.06 ± 0.03 eV. The EA of CO3 has been the subject of many previous experimental studies;22 but our NIPES value is considerably larger than all but one of these experimental estimates.21 However, our value of EA = 4.06 ± 0.03 eV is within experimental error of the value of EA = 4.08 eV, calculated at the CCSD(T)/aug-cc-pVTZ level by Cappa and Elrod in 2001.20
The experimental singlet–triplet gap of CO3, ΔEST, is defined as the difference between the EBE of the X band (EBE = 4.06 ± 0.03 eV) and the EBE of the first resolved peak in the A band (EBE = 4.83 ± 0.03 eV). Therefore, ΔEST = −0.77 ± 0.04 eV = −17.8 ± 0.9 kcal mol−1 is obtained from the NIPE spectrum in Fig. 1.
Vibrational structure can be discerned in both the X and A bands. The ground state X band shows a vibrational progression with a frequency of 1130 cm−1. This frequency is high enough that it is likely to belong to a C–O stretching, rather than to an O–C–O bending mode.
The vibrational mode that appears to be excited in the A band transition has a frequency of 560 cm−1. Its low frequency makes it much more likely to be due to an O–C–O bending mode than to a C–O stretching mode.
The electronic structure of CO3 – qualitative considerations
Understanding the NIPE spectrum of CO3˙− requires understanding the electronic structure of CO3. As already noted, D3hTMM and D3h CO3 are isoelectronic. Therefore, like D3hTMM, D3h CO3 might have been expected to have a triplet ground state. However, as discussed in the previous section, the NIPE spectrum of CO3˙− shows that the ground state of D3h CO3 is a singlet and that ΔEST = −17.8 ± 0.9 kcal mol−1.The reason that TMM has a triplet ground state is that in D3hTMM two electrons occupy two degenerate, e′′, π MOs. The MOs are non-disjoint;35 therefore, as expected from Hund's rule,36 the triplet is the electronic state of lowest energy.16
However, the C–H bonds in D3hTMM are replaced by σ lone pairs of electrons on the three oxygens of CO3. As shown in Fig. 2, the a′2 combination of oxygen lone pair orbitals is antibonding between all three oxygens. Therefore, in the lowest electronic state of D3h CO3, the a′2 MO is left empty.
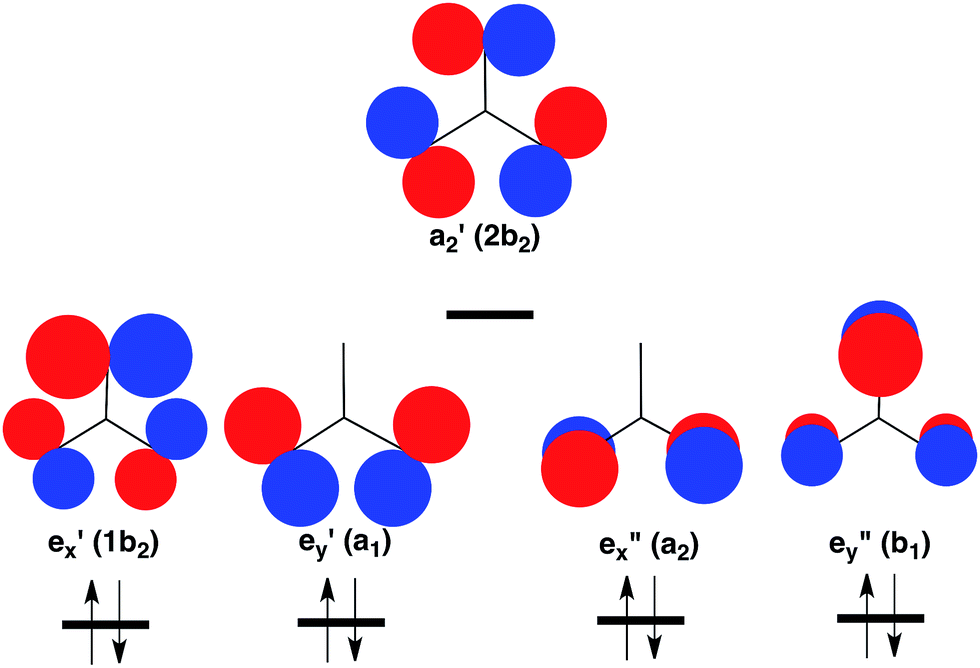 | ||
| Fig. 2 Schematic depiction of the three σ and two π lone-pair MOs of highest energy that are localized on the three oxygens in CO3. Symmetries of these MOs are given at D3h and (C2v) geometries. Of these MOs, a′2 is highest in energy, because it contains antibonding σ interactions between all three oxygens. Therefore, a′2 is left empty and the degenerate pairs of e′ and e′′ MOs are each doubly occupied in the closed-shell singlet ground state of CO3.37 The orbital occupancy in this 1A′1 state is indicated at the bottom of Fig. 2. | ||
The pair of electrons that occupy the a′2 C–H bonding MO in D3hTMM reside in one of the pair of e′′ π MOs in D3h CO3. Consequently, a total of four electrons occupy the e′′, π MOs in D3h CO3, and four more electrons occupy the degenerate pair of e′, σ MOs. This is the reason why the lowest electronic state of D3h CO3 is a closed-shell singlet.
