
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic studies on the addition of hydrogen to iridaepoxide complexes with subsequent elimination of water†
Lauren E.
Doyle
,
Warren E.
Piers
*,
Javier
Borau-Garcia
,
Michael J.
Sgro
and
Denis M.
Spasyuk
University of Calgary, Department of Chemistry, 2500 University Drive N.W., Calgary, Alberta T2N 1N4, Canada. E-mail: wpiers@ucalgary.ca
First published on 27th October 2015
Abstract
Iridium complexes of the PCsp2P ligand in which the donors are linked by 2,3-benzo[b]thiophene groups engage in the cooperative activation of N2O and the resulting iridaepoxides can be treated with dihydrogen to effect elimination of water and regeneration of the starting iridium complex. The mechanism of the steps in this reaction have been investigated using low temperature NMR investigations that reveal H/D exchange processes that point to a highly reactive kinetic product of hydrogen addition to the iridaepoxide. This intermediate is also involved in the water elimination pathway, and model compounds have been synthesized to provide further evidence for the mechanistic proposals for water elimination. The adaptable donor properties of the PCsp2P ligand framework, particularly the anchoring carbene donor, plays a significant role in the ability of these compounds to mediate the transformation of N2O in this way.
Introduction
Tridentate “pincer” ligand frameworks continue to attract interest for supporting complexes of metals from across the periodic table that serve as platforms for small molecule activation and selective bond cleavage.1,2 Their appeal stems from the large number of permutations for EE′E′′ donor arrays, the extensive electronic and steric tunability possible via modification of the linking features of the ligand framework, and the opportunity to design ligands of formal charge ranging from 0 to −3. Furthermore, through incorporation of functions capable of accepting and delivering hydrogen atoms or protons, ligand cooperativity can feature prominently in catalytic cycles mediated by pincer ligand metal complexes.3Recently, we introduced a variation on the venerable PCP4 motif that incorporates a diaryl carbene donor in the central anchoring position of the pincer framework,5 exemplified by ligands I and II.6,7 These ligands are not only quite electron rich, but allow for the possibility of cooperativity through engagement of the M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C unit of the chelate array.8 Furthermore, depending on the metal employed, the carbene unit in the ligand assumes either Schrock-like9–13 or Fischer-like behavior.14,15 Thus, in nickel complexes, the carbene is highly nucleophilic, whereas in iridium manifolds, it is less so. This “continuum of reactivity” can be exploited in the deployment of such ligands for small molecule activation.16
C unit of the chelate array.8 Furthermore, depending on the metal employed, the carbene unit in the ligand assumes either Schrock-like9–13 or Fischer-like behavior.14,15 Thus, in nickel complexes, the carbene is highly nucleophilic, whereas in iridium manifolds, it is less so. This “continuum of reactivity” can be exploited in the deployment of such ligands for small molecule activation.16
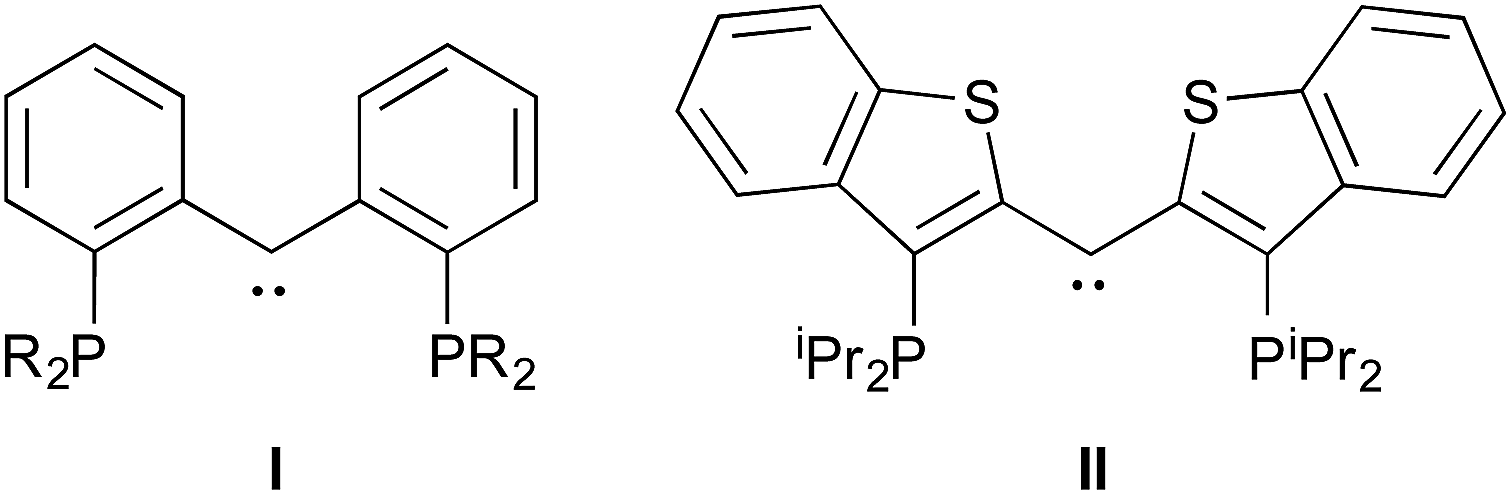
Despite the lowered nucleophilicity of the carbene carbon, the iridium complexes of ligands of type I or II react with N2O to afford “iridaepoxide” complexes in which the oxygen atom has added to the IrC units.7 In compounds of ligand type I, C–C bond activations17 ensue from the initially observed iridaepoxide, which eventually leads to the dismantling of the pincer ligand framework.18 However, the rigidity imparted by the 2,3-benzo[b]thiophene linker in ligand II raises the barrier to this undesired ruination of the ligand framework, allowing for other chemistry to be explored. For instance, reaction of iridaepoxide 1·Cl with H2 gives an iridium dihydride complex, 2·Cltrans, where the two hydride ligands reside in positions trans to the iridaepoxide moiety (Scheme 1). Upon heating to ≈100 °C, this compound eliminates water to return to the iridium carbene complex 3·Cl. Attempts to garner information concerning the mechanism of water loss were to some degree hampered by the fact that dihydride 2·Cltrans is a thermodynamic isomer that we proposed must convert to a kinetic isomer 2·Clcis in which the hydride ligands are cis-disposed with respect to the iridium oxygen bond of the iridaepoxide unit. The reasons for this kinetic preference are not clear, but they may be related to the seminal studies of Eisenberg on the preferred paths for addition of H2 to Vaska's complex along a P–Ir–CO vector instead of P–Ir–Cl.19 Regardless, the kinetic data we obtained by following the loss of water from 2·Cltrans provided information on the loss of H2 from this compound, which is the rate limiting step in the loss of water, and the process by which H2O was eliminated was essentially opaque to these kinetic investigations.7 Here, we provide more evidence for this proposal through a combination of labeling studies, low temperature NMR investigations and model synthesis of likely intermediates in the water elimination path.
 | ||
| Scheme 1 Hydrogen addition and water elimination to iridaepoxide 1·Cl. | ||
Results and discussion
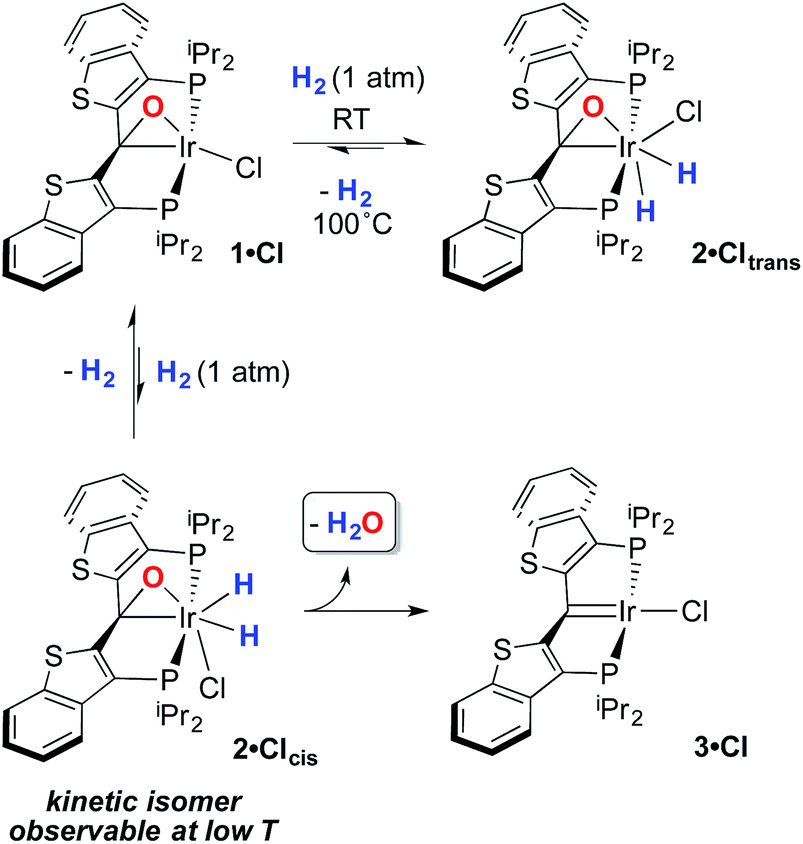
The structure and properties of compound 2·Cltrans were reported in our preliminary communication.7 To summarize, it is stable in toluene solution at temperatures ranging from 25–80 °C, but EXSY spectroscopy indicates that the two diastereotopic hydride ligands (characterized as triplets in the 1H NMR spectrum at −7.58 and −15.22 ppm) undergo exchange even at room temperature on the timescale of these experiments. However, there is no exchange with free H2 at room temperature as measured by exposure of a solution of 2·Cltrans to D2; only when temperatures reach 80–90 °C does label begin to appear in these positions, but as indicated above, water elimination also occurs in this temperature regime.In order to assess the results of this exchange experiment, the spectroscopic signature of 2·Cltrans-d2 was obtained on a separately prepared sample obtained by treatment of 1·Cl with D2. The 31P{1H} NMR chemical shift of 24.1 ppm for the dideuterated isotopologue is almost baseline resolved from that of undeuterated 2·Cltrans at 24.0 ppm in the 162 MHz spectrum. We therefore sought to assess the kinetic isotope effect for the oxidative addition of H2 to 1·Cl by reacting it with 1 atm of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of H2/D2 at room temperature. Surprisingly, rather than the expected mixture of 2·Cltrans and 2·Cltrans-d2, both the 31P{1H} and the 1H{31P} NMR spectra revealed that a near-statistical mixture of the four possible isotopologues (Fig. 1a) was observed (Fig. 1b, middle trace); the presence of the d2 isotopologue was confirmed by 2H and 31P{1H} NMR spectroscopy. Furthermore, the presence of significant quantities of HD was detected in the 1H NMR spectrum (4.47 ppm, 1JHD = 43 Hz, 1:1:1 triplet). This puzzling result was corroborated by a complementary experiment in which the reaction of 1·Cl with pure HD gave the same mixture of four isotopologues (Fig. 1b, bottom trace), along with detectable amounts of H2 in the 1H NMR spectrum (singlet, 4.51 ppm).‡ It was further observed in a separate experiment that equimolar mixtures of 2·Cltrans and 2·Cltrans-d2 exhibited no scrambling of the label to form either of the two 2·Cltrans-d1 isotopomers unless heated to above 80 °C—the temperature regime in which hydrogen/deuterium elimination from 2·Cltrans-dn begins. Since the scrambling to form the four isotopologues of 2·Cltrans occurred in reactions done at room temperature, this requires that the isotope redistribution in the H2/D2 mixture and the pure HD must be occurring prior to final formation of the thermodynamic isomer 2·Cltrans.
:1 mixture of H2/D2 at room temperature. Surprisingly, rather than the expected mixture of 2·Cltrans and 2·Cltrans-d2, both the 31P{1H} and the 1H{31P} NMR spectra revealed that a near-statistical mixture of the four possible isotopologues (Fig. 1a) was observed (Fig. 1b, middle trace); the presence of the d2 isotopologue was confirmed by 2H and 31P{1H} NMR spectroscopy. Furthermore, the presence of significant quantities of HD was detected in the 1H NMR spectrum (4.47 ppm, 1JHD = 43 Hz, 1:1:1 triplet). This puzzling result was corroborated by a complementary experiment in which the reaction of 1·Cl with pure HD gave the same mixture of four isotopologues (Fig. 1b, bottom trace), along with detectable amounts of H2 in the 1H NMR spectrum (singlet, 4.51 ppm).‡ It was further observed in a separate experiment that equimolar mixtures of 2·Cltrans and 2·Cltrans-d2 exhibited no scrambling of the label to form either of the two 2·Cltrans-d1 isotopomers unless heated to above 80 °C—the temperature regime in which hydrogen/deuterium elimination from 2·Cltrans-dn begins. Since the scrambling to form the four isotopologues of 2·Cltrans occurred in reactions done at room temperature, this requires that the isotope redistribution in the H2/D2 mixture and the pure HD must be occurring prior to final formation of the thermodynamic isomer 2·Cltrans.
 | ||
| Fig. 1 (a) Reaction of compound 1·Cl with either a 1:1 mixture of H2/D2 or pure HD leads to the mixture of isotopologues of compound 2·Cltrans. (b) Hydride region of the 1H{31P} NMR spectrum of pure 2·Cltrans (top), the reaction mixture when 1·Cl is treated with H2/D2 (middle) and the reaction mixture when 1·Cl is treated with HD (bottom). Note that, while the outer resonances in each pair are due to the 2·Cltrans-d1 isotopomers, we have not made absolute assignments for these resonances. | ||
Given this conclusion, we postulated that these rapid H/D scrambling processes are mediated by the proposed kinetic isomer 2·Clcis. As briefly discussed previously,7 this kinetic isomer can be observed spectroscopically at low temperatures (−38 to −78 °C) via both 1H and 31P NMR experiments by treatment of 1·Cl with ≈2–3 atm of H2 at −78 °C. As long as the temperature remains below −38 °C, only 1·Cl and 2·Clcis are observed; the thermodynamic isomer only begins to appear if the sample is warmed above this temperature. In the low temperature regime, 1·Cl, H2 and 2·Clcis are in rapid equilibrium, as indicated by a changing ratio of 1·Cl and 2·Clcis and the broadening of the resonances for 2·Clcis in the 31P NMR spectrum as the temperature is raised. At −78 °C, the equilibrium constant for the addition of H2 to 1·Cl to form 2·Clcis is 145 M−1 and the equilibrium constant can be evaluated at various temperatures and treated using a van't Hoff analysis (Fig. S1†). The thermodynamic parameters thus derived indicate that, while enthalpically favored (ΔH° = −4.9(3) kcal mol−1), the entropic penalty (ΔS° = −15(2) cal mol−1 K−1) renders the formation of 2·Clcis at room temperature essentially thermoneutral (ΔG°298 = −0.4(5) kcal mol−1), completely consistent with our qualitative observations. The equilibrium constant measured at −78 °C when substituting deuterium for hydrogen, is larger (Keq(D2) = 590 M−1), and a substantial inverse equilibrium isotope effect (EIE) of 0.37 is obtained. The observation of an inverse EIE in oxidative addition of H2/D2 is expected in this low temperature regimen, and the reasons for this phenomenon have been lucidly rationalized by Parkin and co-workers.20–22
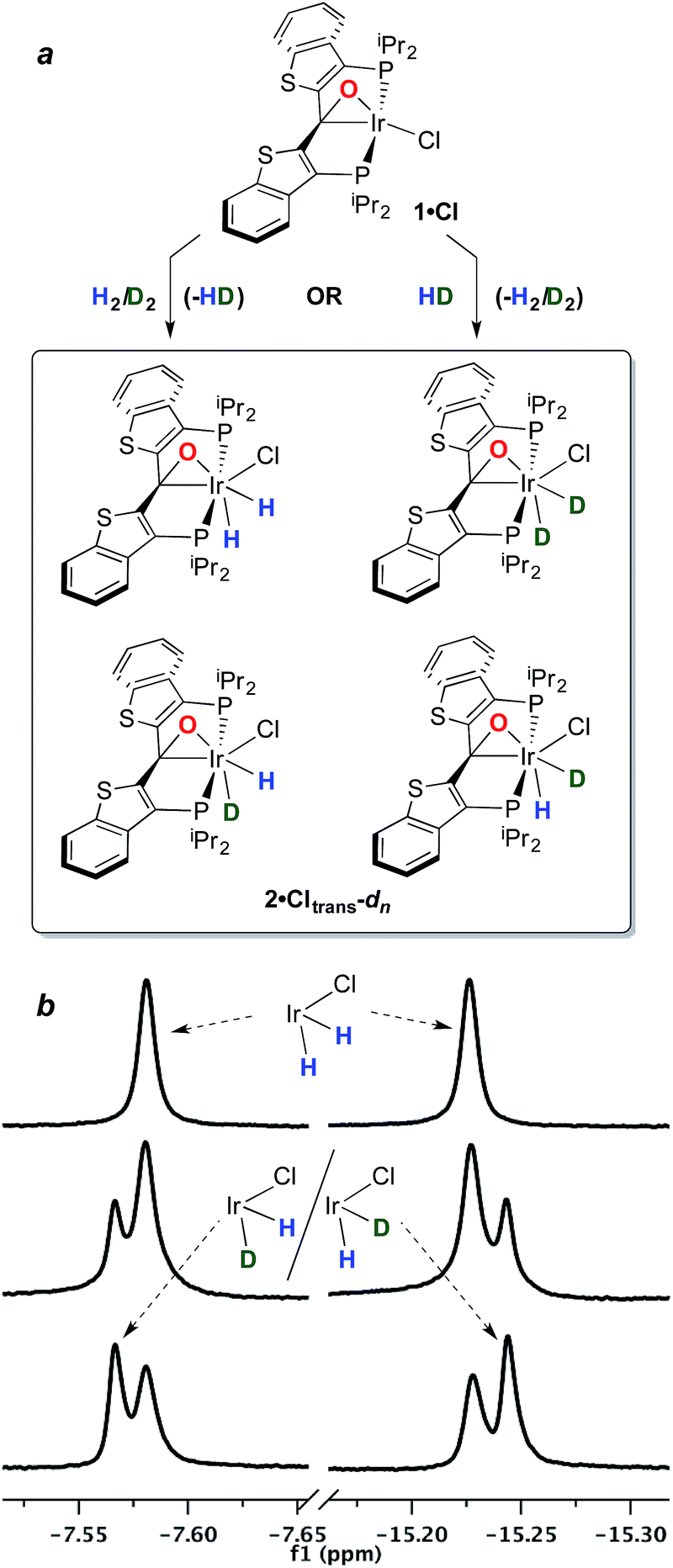
With this behavior in mind, in order to probe for hydrogen/deuterium scrambling in the kinetic dihydrido compound, 1·Cl was treated with a mixture of H2 and D2 (≈1.4:1) at −78 °C. The 243 MHz 31P{1H} spectrum at the top of Fig. 2 is observed initially; here, the broad resonance for iridaepoxide 1·Cl at approximately 25.1 ppm is accompanied by two sharper, baseline resolved signals at 23.25 and 23.09 ppm for the 2·Clcis-d2 and 2·Clcis isotopologues, respectively.§ This spectrum remains constant at this temperature, but as the sample is warmed, the chemical shifts for the isotopologues of 2·Clcis drift downfield and the signals broaden slightly. Furthermore, a new resonance of intermediate chemical shift begins to appear (Fig. 2, −58 °C spectrum). At −38 °C, the amount of 2·Clcis-dn is significantly diminished as the equilibrium shifts to the left and the pattern of the signal is difficult to discern; however, if the sample is re-cooled to −78 °C, and the speciation of the equilibrium shifts towards the addition product, the 31P{1H} NMR spectrum clearly displays signals for all four possible isotopologues of the kinetic isomer 2·Clcis-dn. Also of significance is the observation that the 31P resonances for the separate 2·Clcis-d1 isotopomers coalesce at temperatures above −78 °C into one averaged signal, indicating that these two species are in rapid exchange, likely via an η2-HD complex (III, Fig. 2, inset). Furthermore, in the 1H NMR spectrum obtained for this sample (−78 °C), in addition to the singlet for H2 at 4.55 ppm, the distinct, isotopically shifted 1:1:1 triplet for HD is observed at 4.52 ppm.¶ This experiment clearly establishes 2·Clcis as the agent of the facile isotope scrambling in the iridium dihydrides and the redistribution of label in the headspace gas as observed in the experiments depicted in Fig. 1.
 | ||
| Fig. 2 Reaction of compound 1·Cl with a mixture of H2/D2 at −78 °C, followed by warming to −38 °C and then re-cooling to −78 °C as monitored by 31P{1H} NMR spectroscopy at 243 MHz in CD2Cl2. The equilibrium between 1·Cl and the (H)2 and (D)2 isotopologues of 2·Clcis is apparent in the t = 0 spectrum; warming results in production of the (H)(D) isotopomers, which are resolved in the −78 °C spectrum obtained upon re-cooling to low temperature. | ||
Given the clear implication of the kinetic isomer 2·Clcis in the observed H/D scrambling, and its potential involvement in the process by which water is eliminated,7 we sought to probe the effect on its formation as a function of the X type ligand present in 1·Cl. It is well-established that the exothermicity of hydrogen addition to Vaska's complex is strongly influenced by the nature of the X ligand,23 and that the addition product is favored as the halide is changed from F < Cl < Br < I.24,25 Since compound 1·Cl is phenomenologically similar to Vaska's complex, we postulated that the position of the equilibrium for formation of the kinetic H2 addition product might be influenced by changing this group. We therefore synthesized the hydroxo and bromo derivatives shown in Scheme 2 and measured the equilibrium constants for formation of the dihydrido derivatives 2·OHcis and 2·Brcis. The carbene hydroxo complex 3·OH was prepared using cesium hydroxide as a reagent.15,26 This species was then used to prepare the bromo derivative 3·Brvia treatment with LiBr. Both compounds exhibit characteristic triplet resonances (2JCP = ≈2.5 Hz) in the 13C NMR spectra at 172.2 (3·OH) and 161.4 (3·Br) ppm for the carbene carbon of the PCsp2P ligand. Compound 3·OH was further characterized by X-ray crystallography, the first example of this family of monomeric hydroxo compounds to be structurally characterized; its molecular structure is shown in Fig. 3 along with selected metrical parameters. The ligand adopts a nearly planar geometry, and features an IrC bond distance of 1.943(5) Å. The Ir–O distance of 2.004(4) Å is between those of 1.978(12) Å and 2.020(6) Å found in other four coordinate, monomeric iridium hydroxo compounds.27–29 Both the carbene hydroxo and bromo compounds react smoothly with N2O to yield the corresponding iridaepoxides 1·OH and 1·Br, as shown in Scheme 2 and each of these compounds has also been characterized crystallographically. Fig. 4 shows the molecular structure of one of the two molecules found in the unit cell for the hydroxo complex; metrical parameters are essentially identical for both molecules. The molecular structure of 1·Br can be found in Fig. S2.† Metrical parameters within the series of compounds 1·X are broadly similar, with the exception of those distances and angles pertaining to the X group. Notably, the C(1)–O(1) distance within the iridaepoxide unit is virtually unchanged within these derivatives, indicating little effect on the C–O bond order as X is changed.