In the CO3˙− radical anion one electron occupies the a′2 MO. In the low energy triplet states of neutral CO3 one electron in the closed-shell, singlet, ground state is excited into this MO. However, whether the electron that occupies the a′2 MO in the triplet comes from one of the e′ σ MOs or one of the e′′ π MOs in the singlet is not obvious. The question of the relative energies of the resulting 3E′ and 3E′′ states of CO3 has been addressed by the calculations that are described in a later section of this paper.
Computational results for the lowest singlet state of CO3
The results of our B3LYP, CCSD(T), and CASPT2 calculations on CO3˙− and CO3 are summarized in Table 1. All of these calculations find that in the lowest electronic state of the radical anion the unpaired electron occupies the a′2 MO, so that CO3˙− maintains D3h symmetry.38 The B3LYP and CASPT2 calculations find that the D3h singlet state is also an energy minimum.| Electronic state | B3LYP | CCSD(T) | CASPT2 |
|---|---|---|---|
| a Previous calculations at this level of theory obtained −4.08 eV for the EA of CO3.20 b Artifactual symmetry breaking39–41 results in this state having two imaginary frequencies for distortions that lead to three equivalent C2v minima. These minima have CCSD(T) energies that are 0.9 kcal mol−1 lower than that of the D3h singlet state. c One of three transition structures that connect the D3h singlet to one of the three C2v structures that are the global minima on the potential energy surface for the lowest singlet state of CO3. | |||
| 2A′2 of CO3˙− | −116.4 (−5.05 eV) | −95.4 (−4.13 eV)a | −93.9 (−4.07 eV) |
| 1A′1 (D3h minimum) | 0 | 0b | 0 |
| 1A1 (C2v TS)c | 0.6 | 1.5 | 6.6 |
| 1A1 (C2v minimum containing an O–C–O ring) | −13.4 | −5.5 | −2.2 |
| 3E′x (3B2 = |…a1α2b2α>) | −1.0 → −4.2 | 19.1 → 16.3 | 24.8 → 21.0 |
| 3E′y (3A1 = |…1b2α2b2α>) | −1.0 → −3.6 | 18.6 → 15.5 | 23.7 → 20.8 |
| 3E′′x (3A2 = |…b1α2b2α>) | 1.0 → −1.0 | 19.5 → 17.8 | 20.1 → 19.8 |
| 3E′′y (3B1 = |…a2α2b2α>) | −0.6 → −0.7 | 19.5 → 19.2 | 20.2 → 19.3 |
| 3A′2 (3B2 = |…b1αa2α>) | 11.7 | 28.4 | 35.9 |
However, the CCSD(T) calculations find that the D3h geometry of the 1A′1 state has two imaginary frequencies of 472i cm−1. These correspond to a degenerate pair of e′ vibrations that lead to a trio of slightly distorted structures with C2v symmetry (not to be confused with the cyclic C2v structure with an O–C–O ring and a carbonyl group). The three equivalent C2v structures are 0.9 kcal mol−1 lower in energy than the D3h structure at the CCSD(T)/aug-cc-pVTZ level of theory.
When the basis set is expanded to aug-cc-pVQZ, the energy difference between the D3h and C2v structures drops to only 0.3 kcal mol−1. Since the B3LYP and the CASPT2 calculations both find the D3h structure to be an energy minimum, we believe that the small geometry distortions to structures with C2v symmetry in the CCSD(T) calculations are due to artifactual symmetry breaking in the CCSD(T) wave function for the 1A′1 state at D3h geometries.39–41
If the lowest electronic states of CO3˙− and CO3 both have D3h symmetry, it is possible to assign the vibrational progression in the X band of the NIPE spectrum in Fig. 1 to a symmetrical C–O stretching mode. Only vibrational modes that preserve those symmetry elements that the electronic states of the radical ion and the neutral molecule have in common are seen in NIPE spectra. Therefore, the vibrational progression with a frequency of 1130 cm−1 that is seen in the X band in Fig. 1 must belong to the a′1 vibration of D3h CO3.
On going from the CO3˙− radical anion to neutral CO3 the C–O bond lengths are calculated to shorten by 0.013 Å (CASPT2), 0.015 Å [CCSD(T)], and 0.023 Å (UB3LYP). Consequently, the calculated Franck–Condon factors predict that an a′1 C–O bond stretching vibrational progression should be seen in the X band in Fig. 1. The calculated harmonic frequencies for the a′1 C–O stretching vibration are 1083 cm−1 (CASPT2), 1090 cm−1 [CCSD(T)] and 1140 cm−1 (B3LYP). The B3LYP value differs by only 10 cm−1 from the experimental value of 1130 cm−1.
Since the 1130 cm−1, a′1, C–O bond stretching mode is totally symmetric, it would not have been seen in the IR spectrum of D3h CO3 in matrix isolation. The observed, asymmetric (e′), C–O bond-stretching frequency was reported to be 1165 cm−1.6
As shown in Table 1, and, in agreement with the results of previous calculations11 and experiments,3–7 B3LYP, CCSD(T), and CASPT2 all find that there is a C2v singlet energy minimum, containing an O–C–O ring, that is lower in energy than the D3h singlet. Not unexpectedly, the barrier height that is calculated for ring closure increases as the calculated exothermicity of this reaction decreases.