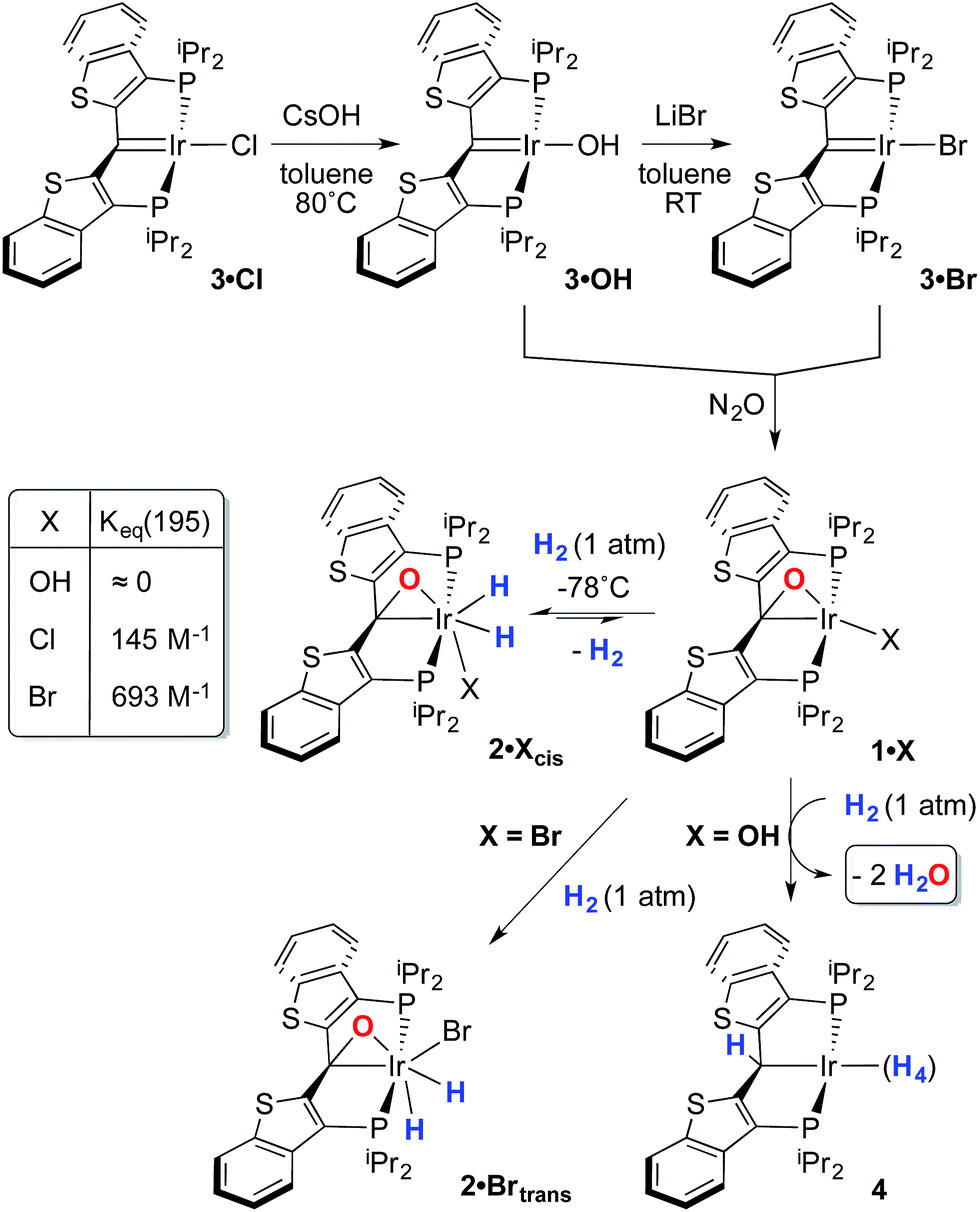 | ||
| Scheme 2 Synthesis of compounds 1·X and addition of H2. | ||
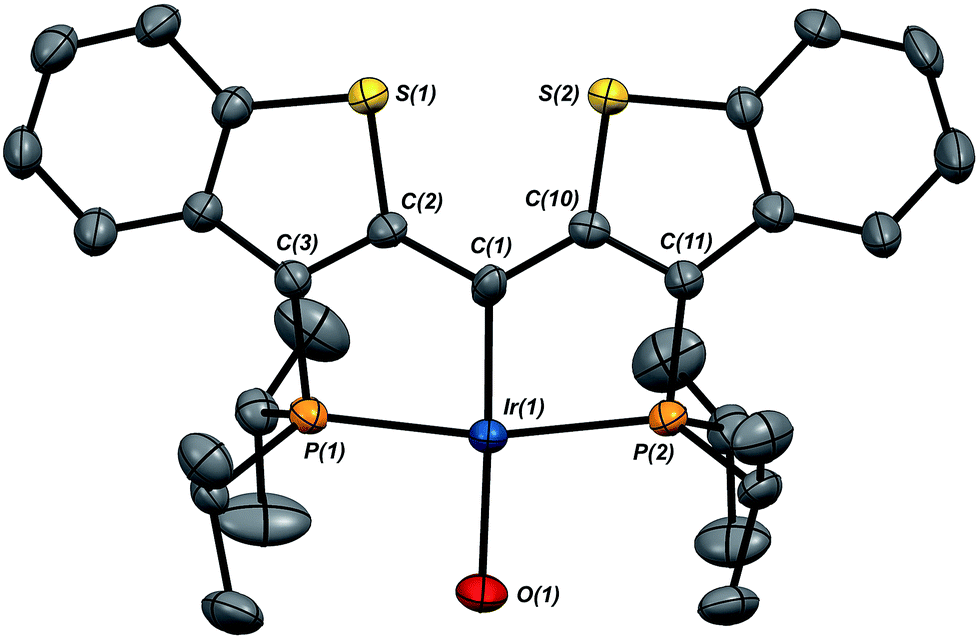 | ||
| Fig. 3 Molecular structure of 3·OH. Hydrogen atoms have been omitted for clarity. Displacement ellipsoids are shown at the 50% probability level. Selected bond lengths (Å): Ir(1)–P(1), 2.2904(13); Ir(1)–P(2), 2.2817(13); Ir(1)–C(1), 1.943(5); Ir(1)–O(1), 2.004(4). Selected bond angles (°): C(1)–Ir(1)–O(1), 175.5(2); P(1)–Ir(1)–P(2), 166.08(5). | ||
 | ||
| Fig. 4 Molecular structure of 1·OH. Hydrogen atoms have been omitted for clarity. Displacement ellipsoids are shown at the 50% probability level. Selected bond lengths (Å): Ir(1)–P(1), 2.3110(10); Ir(1)–P(2), 2.3135(10); Ir(1)–C(1), 2.078(4); Ir(1)–O(1), 2.084(3); Ir(1)–O(2), 1.972(4); C(1)–O(1), 1.359(5). Selected bond angles (°): C(1)–Ir(1)–O(1), 38.12(13); C(1)–O(1)–Ir(1), 70.7(2); Ir(1)–C(1)–O(1), 71.2(2); P(1)–Ir(1)–P(2), 166.23(4). | ||
With these compounds in hand, reactions with H2 were studied at both −78 °C and at ambient temperatures. The hydroxo complex 1·OH exhibits no reaction with H2 at −78 °C, indicating that the equilibrium constant for formation of a hydrogen oxidative addition product, 2·OHcis, analogous to the chloro case discussed above, is very small. When the temperature of this sample is warmed to >45 °C, a reaction ensues that produces two equivalents of water and a polyhydride PCsp3P complex, 4Scheme 2, that is analogous to several such compounds reported in the literature.30 Compound 4 was only stable under an atmosphere of dihydrogen and so was characterized in situ by multinuclear NMR spectroscopy. This was further complicated by the observation of partial deuteration (via the solvent, toluene-d8) in the ligand aromatic and benzylic positions, as well as the Ir–H moieties,30–32 under the high temperature conditions under which it forms. Nevertheless, the observed generation of 4 and water suggests that H2 oxidative addition to 1·OH does occur at higher temperatures, but facile O–H reductive eliminations33,34 ensue that eventually lead to the observed products. In contrast, compound 1·Br exhibits behavior more akin to that of 1·Cl, forming a kinetic isomer at low temperatures and an isolable thermodynamic isomer at higher temperatures (Scheme 2). The equilibrium constant measured for the formation of 2·Brcis is about 4 times larger than that found for the chlorido derivative, indicating that the exothermicity of hydrogen oxidative addition to these compounds follows a similar trend to that observed in the Vaska's complex series.22 The greater stability of 2·Brcis is reflected in the much slower release of water from this derivative. At 110 °C, elimination of H2O is extremely slow, and complete conversion is only observed when heating in o-xylene at 150 °C for 2–3 hours.
While the kinetic isomer 2·Clcis is thus strongly implicated in the observed H/D scrambling, the mechanism by which it mediates this facile process is a matter of some speculation. A further question regarding the relationship, if any, between the label redistribution process and the path by which water elimination from 2·Cl (trans or cis) occurs is also significant. In our initial communication, we postulated that, because the Ir–O bond of the iridaepoxide moiety and an Ir–H bond are now cis to one another in the kinetic isomer, it is plausible that reductive elimination of O–H33,34 could be a first step towards overall release of water from 2·Clcis to form the carbene chloride 3·Cl (Scheme 1).7 We believe that the H/D scrambling observed is consistent with this proposal (Scheme 3). Reductive elimination of O–H from 2·Clcis leads to the hydrido chloride Ir(III) species IV bearing an alcohol function on the anchoring carbon of a PCsp3P pincer ligand. This compound, while not observed here, is directly analogous to the product formed via a single C–H bond activation in the ligand attachment chemistry to iridium for these ligands—lacking the OH function, of course. These five coordinate Ir(III) compounds are highly fluxional,6,30,35,36 and have been shown to bind H2 to form η2-dihydrogen hydrido complexes in which all three hydrogens are in rapid exchange; the specific case involving ligand II is no exception (Scheme 4). Treatment of the carbene chloride complex 3·Cl with an atmosphere of H2 gives an equilibrium mixture of compounds 5·Cl and 6·Cl; in the presence of H2, the equilibrium lies far to the side of dihydrogen complex 6·Cl. This species is characterized by a broad resonance at −5.35 ppm for the η2-H2 ligand and a hydride signal at −21.7 ppm, also broad. When the H2 atmosphere is removed, compound 5·Cl is isolable as an analytically pure orange solid. Treatment of 5·Cl with D2 regenerates 6·Cl as its d4-isotopologue; exchange of D into the benzylic position of the PCsp3P ligand is also observed as would be expected. The rapid exchange process for the hydrogen/deuterium atoms in 6·Cl is also likely to be present in V, providing a valid mechanism for the scrambling observed when IV comes into contact with D2; exchange leads not only to IV-d1, but the HD that is also observed in these reactions (Scheme 3). Oxidative addition of the PCsp3P ligand O–H bond regenerates 2·Clcis as its d1 isotopologue; as the spectra in Fig. 2 demonstrate, the diastereotopic positions in this compound are in rapid exchange even at −68 °C.
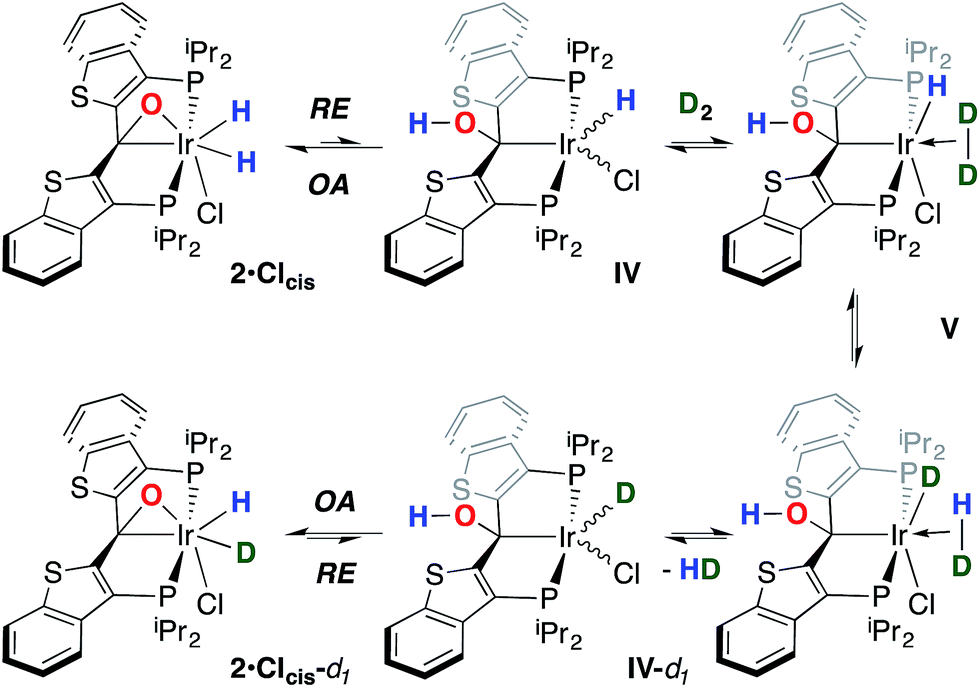 | ||
| Scheme 3 Proposed path for H/D scrambling mediated by 2·Clcis. | ||
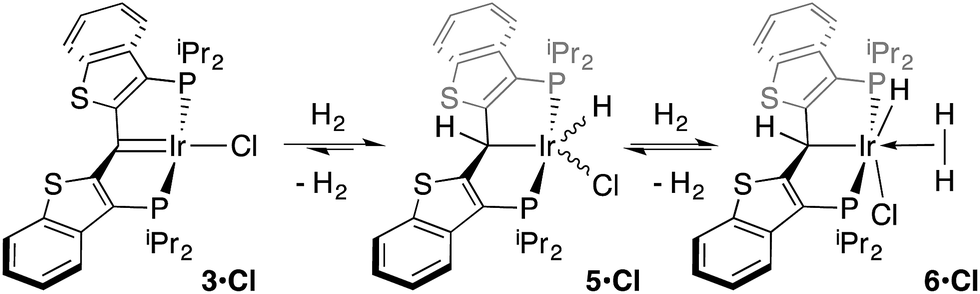 | ||
| Scheme 4 Addition of H2 to 3·Cl. | ||
Overall, this proposal accounts for the statistical redistribution of isotopes observed (as opposed to the pairwise exchange of H2 and D2, for example), but necessitates that reductive elimination/oxidative addition of O–H is rapidly reversible under these conditions. Furthermore, experimental observations suggest that if 2·Clcis is in equilibrium with IV, the equilibrium favors the observed dihydrido species. Close examination of the spectra shown Fig. 2 indicate that, as the sample containing 2·Clcis and 2·Clcis-d2 is warmed from −78 °C to −38 °C, the diproteo isotopologue disappears faster than the dideutero isotopologue, providing qualitative evidence for a normal kinetic isotope effect in the rate limiting step of the process leading to isotope scrambling. This is consistent with expectations for an O–H(D) reductive elimination initial step. It is also consistent with an alternative view of 2·Clcis as an η2-ketone complex (Scheme 5); here, the reversible conversion to IV would be viewed as a 1,2-insertion of the CO double bond into the cis disposed hydrido ligand and the reverse process, β-elimination. The metrical data obtained for the structure of 2·Cltrans, specifically the C–O distance of 1.303(12) Å, and the downfield resonance for the iridaepoxide carbon (99.5 ppm) is consistent with the η2 ketone form contributing significantly to the structural description of this compound. Our experimental data do not distinguish between these two views of such a process, but both, we believe, are reasonable suggestions to explain the observed H/D scrambling.
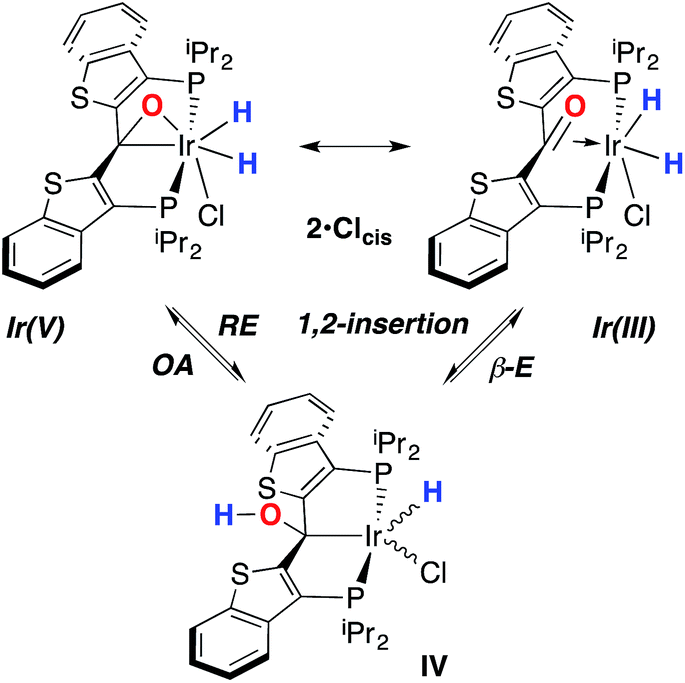 | ||
| Scheme 5 Two views of the conversion of 2·Clcis to IV. | ||
In this mechanistic model, the H/D scrambling observed proceeds via the key species IV and we proposed that the path to water elimination also originates from this species.7 While the H/D scrambling occurs with a very low barrier, since elimination of water only occurs at significantly higher temperatures, the rate limiting step for that process must have a much higher barrier. Cleavage of the C–O bond in IVvia migration of the OH group in IV to give the putative PCsp2P iridium(III) hydroxo hydrido chloride complex VI (Scheme 6) would be expected to have a higher barrier.37,38 Once VI is formed, the facility with which O–H bond reductive elimination occurs in these systems renders this species unstable towards rapid water elimination, giving 3·Cl. Such a scenario is supported by the observation and characterization of model compounds for the six coordinate Ir(III) intermediate VI. For example, the PCsp2P carbene chloride 3·Cl can be treated with an excess of hexachloroethane (C2Cl6) at 50 °C to yield the highly insoluble trichloride complex 7·Cl3 in excellent isolated yield (Scheme 6). The insolubility of this species in most common solvents made full spectroscopic characterization difficult, but the solid obtained is pure by elemental analysis and an X-ray crystal structure analysis confirms its identity (Fig. 5). The iridium center exhibits octahedral geometry and the Ir(1)–C(1) distance of 1.938(8) Å is slightly longer than the distances we have observed for these ligands in most square planar, Ir(I) derivatives. The Ir(1)–Cl(2) bond length (trans to the carbene donor) is significantly longer at 2.428(2) Å than the lengths recorded for the cis chlorides (2.348(2) and 2.356(2) Å).