Because the D3h → C2v ring closure reaction requires mixing of the filled e′ MOs in Fig. 2 with the empty a′2 MO, ring closure is computed to involve passage over a barrier. This orbital mixing, which occurs on an e′ distortion from D3h to C2v symmetry, may be regarded as a second-order Jahn–Teller effect.42
For example, at C2v geometries e′x and a′2 both have b2 symmetry and so can be mixed by an ey distortion from D3h symmetry. From inspection of the MOs in Fig. 2, one can deduce that this mixing reduces the contribution of the AOs on the two oxygens between which O–O bond formation occurs and thus makes the resulting b2 MO much less antibonding than the e′x MO. In fact, the b2 MO that results from the mixing between e′x and a′2 MOs becomes the 2px lone-pair AO on the carbonyl group of the C2v singlet energy minimum.
The large change in geometry that occurs on formation of the cyclic singlet CO3 molecule results in the absence of overlap between its vibrational wave function and the vibrational wave function of the D3h CO3˙− radical anion. Consequently, the Franck–Condon factor for the laser-induced transition from D3h CO3˙− to the C2v energy minimum of singlet CO3 is calculated to be effectively zero. Therefore, the value of EA = 4.06 ± 0.03 eV in the NIPE spectrum corresponds to the energy difference between the D3h equilibrium geometry of CO3˙− and the local D3h energy minimum of neutral singlet CO3, not the global C2v energy minimum, of singlet, CO3.
There are two types of experimental evidence that support this conclusion. The first is that the measured EA is very close to the calculated CCSD(T) and CASPT2 energy differences in Table 1 between the D3h equilibrium geometry of CO3˙− and the local D3h energy minimum of neutral CO3. Second, as already discussed, the vibrational progression of 1130 cm−1 seen in the X band of the NIPE spectrum in Fig. 1 is in good agreement with that predicted by all three levels of theory for the D3h local minimum.
Computational results for the lowest triplet state of CO3
As shown in Table 1, there are two low-lying triplet states in CO3. They are E′, in which the two unpaired electrons occupy the a′2 and e′ σ MOs, and E′′, in which the second unpaired electron occupies the e′′ π MO, instead of the e′ σ MO.A third triplet, 3A′2, which is the ground state of TMM, is calculated to be very high in energy in CO3. In this state the e′′x and e′′y π MOs are each singly occupied, and the a′2 MO is doubly occupied. As shown in Fig. 2, the a′2 MO is strongly O–O antibonding; and its double occupancy in 3A′2 makes this triplet state much higher in energy than either 3E′ or 3E′′, in both of which the a′2 MO is singly occupied.
Whether 3E′ or 3E′′ is lower in energy is not clear from qualitative considerations. As shown in Fig. 2, the e′ MOs are weakly bonding σ MOs; whereas, the e′′ MOs are non-bonding π MOs. On this basis, leaving e′ doubly occupied and having e′′ singly occupied should be favored; so 3E′′ should be lower in energy than 3E′.
On the other hand, two electrons of the same spin cannot simultaneously occupy the same AO. With one electron in the a′2 σ MO, having a second electron of the same spin in an e′ σ MO prevents these two electrons from ever appearing on the same atom; whereas, no such prohibition exists if the second unpaired electron occupies an e′′ π MO. Consequently, although maximization of bonding is expected to favor the 3E′′ state, minimization of electron repulsion should favor the 3E′ state. Which effect dominates cannot be predicted from qualitative considerations; so one has to rely on calculations for the prediction of which triplet state, 3E′ and 3E′′, is lower in energy.
Table 1 shows that 3E′ and 3E′′ are, in fact, calculated to be very close in energy. Both degenerate triplet states are expected to undergo first-order Jahn–Teller distortions to C2v symmetry,43 and the calculated energy differences between the two triplet states at their C2v equilibrium geometries range between 1 and 3 kcal mol−1.
Interestingly, the results, tabulated in Table 1, reveal that the CCSD(T) and CASPT2 calculations differ as to which triplet state is predicted to be lower in energy. The CCSD(T) calculations predict that the C2v triplet, formed by exciting an electron from the pair of e′ σ MOs into the a′2 σ MO, is lower in energy than the triplet that is formed by exciting an electron from the pair of e′′ π MOs into the a′2 MO. B3LYP makes the same prediction as CCSD(T). However, it should be noted that B3LYP erroneously predicts that both triplet states are lower in energy than the D3h1A′1 state (Table 1).
CCSD(T) and B3LYP both predict that the 3E′ and 3E′′ states have very similar energies at their respective D3h geometries. However, as would be expected, removing an electron from one of the e′ σ MOs results in a larger Jahn–Teller distortion than removing an electron from one of the e′′ π MOs. B3LYP, CCSD(T), and CASPT2 calculations all find that this is, in fact, the case. The larger energy lowering of the 3E′ state by a first-order Jahn–Teller distortion leads to the prediction by both CCSD(T) and B3LYP that the C2v distorted 3E′ state is the lowest energy triplet state of CO3 by 2–3 kcal mol−1.
In contrast to CCSD(T), CASPT2 places 3E′′ well below 3E′ at their respective D3h geometries. Even though the first-order Jahn–Teller distortion to C2v symmetry stabilizes 3E′ more than 3E′′, the energetic advantage of 3E′′ over 3E′ at their respective D3h geometries prevails; and the C2v distorted 3E′′ (3A2) state is calculated to be lower in energy than the C2v distorted 3E′ (3A1) state by 1–2 kcal mol−1.
Which triplet state is lower in energy, 3E′ (3A1) or 3E′′ (3A2)?
Which method, CCSD(T) or CASPT2, gives the correct answer to the question of what is the lowest triplet state of CO3, 3E′ → 3A1 or 3E′′ → 3A2? As described in the following paragraphs, the NIPE spectrum in Fig. 1 indicates that the CCSD(T) prediction is correct; and, although 3A1 and 3A2 are very close in energy, 3A1 is the lower energy of these two triplet states.This conclusion follows from the vibrational progression seen in the low energy portion of the triplet peak. As already noted, this region of the NIPE spectrum shows a progression of 560 cm−1. This vibrational frequency is too low to be associated with C–O stretching, but is the right size to be due to O–C–O bending. Our calculations indicate that only 3A1 should show an O–C–O bending progression, so it must be the lower energy of the two closely-spaced triplet states.
The conclusion that only 3A1 should show an O–C–O bending progression follows from the calculated geometries of 3A1 and 3A2 and is supported by our simulations of the vibrations in the peaks due to 3A1 and 3A2 in the NIPE spectra of CO3˙−. Table 2 gives the bond lengths and the unique bond angle of the C2v minima to which CCSD(T) and CASPT2 both predict that 3E′ and 3E′′ distort. It is clear that both the bond lengths and the bond angles of the 3A1 minima of the distorted 3E′ state deviate significantly from the equality they have at D3h geometries. However, the bond angles of the 3A2 minima of the distorted 3E′′ state are calculated to remain much more nearly equal after Jahn–Teller distortions.
| Bond length, or bond angle | B3LYP | CCSD(T) | CASPT2 | |||
|---|---|---|---|---|---|---|
| 3A1 | 3A2 | 3A1 | 3A2 | 3A1 | 3A2 | |
| R(C–O1) | 1.311 | 1.338 | 1.321 | 1.334 | 1.325 | 1.315 |
| R(C–O2) = R(C–O3) | 1.257 | 1.245 | 1.259 | 1.254 | 1.259 | 1.267 |
| O2–C–O3 | 113.6° | 122.0° | 113.8° | 119.2° | 114.2° | 119.7° |
| O1–C–O2 = O1–C–O3 | 123.2° | 119.0° | 123.1° | 120.4° | 122.9° | 120.1° |
The difference between the geometries of the C2v minima for the two triplet states is a consequence of the difference between the MOs that are occupied in these two states. In the 3A1 state an electron, which occupies the 1b2 σ MO in the D3h1A′1 ground state, is removed and placed in the 2b2 σ MO. As shown in Fig. 2, this electronic excitation results in the O1–O2 and O1–O3 σ bonding interactions in the 1b2 MO being replaced by σ antibonding interactions between all of the oxygens in the 2b2 MO. Consequently, the O1–C–O2 and O1–C–O3 bond angles in 3A1 are calculated to be larger than 120°; so the O2–C–O3 bond angle is predicted to be much less than 120° in this state.
In the 3A2 state an electron, which occupies the b1 π MO in the D3h1A′1 ground state, is removed and placed in the 2b2 σ MO. The antibonding O1–O2 and O1–O3 π interactions in b1 are lost, as is the bonding O2–O3 π interaction. Consequently, the O1–C–O2 and O1–C–O3 angles in the 3A2 state are expected to be less than 120°, and the O2–C–O3 angle is expected to be greater than 120°.
These qualitative expectations are fulfilled at the B3LYP level of theory. However, because the 1,3-interactions between the oxygens in 3A2 involve π, rather than σ AOs, the deviations of the B3LYP bond angles from 120° are about three times smaller in 3A2 than in 3A1. In fact, the π interactions in 3A2 are so small that, in the optimized CCSD(T) and CASPT2 geometries, the deviations of the bond angles from 120° are not only less than 1° but they actually deviate from 120° in the opposite direction from the B3LYP bond angles.
In NIPE spectra progressions are only seen in vibrational modes that affect the geometrical parameters by which an electronic state differs from the radical anion from which the electronic state is formed.34 The calculated O–C–O bond angles in the 3A1 state of CO3 differ significantly from those in the D3h equilibrium geometry of the 2A′2 ground state of CO3˙−. Therefore, one would expect to see a long vibrational progression in O–C–O bending in the band for formation of the 3A1 state of CO3 in the NIPE spectrum of CO3˙−.
On the other hand, the calculated O–C–O bond angles in the 3A2 state of CO3 are very close to those in the D3h equilibrium geometry of the 2A′2 radical anion. Therefore, one would not expect to see a long vibrational progression in O–C–O bending in the band for formation of the 3A2 state of CO3 in the NIPE spectrum of CO3˙−. The only long vibrational progression that should appear in the band for formation of the 3A2 state is one in C–O bond stretch, since the C–O bond lengths in the C2v equilibrium geometry of 3A2 in Table 2 differ from those in the D3h equilibrium geometry of the radical anion.44
Fig. 1 shows that a vibrational progression of 560 cm−1 in O–C–O bending is found in the band for formation of the lowest triplet state of CO3 in the NIPE spectrum of CO3˙−. As discussed above, such a progression is expected to be seen in the 3A1 state of CO3, but not in the 3A2 state. Thus, it follows that the lowest triplet state of CO3 is the 3A1 state, in which one unpaired electron resides in the 1b2 MO and the other resides in the 2b2 MO.
This qualitative conclusion is supported by both B3LYP and CCSD(T) simulations of the triplet region of the NIPE spectrum of CO3˙−. Using the Franck–Condon factors (FCFs) calculated with ezSpectrum,32Fig. 3 shows how the NIPE spectrum of CO3˙− is predicted to appear, based on the results obtained with B3LYP calculations.