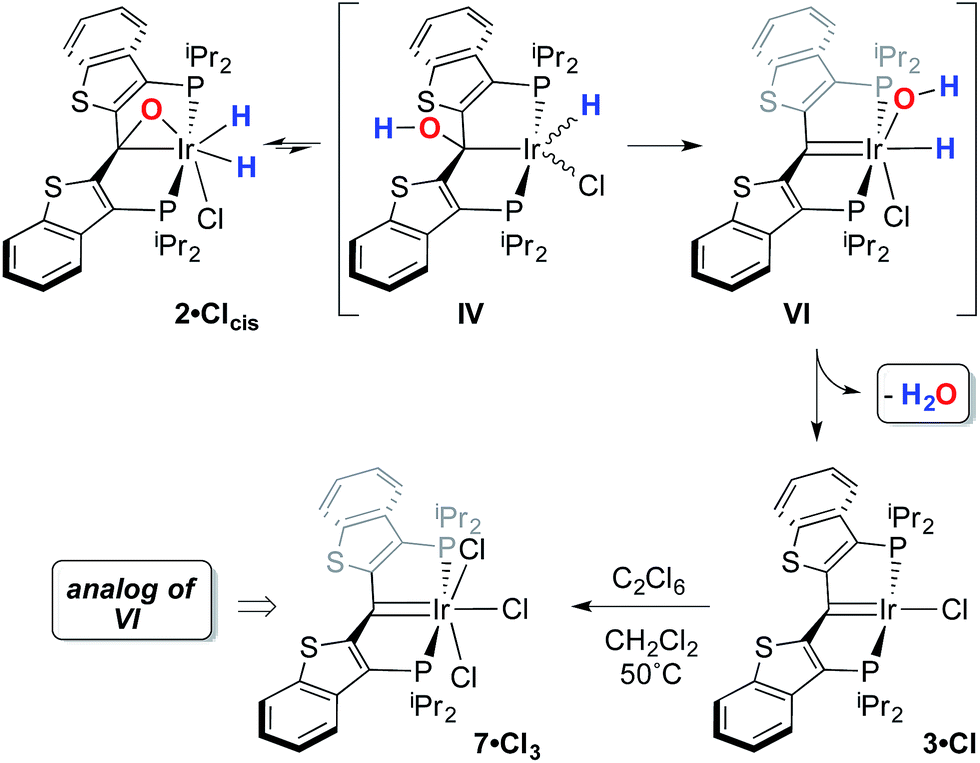 | ||
| Scheme 6 Proposed mechanism for water elimination from 2·Clcis. | ||
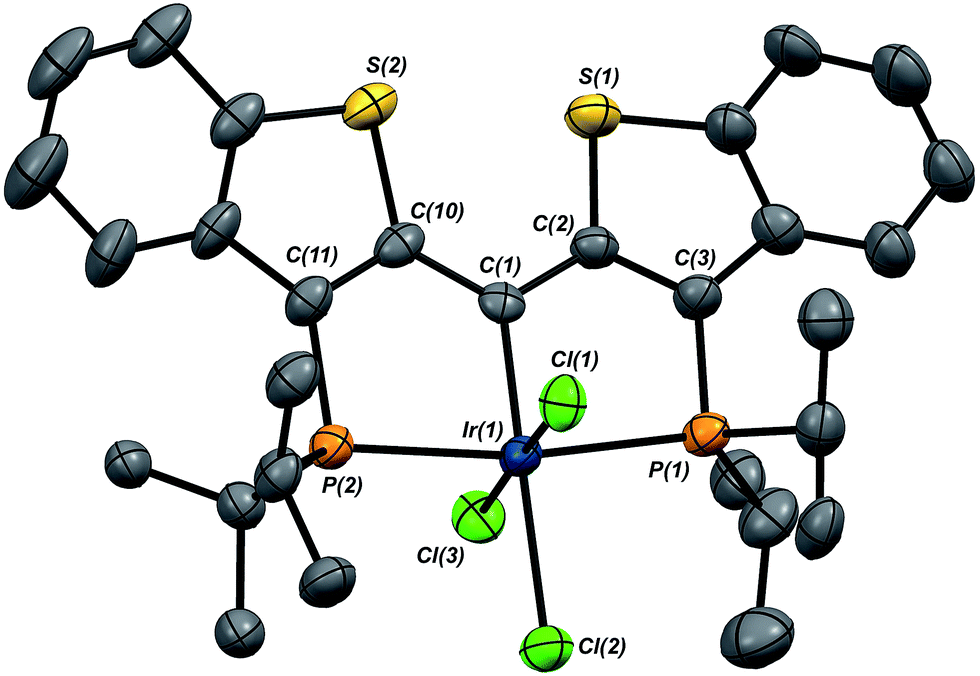 | ||
| Fig. 5 Molecular structure of 7·Cl3. Hydrogen atoms have been omitted for clarity. Displacement ellipsoids are shown at the 50% probability level. Selected bond lengths (Å): Ir(1)–P(1), 2.343(2); Ir(1)–P(2), 2.346(2); Ir(1)–C(1), 1.938(8); Ir(1)–Cl(1), 2.348(2); Ir(1)–Cl(2), 2.428(2); Ir(1)–Cl(3), 2.356(2). Selected bond angles (°): C(1)–Ir(1)–Cl(1), 92.0(2); C(1)–Ir(1)–Cl(2), 178.6(2); C(1)–Ir(1)–Cl(3), 90.3(2); P(1)–Ir(1)–P(2), 170.47(8). | ||
A second, more soluble member of this family of compounds can be made by treatment of the iridaepoxide complex 1·Cl with two equivalents trimethylsilyl triflate. This reaction proceeds smoothly at room temperature to form the PCsp2P iridium(III) bis triflato chloride compound 7·(OTf)2Cl (Scheme 7) along with one equivalent of hexamethyldisiloxane. Interestingly, use of only one equivalent of TMSOTf yields a 1:1 mixture of 1·Cl and 7·(OTf)2Cl, indicating that reaction of the second equivalent of TMSOTf with the product of the reaction of 1·Cl with the first equivalent is very rapid. In any case, because of its greater solubility, compound 7·(OTf)2Cl was fully characterized spectroscopically and features a highly downfield shifted signal at 219.1 ppm (the 2JCP is unresolved) for the carbene carbon, and two distinct resonances at −78.8 and −79.1 ppm in the 19F NMR spectrum, indicating that the two triflate ions are diastereotopic. A solid state structural analysis on single crystals of this species confirms the geometry of the complex as drawn in Scheme 7; the molecular structure is given in Fig. 6. The structure is similar to that of trichloride 7·Cl3 but the ligand backbone exhibits some torsion between the planes of the benzothiophene linkers, possibly due to the more bulky triflate ligands interacting with the phosphine iso-propyl groups. The Ir(1)–C(1) distance in 7·(OTf)2Cl is slightly longer at 1.948(7) Å and the differences in the Ir(1)–O(1) (2.197(5)) and O(4) (2.131(5)) distances are less pronounced than the analogous Ir–Cl distances in 7·Cl3.
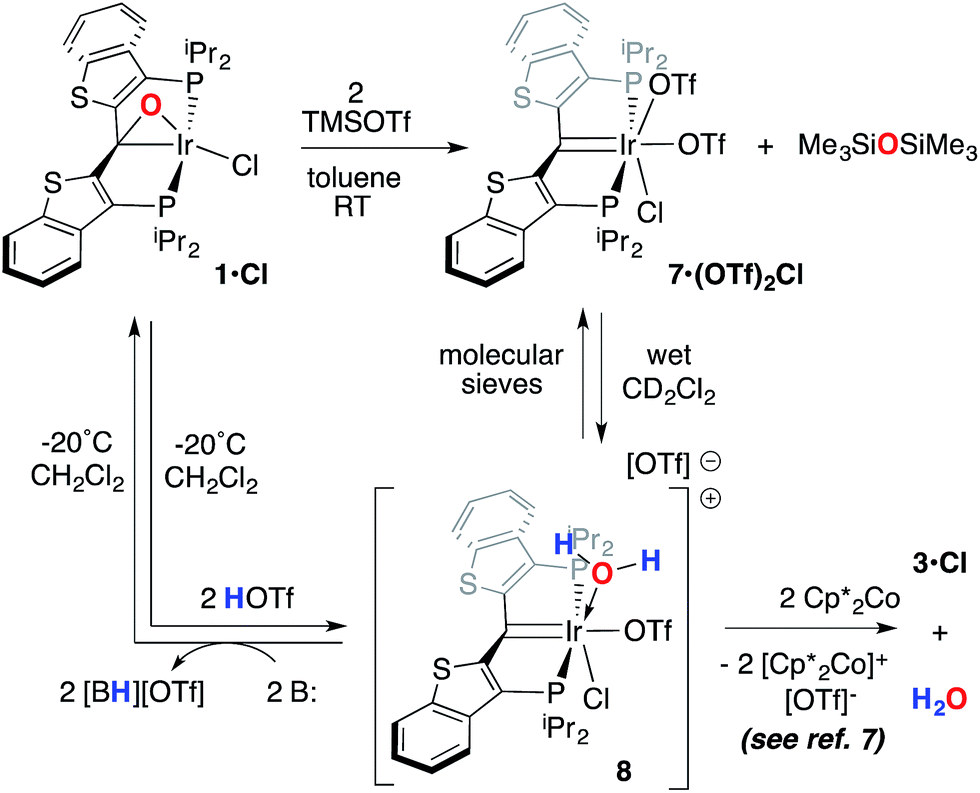 | ||
| Scheme 7 Model reactions for water elimination. | ||
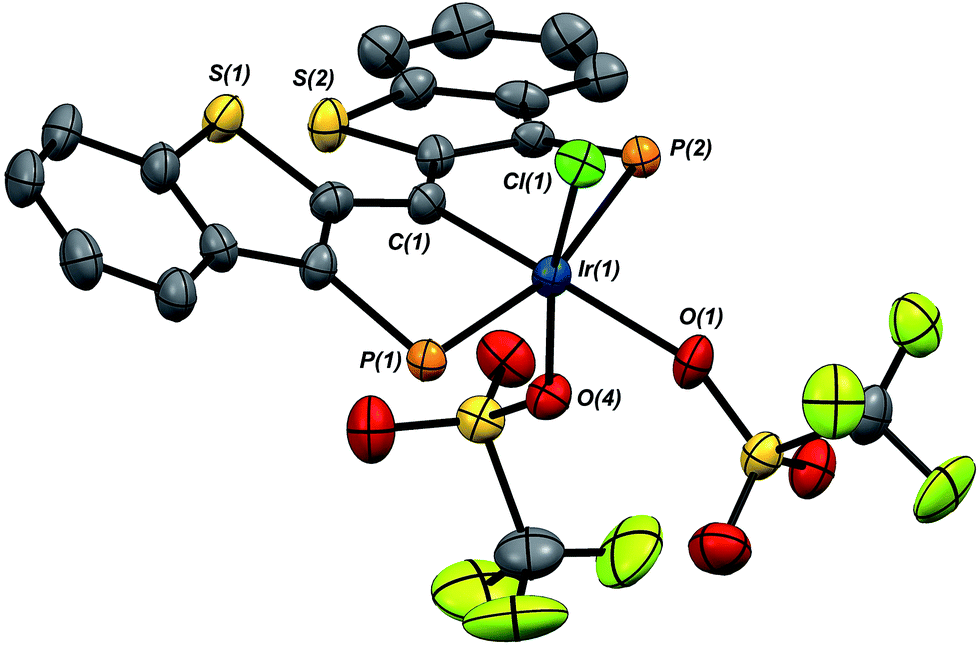 | ||
| Fig. 6 Molecular structure of 7·(OTf)2Cl. Hydrogen atoms and the iso-propyl groups on phosphorus have been omitted for clarity. Displacement ellipsoids are shown at the 50% probability level. Selected bond lengths (Å): Ir(1)–P(1), 2.3797(17); Ir(1)–P(2), 2.3772(17); Ir(1)–C(1), 1.948(7); Ir(1)–Cl(1), 2.3035(17); Ir(1)–O(1), 2.197(5); Ir(1)–O(4), 2.131(5). Selected bond angles (°): C(1)–Ir(1)–Cl(1), 96.5(2); C(1)–Ir(1)–O(1), 177.7(3); C(1)–Ir(1)–O(4), 93.9(2); P(1)–Ir(1)–P(2), 164.76(6). | ||
Compound 7·(OTf)2Cl is significant because it is constitutionally related to the compound we obtained upon double protonation of 1·Cl with triflic acid.7 This product, 8 in Scheme 7, although obtained very cleanly upon treatment of 1·Cl with HOTf at −20 °C in methylene chloride, was not previously isolable and so a precise structural assignment was not possible at the time of the original report.7 Since 7·(OTf)2Cl is related to this species by the addition or elimination of one equivalent of water, we attempted to generate 8 through treatment of 7·(OTf)2Cl with stoichiometric amounts of water. These experiments met with mixed success, but we found that stirring 7·(OTf)2Cl in CD2Cl2 that had not undergone our normal rigorous drying procedure, eventually generated clean samples of compound 8. Water free 7·(OTf)2Cl was slowly regenerated by adding a few activated molecular sieves to this sample; this interconversion is demonstrated by the series of 31P NMR spectra shown in Fig. S3.† We were also able to generate 8 by doubly protonating 1·Cl with HOTf in wet CD2Cl2 and this procedure led to the growth and isolation of single crystals; the X-ray structural determination confirmed the structure of this species as that shown in Scheme 7. Fig. 7 depicts the molecular structure of 8, along with an outer sphere triflate anion from an adjacent unit cell, which exhibits hydrogen bonding with one of the aquo ligand protons as shown. Here, it is the triflate anion located cis to the carbene carbon in 7·(OTf)2Cl that has been formally displaced by the aquo ligand. Presumably the mechanism of this substitution is dissociative in nature, and this geometry for 8 represents the thermodynamically most stable product, with the weakly donating triflate anion occupying the position trans to the carbene carbon.