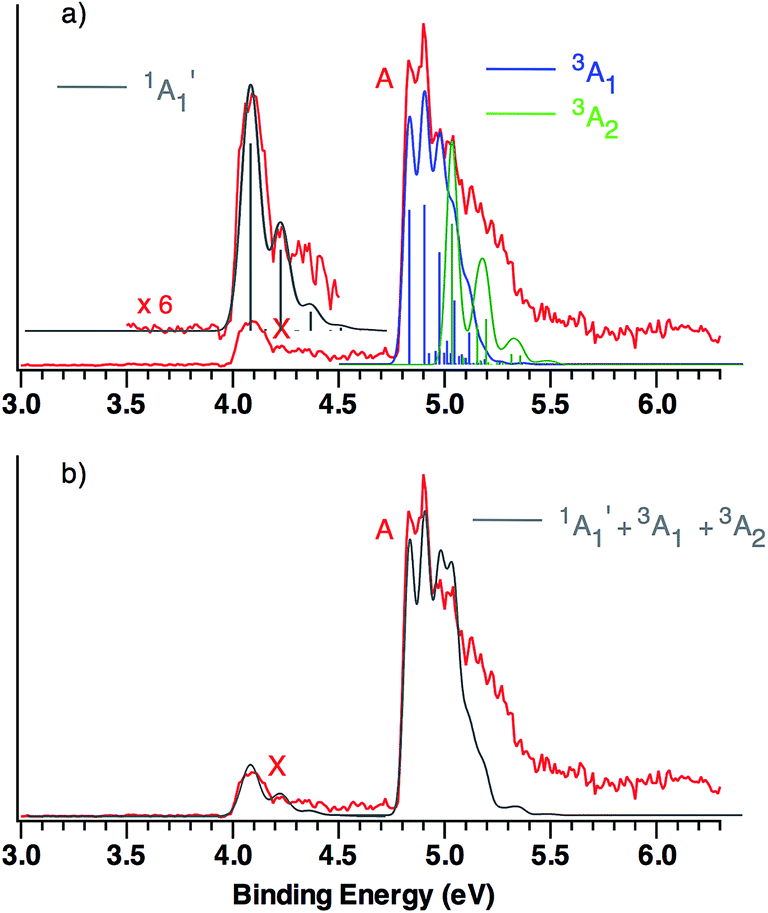 | ||
| Fig. 3 (a) B3LYP/aug-cc-pVTZ calculated vibrational structure in the NIPE spectrum of CO3˙−, superimposed on the experimental NIPE spectrum (red). The positions of the bands in the calculated stick spectrum for 1A′1 (grey), 3A1 (blue), and 3A2 (green) have been adjusted, in order to align the 0–0 bands in the calculated spectrum with the 0–0 bands in the observed spectrum. The calculated spectrum, using Gaussian line shapes with, respectively, 100, 60, and 60 meV full widths at half maxima for each stick in 1A′1, 3A1, and 3A2, is also shown. (b) The computed NIPE spectrum (grey), calculated from the sum of the convoluted contributions of the singlet and two triplets in Fig. 3a, superimposed on the experimental 193 nm spectrum (red). | ||
The predicted vibrational structure for the triplet region of the NIPE spectrum, based on the results of CCSD(T) calculations, has a very similar appearance to the B3LYP-based simulation in Fig. 3. The CCSD(T) simulations are provided in Fig. S1 and S2 of the ESI† of this manuscript, and the vibrational mode assignments are given in Fig. S1.† Both the B3LYP and CCSD(T) simulations confirm that the vibrational progressions in the triplet region of the experimental NIPE spectrum are dominated by the O–C–O bending mode in 3A1 and by C–O bond stretch in 3A2.
Comparison of the simulated spectra for both triplet states with the actual NIPE spectrum suggests aligning the 0–0 band of 3A2 with the fourth resolved peak (EBE = 5.03 eV) in band A, which leads to the conclusion that the 3A2 state is 0.20 eV (4.6 kcal mol−1) higher in energy than 3A1. This ordering of the two triplet states is in accordance with the results of both the B3LYP and CCSD(T) calculations (Table 1). However, an energy difference of 4.6 kcal mol−1 would be about twice the size of the energy differences of, respectively, 2.6 and 2.3 kcal mol−1 that are predicted by these two types of calculations.
An alternative alignment of the 0–0 band of 3A2 with the third resolved peak (EBE = 4.97 eV) in band A is shown in Fig. S3 of the ESI.† This alignment makes the 3A2 state only 0.14 eV (3.2 kcal mol−1) higher in energy than 3A1, which is in better agreement with the energy differences between these two states, computed by both B3LYP and CCSD(T). However, comparison of Fig. S3† with Fig. 3, shows that the alignment in Fig. S3† fits the observed intensities of the peaks in the experimental NIPE spectrum less well than the alignment in Fig. 3.
The simulated vibrational structure for formation of the singlet ground state of CO3 is also shown in Fig. 3. The simulation reproduces well the observed vibrational progression in the singlet ground state and confirms the conclusion that this progression is due to the symmetric C–O stretching.
The simulations, based on the calculated FCFs, for formation of the singlet and two triplet states of CO3 from the 2A′2 state of CO3˙−, provide a good fit to the experimental NIPE spectrum of CO3˙− up to 5.1 eV. There appears to be a shoulder at EBE ∼ 5.3 eV in the experimental spectrum, which might be due to formation of the third, low-lying triplet state, 3A′2, which is predicted by the CCSD(T) calculations to have EA = 5.37 eV.
The similarity between the calculated and experimental NIPE spectra of CO3˙− in Fig. 3 provides evidence that our assignments of the peaks in the experimental NIPE spectra are correct and that, as predicted by both B3LYP and CCSD(T), the 3A1 state of CO3 is lower in energy than the 3A2 state.
Conclusions
We report the first NIPE spectrum of CO3˙−. The spectrum shows that, substitution of the three oxygens in CO3 for the three CH2 groups in TMM results in a change in the ground state, going from 3A′2 and ΔEST = 16.2 kcal mol−1 in TMM15 to 1A′1 and ΔEST = −17.8 ± 0.9 kcal mol−1 in CO3. The NIPE spectrum also provides the first measurement of EA = 4.06 ± 0.03 eV in D3h CO3. Qualitative MO analysis and quantitative electronic structure calculations confirm that the ground state of CO3 is a singlet and reveal which of the two closely-spaced triplet excited states is lower in energy. The CCSD(T) and CASPT2 calculations reproduce the experimental EA and ΔEST values of CO3 rather well.The combined results of our experiments and calculations contribute fundamental information about the electronic structure of CO3, a molecule that is not only of interest because it is isoelectronic with both TMM and OXA, but that is also important in both atmospheric chemistry and astrochemistry.12–14,45
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
The calculations at UNT were supported by Grant B0027 from the Robert A. Welch Foundation. The NIPES research at PNNL was supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences and Biosciences and was performed at the EMSL, a national scientific user facility sponsored by DOE's Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory, which is a multiprogram national laboratory operated for DOE by Battelle.Notes and references
- D. Katakis and H. Taube, J. Chem. Phys., 1962, 36, 416 CrossRef CAS.
- A.-Y. Ung and H. I. Schiff, Can. J. Chem., 1966, 44, 1981 CrossRef CAS.
- N. G. Moll, D. R. Clutter and W. E. Thompson, J. Chem. Phys., 1966, 45, 4469 CrossRef CAS.
- E. Weissberger, W. H. Breckenridge and H. Taube, J. Chem. Phys., 1967, 47, 1764 CrossRef CAS.
- M. E. Jacox and D. E. Milligan, J. Chem. Phys., 1971, 54, 919 CrossRef CAS.
- C. S. Jamieson, A. M. Mebel and R. I. Kaiser, ChemPhysChem, 2006, 7, 2508 CrossRef CAS PubMed.
- B. Sivaraman, B. N. R. Sekhar, D. Fulvio, A. Hunniford, B. McCullough, M. E. Palumbo and N. Mason, J. Chem. Phys., 2013, 139, 074706 CrossRef PubMed.
- (a) B. M. Gimarc and T. S. Chou, J. Chem. Phys., 1968, 49, 4043 CrossRef CAS; (b) J. F. Olsen and L. Burnelle, J. Am. Chem. Soc., 1969, 91, 7286 CrossRef CAS; (c) M. Cornille and J. Horsley, Chem. Phys. Lett., 1970, 6, 373 CrossRef CAS; (d) J. R. Sabin and H. Kim, Chem. Phys. Lett., 1971, 11, 593 CrossRef CAS; (e) J. A. Pople, U. Seeger, R. Seeger and P. v. R. Schleyer, J. Comput. Chem., 1980, 1, 199 CrossRef CAS; (f) J. S. Francisco and I. H. Williams, Chem. Phys., 1985, 95, 373 CrossRef CAS.
- P. LaBonville, R. Kugel and J. R. Ferraro, J. Chem. Phys., 1977, 67, 1477 CrossRef CAS.
- W. J. van de Guchte, J. P. Zwart and J. J. C. Mulder, J. Mol. Struct.: THEOCHEM, 1987, 152, 213 CrossRef.
- (a) M. A. Castro, S. Canuto and A. M. Simas, Chem. Phys. Lett., 1991, 177, 98 CrossRef CAS; (b) R. D. J. Froese and J. D. Goddard, J. Phys. Chem., 1993, 97, 7484 CrossRef CAS; (c) T. Kowalczyk and A. I. Krylov, J. Phys. Chem. A, 2007, 111, 8271 CrossRef CAS; (d) Y. Liu, I. B. Bersuker, W. Zou and J. E. Boggs, J. Chem. Theory Comput., 2009, 5, 2679 CrossRef CAS; (e) C. Qin and T.-Y. Soo, J. Mol. Struct.: THEOCHEM, 2009, 897, 32 CrossRef CAS; (f) F. Grein, J. Chem. Phys., 2013, 138, 204305 CrossRef PubMed; (g) A. S. Averyanov, Y. G. Khait and Y. V. Puzanov, J. Mol. Struct.: THEOCHEM, 1999, 459, 95 CrossRef CAS.
- A. M. Mebel, M. Hayashi, V. V. Kislov and S. H. Lin, J. Phys. Chem. A, 2004, 108, 7983 CrossRef CAS.
- Review: R. I. Kaiser and A. M. Mebel, Chem. Phys. Lett., 2008, 465, 1 CrossRef CAS.
- C. J. Bennett, C. Jamieson, A. M. Mebel and R. I. Kaiser, Phys. Chem. Chem. Phys., 2004, 6, 735 RSC.