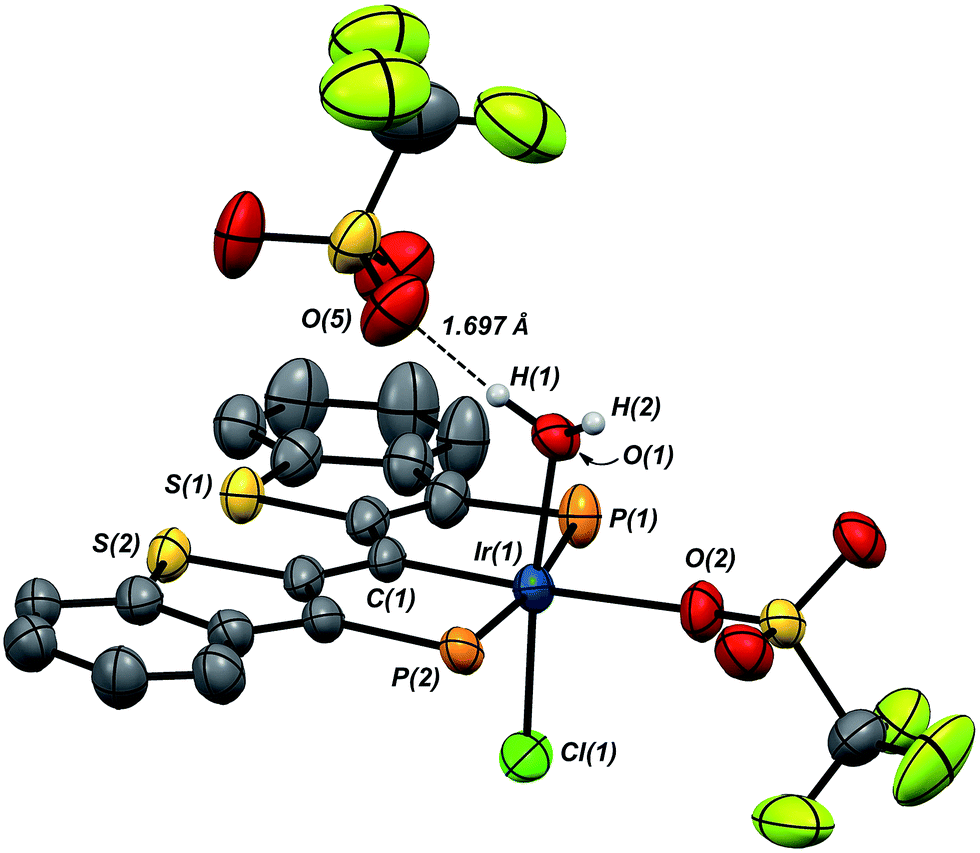 | ||
| Fig. 7 Molecular structure of 8. Hydrogen atoms, except for those on O(1), and the iso-propyl groups on phosphorus have been omitted for clarity. Displacement ellipsoids are shown at the 50% probability level. Selected bond lengths (Å): Ir(1)–P(1), 2.3642(13); Ir(1)–P(2), 2.3648(12); Ir(1)–C(1), 1.960(4); Ir(1)–O(1), 2.098(3); Ir(1)–O(2), 2.196(3); Ir(1)–Cl(1), 2.3200(12). Selected bond angles (°): C(1)–Ir(1)–O(1), 93.25(15); C(1)–Ir(1)–O(2), 175.46(16); O(1)–Ir(1)–O(2), 84.85(13); P(1)–Ir(1)–P(2), 171.34(4). | ||
As we reported previously7 (and also shown in Scheme 7), compound 8 can be reduced with two equivalents of Cp*2Co to generate one equivalent of water and 1·Cl. We made some attempts to deprotonate 8 using Et3N or PhNH2 to access a PCsp2P Ir(III)X3 compound incorporating a hydroxo ligand, but both of these bases simply returned back 1·Cl exclusively. These results collectively show that, if a derivative of 7 with a hydroxo ligand (akin to VI, for example) is generated, it is highly reactive and is prone to either O–H reductive elimination or transfer of the OH group from the metal to the carbene carbon (to give a complex related to IV).
Experimental
For general experimental details, see the ESI.†Synthesis of PCP(O)IrOH 1·OH
IrOH 3·OH and dissolved in ca. 3 mL toluene. The solution was degassed and subsequently placed under 1 atm N2O gas. The solution was heated to 100 °C for 2 hours during which a colour change from deep brown to bright red was observed. The volatile components were removed in vacuo and the product was isolated as a red solid in 94% yield (24 mg, 0.033 mmol). Single crystals were grown by slow evaporation of a toluene solution. Co-crystallization of one toluene molecule per two molecules of 1·OH was observed in the unit cell
Synthesis of PCP(O)IrBr 1·Br
A 50 mL thick-walled glass reactor vessel was charged with 10 mg (0.013 mmol) of PCPIrBr 3·Br and dissolved in ca. 1 mL toluene. The solution was degassed and subsequently placed under 1 atm N2O gas. The solution was heated to 100 °C for 2 hours during which a colour change from deep brown to bright red was observed. The same reaction also proceeds at room temperature over the course of 16 hours. The volatile components were removed in vacuo and the product was recrystallized from a toluene/hexanes (ca. 1:3) mixture at −25 °C. The product was isolated as a bright red crystalline solid in 98% yield (10 mg, 0.013 mmol). Single crystals were obtained by slow evaporation of a toluene/hexanes mixture resulting in co-crystallization of one molecule of toluene. 1H NMR (600 MHz, toluene-d8) δ 7.71 (d, 3JHH = 8.1 Hz, 2H, ArH), 7.42 (d, 3JHH = 8.1 Hz, 2H, ArH), 7.15 (t, 3JHH = 7.5 Hz, 2H, ArH), 7.06 (t, 3JHH = 7.6 Hz, 2H, ArH), 3.47 (sept, 3JHH = 7.1 Hz, 2H, CH(CH3)2), 2.80 (m, 2H, CH(CH3)2), 1.54 (dvt, 3JHH = 7.6 Hz, JHP = 8.3 Hz, 6H, CH(CH3)2), 1.30 (dvt, 3JHH = JHP = 7.8 Hz, 6H, CH(CH3)2), 1.25 (dvt, 3JHH = JHP = 7.1 Hz, 6H, CH(CH3)2), 1.13 (dvt, 3JHH = JHP = 7.7 Hz, 6H, CH(CH3)2). 13C{1H} NMR (151 MHz, toluene-d8) δ 159.0 (vt, J = 11.1 Hz, aryl C), 143.7 (vt, J = 3.9 Hz, aryl C), 138.8 (s, aryl C), 126.5 (vt, J = 16.9 Hz, aryl C), 125.2 (s, aryl CH), 124.7 (s, aryl CH), 123.9 (s, aryl CH), 123.5 (s, aryl CH), 64.5 (vt, J = 2.4 Hz, C(O)Ir), 24.4 (vt, JCP = 15.9 Hz, CH(CH3)2), 24.3 (vt, JCP = 13.2 Hz, CH(CH3)2), 19.8 (s, CH(CH3)2), 19.7 (vt, JCP = 3.2 Hz, CH(CH3)2), 19.7 (s, CH(CH3)2), 19.2 (s, CH(CH3)2). 31P{1H} NMR (243 MHz, toluene-d8) δ 25.0 (s). Elemental anal. calcd (%) for C36H44BrIrOP2S2 (1·Br + toluene): C 48.53; H 4.98. Found: C 48.83; H 4.95.
Synthesis of PCP(O)Ir(H)2Br 2·Brtrans
A J-Young NMR tube was charged with 7 mg (0.009 mmol) of PCP(O)IrBr 1·Br dissolved in ca. 0.5 mL toluene. The solution was degassed and subsequently placed under 1 atm H2 gas. The solution was allowed to mix for 30 minutes at room temperature during which a colour change from bright red to pale yellow was observed. The volatile components were removed in vacuo and the pale yellow-brown solid was recrystallized from a toluene/hexanes (ca. 1:3) mixture at −25 °C. The product was isolated as a pale yellow-brown solid in 99% yield (7 mg, 0.009 mmol). 1H NMR (600 MHz, toluene-d8) δ 7.57 (d, 3JHH = 8.1 Hz, 2H, ArH), 7.39 (d, 3JHH = 8.0 Hz, 2H, ArH), 7.14 (t, 3JHH = 7.8 Hz, 2H, ArH), 7.02 (t, 3JHH = 7.6 Hz, 2H, ArH), 3.19 (sept, 3JHH = 7.0 Hz, 2H, CH(CH3)2), 2.26 (m, 2H, CH(CH3)2), 1.41 (dvt, 3JHH = 8.9 Hz, JHP = 7.1 Hz, 6H, CH(CH3)2), 1.16 (m, 12H, CH(CH3)2), 0.68 (dvt, 3JHH = 8.7 Hz, JHP = 7.1 Hz, 6H, CH(CH3)2), −8.29 (t, 2JHP = 10.5 Hz, 1H, IrH), −15.42 (t, 2JHP = 10.5 Hz, 1H, IrH). 13C{1H} NMR (151 MHz, toluene-d8) δ 161.8 (vt, JCP = 11.2 Hz, aryl C), 143.7 (vt, JCP = 4.0 Hz, aryl C), 138.1 (vt, JCP = 1.9 Hz, aryl C), 133.2 (vt, JCP = 18.2 Hz, aryl C), 125.3 (s, aryl CH), 125.1 (s, aryl CH), 123.9 (s, aryl CH), 123.8 (s, aryl CH), 99.0 (t, JCP = 3.5 Hz, C(O)Ir), 25.6 (vt, JCP = 14.0 Hz, CH(CH3)2), 22.1 (vt, JCP = 17.3 Hz, CH(CH3)2), 19.5 (s, CH(CH3)2), 19.2 (s, CH(CH3)2), 19.1 (vt, JCP = 3.9 Hz, CH(CH3)2), 19.0 (s, CH(CH3)2). 31P{1H} NMR (243 MHz, toluene-d8) δ 22.8 (s). HRMS (APCI) calculated for C29H38BrIrOP2S2 (M+): 799.0568, found 799.0543.
Synthesis of PCP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) IrOH 3·OH
IrOH 3·OH
A 50 mL sealed glass reactor vessel was charged with 50 mg (0.068 mmol) of PCPIrCl 3·Cl and dissolved in ca. 10 mL toluene. A large excess of CsOH was added to the solution and the mixture was heated at 80 °C for 48 hours. The solution was then cooled to room temperature and filtered through Celite. The solvent was removed in vacuo and the product was isolated as a deep brown solid in 98% yield (48 mg, 0.067 mmol). X-ray quality crystals were obtained by slow evaporation from hexanes. 1H NMR (600 MHz, toluene-d8) δ 8.33 (d, 3JHH = 8.1 Hz, 2H, ArH), 7.84 (t, 3JHH = 7.6 Hz, 2H, ArH), 7.13 (d, 3JHH = 8.2 Hz, 2H, ArH), 7.01 (t, 3JHH = 7.6 Hz, 2H, ArH), 4.18 (t, 3JHP = 4.5 Hz, 1H, IrOH), 3.07 (septvt, 3JHH = 6.9 Hz, JHP = 1.8 Hz, 4H, CH(CH3)2), 1.55 (dvt, 3JHH = JHP = 7.7 Hz, 12H, CH(CH3)2), 1.26 (dvt, 3JHH = JHP = 7.4 Hz, 12H, CH(CH3)2). 13C{1H} NMR (151 MHz, toluene-d8) δ 188.9 (vt, JCP = 22.0 Hz, aryl C), 172.2 (t, JCP = 2.3 Hz, CIr), 145.3 (vt, JCP = 3.9 Hz, aryl C), 142.6 (s, aryl C), 138.0 (vt, JCP = 18.9 Hz, aryl C), 127.2 (s, aryl CH), 126.6 (s, aryl CH), 124.75 (s, aryl CH), 122.9 (s, aryl CH), 24.5 (vt, JCP = 13.5 Hz, CH(CH3)2), 21.0 (vt, JCP = 3.2 Hz, CH(CH3)2), 20.7 (s, CH(CH3)2). 31P{1H} NMR (243 MHz, toluene-d8) δ 34.5 (s). Elemental anal. calcd (%) for C29H37IrOP2S2: C 48.38; H 5.18. Found: 48.58; H 5.20.
Synthesis of PCPIrBr 3·Br
A large excess of LiBr was added to a solution of PCPIrOH 3·OH (80 mg, 0.111 mmol) in ca. 3 mL toluene. The mixture was allowed to stir for 1 hour at room temperature after which the dark brown solution was filtered through Celite and the solvent was removed in vacuo. The desired product was isolated as a dark brown solid in 97% yield (84 mg, 0.107 mmol). 1H NMR (600 MHz, CD2Cl2) δ 8.80 (d, 3JHH = 8.2 Hz, 2H, ArH), 8.38 (t, 3JHH = 7.6 Hz, 2H, ArH), 7.44 (d, 3JHH = 8.0 Hz, 2H, ArH), 7.27 (t, 3JHH = 7.7 Hz, 2H, ArH), 3.71 (septvt, 3JHH = 7.1 Hz, JHP = 2.1 Hz, 4H, CH(CH3)2), 1.65 (dvt, 3JHH = 8.8, JHP = 7.2 Hz, 12H, CH(CH3)2), 1.47 (dvt, 3JHH = JHP = 7.4 Hz, 12H, CH(CH3)2). 13C{1H} NMR (151 MHz, CD2Cl2) δ 185.0 (vt, JCP = 22.6 Hz, aryl C), 161.4 (t, 2JCP = 2.8 Hz, CIr), 147.7 (vt, JCP = 3.7 Hz, aryl C), 143.5 (vt, JCP = 16.3 Hz, aryl C), 141.9 (s, aryl C), 127.8 (s, aryl CH), 127.4 (s, aryl CH), 126.5 (s, aryl CH), 125.5 (s, aryl CH), 24.2 (vt, JCP = 14.0 Hz, CH(CH3)2), 21.4 (vt, JCP = 2.4 Hz, CH(CH3)2), 21.1 (s, CH(CH3)2). 31P{1H} NMR (243 MHz, CD2Cl2) δ 36.3 (s). HRMS (APCI) calculated for C29H36BrIrP2S2 (M + H)+: 783.0619, found 783.0608.
Synthesis of PCPIrH44
PCP(O)IrOH 1·OH was dissolved in toluene-d8 in a J-Young NMR tube and subsequently degassed and placed under 1 atm H2. The mixture was then heated to 100 °C for 2 hours during which a colour change from red to yellow was observed. The product, PCPIrH44, is unstable outside of an H2 atmosphere and was therefore characterized by NMR spectroscopy in situ without isolation. During the hydrogenation process, partial deuteration of the aromatic, benzylic and iridium centered hydrogens was observed, making NMR assignments difficult. 1H NMR (600 MHz, toluene-d8) δ 7.56 (d, 3JHH = 7.9 Hz, 2H, ArH), 7.49 (d, 3JHH = 8.0 Hz, 2H, ArH), 7.20 (t, 3JHH = 7.5 Hz, 2H, ArH), 7.04 (m, 2H, ArH), 5.95 (s, 1H, IrCH), 2.17 (m, 2H, CH(CH3)2), 1.97 (sept, 3JHH = 6.7 Hz, 2H, CH(CH3)2), 1.10 (m, 12H, CH(CH3)2), 0.79 (m, 12H, CH(CH3)2), −9.63 (t, 2JHP = 10.3 Hz, 4H, IrH4). 13C{1H} NMR (151 MHz, toluene-d8) δ 173.3 (vt, JCP = 19.0 Hz, aryl C), 146.2 (vt, JCP = 4.2 Hz, aryl C), 137.1 (s, aryl C), 134.6 (vt, JCP = 23.5 Hz, aryl C), 124.6 (s, aryl CH), 124.2 (s, aryl CH), 123.6 (s, aryl CH), 122.0 (s, aryl CH), 28.5 (vt, JCP = 15.5 Hz, CH(CH3)2), 24.5 (vt, JCP = 17.9 Hz, CH(CH3)2), 21.7 (vt, JCP = 4.1 Hz, CH(CH3)2), 21.3 (vt, JCP = 3.5 Hz, CH(CH3)2), 19.4 (s, CH(CH3)2), 19.2 (s, CH(CH3)2), 11.7 (s, IrCH). 31P{1H} NMR (243 MHz, toluene-d8) δ 49.0 (s).Synthesis of PCPIrCl37·Cl3
PCPIrCl 3·Cl (27 mg, 0.037 mmol) was dissolved in ca. 1 mL CH2Cl2 in a 50 mL sealed glass reactor vessel. Hexachloroethane (9 mg, 0.038 mmol) was added to the solution as a solid and the mixture was heated to 50 °C for 1 hour. During this time, a colour change from dark brown to almost colourless was observed as well as a large amount of green-black precipitate. The solvent was decanted and the precipitate was washed three times with hexanes. The product was dried in vacuo and isolated as a green-black solid in 95% yield (28 mg, 0.035 mmol). This reaction can also be done with toluene as the solvent, resulting in bright red single crystals co-crystallized with toluene. 1H NMR (500 MHz, CD2Cl2) δ 8.24 (d, 3JHH = 8.4 Hz, 2H, ArH), 7.96 (d, 3JHH = 8.5 Hz, 2H, ArH), 7.81 (t, 3JHH = 7.7 Hz, 2H, ArH), 7.57 (t, 3JHH = 7.8 Hz, 2H, ArH), 3.61 (m, 4H, CH(CH3)2), 1.60 (dvt, 3JHH = JHP = 7.8 Hz, 12H, CH(CH3)2), 1.54 (dvt, 3JHH = JHP = 7.7 Hz, 12H, CH(CH3)2). 31P{1H} NMR (203 MHz, CD2Cl2) δ −0.3 (s). Elemental anal. calcd (%) for C29H36Cl3IrP2S2: C 43.04; H 4.48. Found: C 43.01; H 4.55.
Synthesis of PCPIr(OTf)2Cl 7·(OTf)2Cl
TMSOTf (12.0 μL, 0.066 mmol) was dissolved in 0.5 mL toluene and added dropwise to a solution of PCP(O)IrCl 1·Cl (25 mg, 0.033 mmol) in 0.5 mL toluene in a 4 mL vial at room temperature. A colour change from bright red to a darker red-brown colour was observed. The vial was left to sit without stirring at room temperature for 24 hours during which large green crystals were formed. The solution was decanted and the crystals were washed with hexanes and pumped to dryness. The desired product co-crystallizes with one molecule of toluene. 7·(OTf)2Cl was isolated as a dark green crystalline solid in 83% yield (31 mg, 0.027 mmol). When dissolved in CD2Cl2, 7·(OTf)2Cl appears as a dark red-purple solution. 1H NMR (500 MHz, CD2Cl2) δ 8.30 (d, 3JHH = 8.4 Hz, 2H, ArH), 8.05 (d, 3JHH = 8.2 Hz, 2H, ArH), 7.80 (t, 3JHH = 7.7 Hz, 2H, ArH), 7.66 (t, 3JHH = 7.7 Hz, 2H, ArH), 3.56 (m, 2H, CH(CH3)2), 3.52 (m, 2H, CH(CH3)2), 1.60 (dvt, 3JHH = JHP = 8.2 Hz, 6H, CH(CH3)2), 1.55 (dvt, 3JHH = JHP = 7.7 Hz, 6H, CH(CH3)2), 1.45 (dvt, 3JHH = JHP = 8.0 Hz, 6H, CH(CH3)2), 1.43 (dvt, 3JHH = JHP = 8.5 Hz, 6H, CH(CH3)2). 13C{1H} NMR (126 MHz, CD2Cl2) δ 219.1 (s, CIr), 168.8 (vt, JCP = 15.8 Hz, aryl C), 158.7 (vt, JCP = 13.8 Hz, aryl C), 150.8 (vt, JCP = 4.1 Hz, aryl C), 140.9 (s, aryl C), 131.3 (s, aryl CH), 128.5 (s, aryl CH), 128.2 (s, aryl CH), 125.5 (s, aryl CH), 119.3 (q, 1JCF = 319.1 Hz, CF3SO3), 117.0 (q, 1JCF = 318.9 Hz, CF3SO3), 25.9 (vt, JCP = 11.0 Hz, CH(CH3)2), 24.0 (vt, JCP = 12.7 Hz, CH(CH3)2), 21.2 (s, CH(CH3)2), 21.1 (s, CH(CH3)2), 20.5 (s, CH(CH3)2), 19.3 (s, CH(CH3)2). 31P{1H} NMR (203 MHz, CD2Cl2) δ 19.2 (s). 19F NMR (471 MHz, CD2Cl2) δ −78.7 (s, CF3SO3), −79.1 (s, CF3SO3). Elemental anal. calcd (%) for C38H44ClF6IrO6P2S4 (7·(OTf)2Cl + toluene): C 40.44; H 3.93. Found: C 40.14; H 4.13.
Low temperature addition of H2, D2 or HD to 1·Cl
All reactions involving the addition of 1 atm of H2, D2 or HD gas were performed by degassing the reaction mixture in a J-Young NMR tube, followed by addition of 1 atm of the gas at room temperature. Reactions carried out under 2–3 atm of a gas were performed by degassing the reaction mixture in a J-Young NMR tube and submerging the entire length of the tube in liquid nitrogen (−196 °C) while open to the gas. Once sealed, the sample was warmed to the desired temperature (−78 to −38 °C).Low temperature addition of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 H2/D2 to 1·Cl
:1 H2/D2 to 1·Cl
A 1:1 mixture of H2:D2 was formed by first fully evacuating a double manifold vacuum line and then introducing H2 (300 mmHg) followed by D2 (300 mmHg). This mixture was allowed to equilibrate for a minimum of 6 hours after which a previously degassed sample in a J-Young NMR tube was opened to allow the mixture in. The same procedure was followed using 420 mmHg H2 for a 1.4:1 mixture of H2/D2.