- (a) P. G. Wenthold, J. Hu, R. R. Squires and W. C. Lineberger, J. Am. Chem. Soc., 1996, 118, 475 CrossRef CAS; (b) P. G. Wenthold, J. Hu, R. R. Squires and W. C. Lineberger, J. Am. Soc. Mass Spectrom., 1999, 10, 800 CrossRef CAS.
- (a) T. Ichino, S. M. Villano, A. J. Gianola, D. J. Goebbert, L. Velarde, A. Sanov, S. J. Blanksby, X. Zhou, D. A. Hrovat, W. T. Borden and W. C. Lineberger, Angew. Chem., Int. Ed., 2009, 48, 8509 CrossRef CAS PubMed; (b) T. Ichino, S. M. Villano, A. J. Gianola, D. J. Goebbert, L. Velarde, A. Sanov, S. J. Blanksby, X. Zhou, D. A. Hrovat, W. T. Borden and W. C. Lineberger, J. Phys. Chem. A, 2011, 115, 1634 CrossRef CAS PubMed.
- P. G. Wenthold, J. B. Kim and W. C. Lineberger, J. Am. Chem. Soc., 1997, 119, 1354 CrossRef CAS.
- (a) Q. Fu, J. Yang and X.-B. Wang, J. Phys. Chem. A, 2011, 115, 3201 CrossRef CAS PubMed; (b) B. Chen, D. A. Hrovat, S. H. M. Deng, J. Zhang, X.-B. Wang and W. T. Borden, J. Am. Chem. Soc., 2014, 136, 3589 CrossRef CAS PubMed; (c) R. C. Fort Jr, S. J. Getty, D. A. Hrovat, P. M. Lahti and W. T. Borden, J. Am. Chem. Soc., 1992, 114, 7549 CrossRef.
- D. A. Hrovat, G.-L. Hou, X.-B. Wang and W. T. Borden, J. Am. Chem. Soc., 2015, 137, 9094 CrossRef CAS PubMed.
- C. D. Cappa and M. J. Elrod, Phys. Chem. Chem. Phys., 2001, 3, 2986 RSC.
- R. G. Ewing and M. Waltman, Int. J. Mass Spectrom., 2010, 296, 53 CrossRef CAS.
- (a) D. E. Hunton, M. Hofmann, T. G. Lindeman and A. W. Castleman Jr, J. Chem. Phys., 1985, 82, 134 CrossRef CAS; (b) J. T. Snodgrass, C. M. Roehl, A. M. van Koppen, W. E. Palke and M. T. Bowers, J. Chem. Phys., 1990, 92, 5935 CrossRef CAS; (c) J. F. Hiller and M. L. Vestal, J. Chem. Phys., 1980, 72, 4713 CrossRef CAS; (d) S. E. Novich, P. Engelking, P. L. Jones, J. H. Futrell and W. C. Lineberger, J. Chem. Phys., 1979, 70, 2652 CrossRef; (e) I. Dotan, J. A. Davidson, G. E. Streit, D. L. Albritton and F. C. Fehsenfeld, J. Chem. Phys., 1977, 67, 2874 CrossRef CAS; (f) S. P. Hong, S. B. Woo and E. M. Helmy, Phys. Rev. A, 1977, 15, 1563 CrossRef CAS; (g) M. L. Vestal and G. H. Mauclaire, J. Chem. Phys., 1977, 67, 3758 CrossRef CAS; (h) T. O. Tiernan and R. L. C. Wu, Adv. Mass Spectrom., 1978, 7, 136 Search PubMed.
- X.-B. Wang and L.-S. Wang, Rev. Sci. Instrum., 2008, 79, 073108 CrossRef PubMed.
- Assuming the last phase to generate CO3˙− from the Na2CO3 aqueous solutions in the electrospray ionization process is nanodroplets of waters containing one CO32− solute, then we can estimate the ratio of CO32− over HCO3− is about 0.1% from the known carbonic acid dissociation constants. Because CO3˙− is derived from autodetachment of CO32−, a weak intensity of CO3˙− ion beam is expected.
- B3LYP is a combination of Becke's 3-parameter hybrid exchange functional ( A. D. Becke, J. Chem. Phys, 1993, 98, 5648 CrossRef CAS ) with the electron correlation functional of Lee, Yang, and Parr ( C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef ).
- (a) G. D. Purvis and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910 CrossRef CAS; (b) K. Raghavachari, G. W. Trucks, J. A. Pople and M. H. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479 CrossRef CAS.
- K. Andersson, P.-Å. Malmqvist and B. O. Roos, J. Chem. Phys., 1992, 96, 1218 CrossRef CAS.
- W. T. Borden and E. R. Davidson, Acc. Chem. Res., 1996, 29, 87 CrossRef.
- (a) T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007 CrossRef; (b) R. A. Kendall, T. H. Dunning Jr and R. J. Harrison, J. Chem. Phys., 1992, 96, 6769 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- K. Andersson, F. Aquilante, M. Barysz, A. Bernhardsson, M. R. A. Blomberg, Y. Carissan, D. L. Cooper, M. Cossi, L. DeVico, N. Ferré, M. P. Fülscher, A. Gaenko, L. Gagliardi, G. Ghigo, C. de Graaf, S. Gusarov, B. A. Hess, D. Hagberg, A. Holt, G. Karlström, R. Lindh, P.-Å. Malmqvist, T. Nakajima, P. Neogrády, J. Olsen, T. Pedersen, M. Pitonak, J. Raab, M. Reiher, B. O. Roos, U. Ryde, B. Schimmelpfennig, M. Schütz, L. Seijo, L. Serrano-Andrés, P. E. M. Siegbahn, J. Stålring, T. Thorsteinsson, V. Veryazov and P.-O. Widmark, MOLCAS version 7, Lund University, Sweden, 2008 Search PubMed.