Measurement of equilibrium experiments
In order to dissolve a sufficient amount of H2 in solution without forming any of the thermodynamic isomer 2·Xtrans, H2 was added to a solution of 1·X (15 mM) in 0.5 mL CD2Cl2 at −196 °C and warmed to −78 °C in a dry ice/acetone bath. The sample was then quickly transferred to the NMR spectrometer, which had previously been cooled to −78 °C. A 31P{1H} experiment (243 MHz, d1 = 2 seconds, 16 scans) was then performed at each temperature and integrations were used to determine relative concentrations of the iridium complexes. To measure the concentration of H2 dissolved in solution during the thermodynamic van't Hoff equilibrium experiments, the reaction conditions were duplicated using an internal standard. Pentachlorobenzene (1.3 mg, 0.005 mmol, 10 mM) was dissolved in 0.5 mL CD2Cl2 in a J-Young NMR tube which was subsequently degassed, charged with H2 gas and introduced into the NMR spectrometer at −78 °C as described above. A 1H NMR experiment (600 MHz, d1 = 20 seconds, 8 scans) was performed at each temperature and integrations were used to determine the concentration of H2 in solution; a correction of 1.33 was used to account for the ≈25% parahydrogen that is undetectable by 1H NMR spectroscopy.39,40 As the concentration of D2 gas in solution could not be easily measured using NMR spectroscopy, it was estimated based on reported isotope fractionation factors.41 Thus, the concentration of D2 in solution used for these calculations is 1.1 × [H2] at a given temperature.Conclusions
The PCsp2P ligand systems exemplified by structures I and II are a unique class of PCP pincer ligands in that the anchoring CM donor is capable of entering into cooperative reactivity with small molecule substrates. The reason for this partly stems from the generally reactive nature of metal carbenes, but here it is also due to the adaptable nature of the character of the CM unit in diaryl carbenes. This class of carbenes is somewhat special in that they lie somewhere between the extremes of Fischer type and Schrock type carbenes.16,42 Although not strongly so, aryl substituents are π-donating and, in the case of ligands I and II, this results in an ability to function as both “Schrock-like” and “Fischer-like”, depending on the nature of the metal center.
This adaptability is clearly manifested in the comparative behavior of the iridium6 and nickel9 complexes of ligand I, but is also potentially a factor within the iridium manifold in the chemistry discussed herein. For example, the proposed transformation of IV into VI as shown in Scheme 6 is illustrative of this notion. Although IV can be clearly formulated as an Ir(III) derivative, transfer of the OH group to the iridium center is in some sense an oxidative addition reaction. But because the PCsp2P ligand that forms in this step can take on Fischer-like character, the iridium center is still formally Ir(III) or, put another way, the ligand enables the stabilization of highly reactive Ir(V) compounds (7·X3) or intermediates (VI). In so doing, the iridium complexes of ligand II provide a pathway for the overall hydrogenation of N2O to water and dinitrogen.
The mechanistic details of this process have been probed by a combination of deuterium labeling experiments and low temperature NMR spectroscopic investigations. While developing this chemistry from an essentially stoichiometric process to a catalytic one would be desirable,43 the reactions of the iridium-based products and catalytic intermediates with hydrogen have precluded us from achieving this goal as of yet. The results of these investigations, however, do provide the basis for further ligand modifications that are currently being pursued.
Acknowledgements
Funding for this work was provided by NSERC of Canada in the form of a Discovery Grant and an Accelerator Supplement to W. E. P. W. E. P. also thanks the Canada Research Chair secretariat for a Tier I CRC (2013–2020). L. E. D. thanks NSERC for a CGSD scholarship and Alberta Innovates Technology Futures for a Graduate Student Scholarship.Notes and references
- Topics in Organometallic Chemistry: Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer, Heidelberg, 2013 Search PubMed.
- Pincer and Pincer-Type Complexes: Applications in Organic Synthesis and Catalysis, ed. Kalman J. Szabo and Ola F. Wendt, Wiley, Weinheim, Germany, 2014 Search PubMed.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- D. Gelman and R. Romm, in Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer, Berlin Heidelberg, 2013, vol. 40, ch. 10, pp. 289–317 Search PubMed.
- C. C. Roberts, D. M. Matías, M. J. Goldfogel and S. J. Meek, J. Am. Chem. Soc., 2015, 137, 6488–6491 CrossRef CAS PubMed.
- R. J. Burford, W. E. Piers and M. Parvez, Organometallics, 2012, 31, 2949–2952 CrossRef CAS.
- L. E. Doyle, W. E. Piers and J. Borau-Garcia, J. Am. Chem. Soc., 2015, 137, 2187–2190 CrossRef CAS PubMed.
- H. D. Empsall, E. M. Hyde, R. Markham, W. S. McDonald, M. C. Norton, B. L. Shaw and B. Weeks, J. Chem. Soc., Chem. Commun., 1977, 589–590 RSC.
- D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776–11779 CrossRef CAS PubMed.
- P. Cui, C. C. Comanescu and V. M. Iluc, Chem. Commun., 2015, 51, 6206–6209 RSC.
- C. C. Comanescu and V. M. Iluc, Organometallics, 2014, 33, 6059–6064 CrossRef CAS.
- C. C. Comanescu, M. Vyushkova and V. M. Iluc, Chem. Sci., 2015, 6, 4570–4579 RSC.
- J. Borau-Garcia, D. V. Gutsulyak, R. J. Burford and W. E. Piers, Dalton Trans., 2015, 44, 12082–12085 RSC.
- R. J. Burford, W. E. Piers and M. Parvez, Eur. J. Inorg. Chem., 2013, 3826–3830 CrossRef CAS.
- R. J. Burford, W. E. Piers, D. H. Ess and M. Parvez, J. Am. Chem. Soc., 2014, 136, 3256–3263 CrossRef CAS PubMed.
- M. T. Whited and R. H. Grubbs, Acc. Chem. Res., 2009, 42, 1607–1616 CrossRef CAS PubMed.
- M. Gozin, A. Weisman, Y. Bendavid and D. Milstein, Nature, 1993, 364, 699–701 CrossRef CAS.
- J. D. Smith, J. Borau-Garcia, W. E. Piers and D. Spasyuk, Can. J. Chem., 2015, 1–4, DOI:10.1139/cjc-2015-0251.
- P. P. Deutsch and R. Eisenberg, Chem. Rev., 1988, 88, 1147–1161 CrossRef CAS.
- T. Hascall, D. Rabinovich, V. J. Murphy, M. D. Beachy, R. A. Friesner and G. Parkin, J. Am. Chem. Soc., 1999, 121, 11402–11417 CrossRef CAS.
- G. Parkin, Acc. Chem. Res., 2009, 42, 315–325 CrossRef CAS PubMed.
- G. Parkin, J. Labelled Compd. Radiopharm., 2007, 50, 1088–1114 CrossRef CAS.
- L. Vaska, Acc. Chem. Res., 1968, 1, 335–344 CrossRef CAS.
- F. Abu-Hasanayn, K. Krogh-Jespersen and A. S. Goldman, Inorg. Chem., 1993, 32, 495–496 CrossRef CAS.
- F. Abu-Hasanayn, A. S. Goldman and K. Krogh-Jespersen, Inorg. Chem., 1994, 33, 5122–5130 CrossRef CAS.
- K. A. Woerpel and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 7888–7889 CrossRef CAS.
- C. A. Miller, T. S. Janik, C. H. Lake, L. M. Toomey, M. R. Churchill and J. D. Atwood, Organometallics, 1994, 13, 5080–5087 CrossRef CAS.
- D. Sieh, M. Schlimm, L. Andernach, F. Angersbach, S. Nückel, J. Schöffel, N. Šušnjar and P. Burger, Eur. J. Inorg. Chem., 2012, 2012, 444–462 CrossRef CAS.
- B. J. Truscott, D. J. Nelson, C. Lujan, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2013, 19, 7904–7916 CrossRef CAS PubMed.
- T. J. Hebden, K. I. Goldberg, D. M. Heinekey, X. Zhang, T. J. Emge, A. S. Goldman and K. Krogh-Jespersen, Inorg. Chem., 2010, 49, 1733–1742 CrossRef CAS PubMed.
- J. Zhou and J. F. Hartwig, Angew. Chem., Int. Ed., 2008, 47, 5783–5787 CrossRef CAS PubMed.
- V. M. Iluc, A. Fedorov and R. H. Grubbs, Organometallics, 2011, 31, 39–41 CrossRef.
- O. Blum and D. Milstein, Angew. Chem., Int. Ed. Engl., 1995, 34, 229–231 CrossRef CAS.
- D. Morales-Morales, D. W. Lee, Z. Wang and C. M. Jensen, Organometallics, 2001, 20, 1144–1147 CrossRef CAS.
- M. Findlater, K. M. Schultz, W. H. Bernskoetter, A. Cartwright-Sykes, D. M. Heinekey and M. Brookhart, Inorg. Chem., 2012, 51, 4672–4678 CrossRef CAS PubMed.
- M. R. Kita and A. J. M. Miller, J. Am. Chem. Soc., 2014, 136, 14519–14529 CrossRef CAS PubMed.
- J. Choi, Y. Choliy, X. Zhang, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2009, 131, 15627–15629 CrossRef CAS PubMed.
- S. Kundu, J. Choi, D. Y. Wang, Y. Choliy, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2013, 135, 5127–5143 CrossRef CAS PubMed.
- Y. A. Ustynyuk and I. P. Gloriozov, Russ. Chem. Bull., 2009, 58, 1841–1846 CrossRef CAS.
- R. Eisenberg, Acc. Chem. Res., 1991, 24, 110–116 CrossRef CAS.
- J. Muccitelli and W.-Y. Wen, J. Solution Chem., 1978, 7, 257–267 CrossRef CAS.
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2000, 34, 18–29 CrossRef.
- W. B. Tolman, Angew. Chem., Int. Ed., 2010, 49, 1018–1024 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Description of general methods, Fig. S1–3, NMR spectra, crystallographic data for 3·OH, 1·OH, 1·Br, 7·Cl3, 7·(OTf)2Cl and 8. CCDC 1423154–1423159. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03575a |
| ‡ The HD employed was 96 mol%, 98 atom% D, so small amounts of H2 are present; however more H2 is present in these samples than would be expected from the source gas. |
| § The isotopologues are found in a nearly 1:1 ratio due to the excess of H2 in the H2/D2 mixture used. |
| ¶ While patterns reminiscent of those shown in Fig. 1 for the hydride ligands appear in the upfield region, they are not as well resolved as those found for the trans isomer. For 2·Clcis, the 31P NMR spectrum is more suited for resolving the isotopologues. |
| This journal is © The Royal Society of Chemistry 2016 |