- V. A. Mozhayskiy and A. I. Krylov, ezSpectrum, version 3.0, see http://iopenshell.usc.edu/downloads Search PubMed.
- (a) J. Franck, Trans. Faraday Soc., 1926, 21, 536 RSC; (b) E. Condon, Phys. Rev., 1926, 28, 1182 CrossRef CAS.
- Reviews: (a) L. C. Lineberger and W. T. Borden, Phys. Chem. Chem. Phys., 2011, 13, 11792 RSC; (b) W. T. Borden, in Fifty Years of the James Flack Norris Award, ed. E. T. Strom and V. Mainz, American Chemical Society, Washington, DC, in press Search PubMed.
- (a) W. T. Borden and E. R. Davidson, J. Am. Chem. Soc., 1977, 99, 4587 CrossRef CAS; (b) W. T. Borden, in Diradicals, ed. W. T. Borden, Wiley-Interscience, New York, 1982, pp. 1–72 Search PubMed.
- Review: W. Kutzelnigg, Angew. Chem., Int. Ed. Engl., 1996, 35, 573 CrossRef CAS and references therein.
- Because the a′′2 π lone-pair MO contains bonding interactions between the 2p–π lone-pairs on the oxygens and the 2p–π AO on the carbon, it is considerably lower in energy than the σ and π lone-pair MOs on oxygen that are shown in Fig. 2.
- As found previously,20 a C2v structure for CO3˙− is computed to be higher in energy than the D3h structure.
- Review: E. R. Davidson and W. T. Borden, J. Phys. Chem., 1983, 87, 4783 CrossRef CAS.
- For more recent discussions of artifactual symmetry breaking in D3h molecules, see (a) W. Eisfeld and K. Morokuma, J. Chem. Phys., 2000, 113, 5587 CrossRef CAS; (b) C. E. Miller and J. W. Francisco, J. Phys. Chem. A, 2001, 105, 1662 CrossRef CAS; (c) Ref. 21 .
- Artifactual symmetry breaking leads to another phenomenon that can be seen in the results given in Table 1 for the triplet states. At D3h geometries the E′x and E′y components of the 3E′ state should have identical energies, as should the E′′x and E′′y components of the 3E′′ state. The fact that the two components of each degenerate state do not have exactly the same energies in Table 1 can be viewed as a consequence of the fact that, in general, electronic structure codes impose only C2v symmetry on the MOs used to construct D3h wave functions. Consequently, a wave function that nominally has Ex symmetry actually contains an A2 contaminant; and a wave function that nominally has Ey symmetry actually contains an A1 contaminant. Although the Ex and Ey components of such wavefunctions do have the same energies, the A2 and A1 contaminants do not; and this leads to the energy differences at D3h geometries between the “Ex” and “Ey” states in Table 1.39a.
- See, inter alia, (a) U. Öpik and M. H. L. Pryce, Proc. R. Soc. London, Ser. A, 1957, 238, 425 CrossRef; (b) R. F. W. Bader, Can. J. Chem., 1962, 40, 1164 CrossRef CAS; (c) R. G. Pearson, J. Am. Chem. Soc., 1969, 91, 4947 CrossRef CAS; (d) R. G. Pearson, J. Mol. Struct., 1983, 103, 25 CrossRef. For a recent review see: (e) I. B. Bersuker, Chem. Rev., 2013, 113, 1351 CrossRef CAS PubMed.
- H. A. Jahn and E. Teller, Proc. R. Soc. London, Ser. A, 1937, 161, 220 CrossRef CAS.
- A similar situation occurs in the lowest energy triplet state of (CO)5, which undergoes a first-order Jahn–Teller distortion from the D5h geometry of the radical anion. The expected C–C bond stretching progression is, in fact, seen in the NIPE spectrum of (CO)5˙− in the band for formation of the lowest triplet state of (CO)5. X. Bao, D. A. Hrovat, W. T. Borden and X.-B. Wang, J. Am. Chem. Soc., 2013, 135, 4291 CrossRef CAS PubMed.
- C. J. Bennett, C. S. Jamieson and R. I. Kaiser, Phys. Chem. Chem. Phys., 2010, 12, 4032 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized geometries, electronic energies, zero-point vibrational energies, and imaginary frequencies for all of the CO3 electronic states discussed in this manuscript, computed with the aug-cc-pVTZ basis set, using B3LYP, CCSD(T), and (16/13)CASPT2 calculations. Fig. S1 contains the CCSD(T) simulation of the triplet region of the NIPE spectrum of (CO)3˙− and the assignment of the vibrational progressions in it; Fig. S2 shows the calculated NIPE spectrum, with the lines in the stick spectrum in Fig. S1, convoluted with Gaussians; and Fig. S3 shows how the appearance of the simulation in Fig. 3 is modified by choosing a different value of the energy difference between the 0–0 bands in the two lowest energy triplet states (9 pages). See DOI: 10.1039/c5sc03542b |
| This journal is © The Royal Society of Chemistry 2016 |