
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel(II)-catalyzed enantioselective cyclopropanation of 3-alkenyl-oxindoles with phenyliodonium ylide via free carbene†
Jing
Guo
a,
Yangbin
Liu
a,
Xiangqiang
Li
a,
Xiaohua
Liu
*a,
Lili
Lin
a and
Xiaoming
Feng
*ab
aKey Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: liuxh@scu.edu.cn; xmfeng@scu.edu.cn; Fax: +86 28 85418249; Tel: +86 28 85418249
bCollaborative Innovation Center of Chemical Science and Engineering, Tianjin, China
First published on 4th January 2016
Abstract
A chiral Lewis acid-promoted enantioselective cyclopropanation using phenyliodonium ylide as the carbene precursor was developed. A variety of spirocyclopropane-oxindoles with contiguous tertiary and all carbon quaternary centers were obtained in excellent outcomes (up to 99% yield, >19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r., up to 99% ee). EPR spectroscopy study supported a stepwise biradical mechanism.
:1 d.r., up to 99% ee). EPR spectroscopy study supported a stepwise biradical mechanism.
Introduction
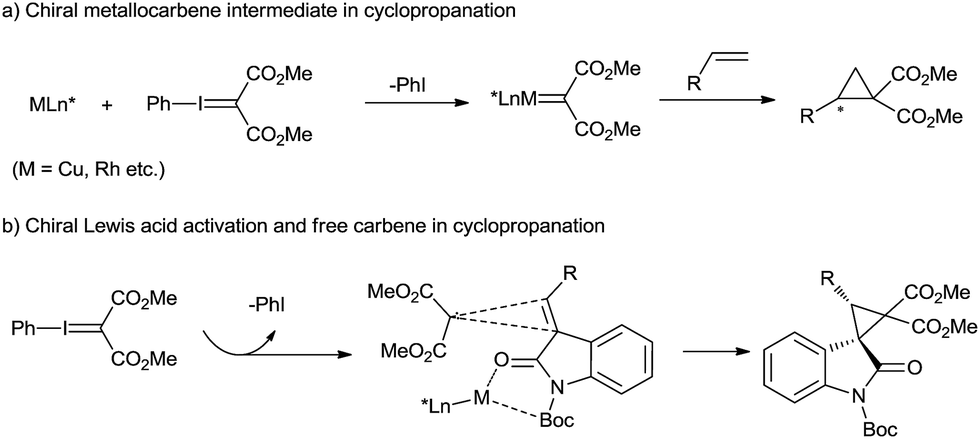
The catalytic asymmetric cyclopropanation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond provides efficient access to optically active cyclopropane derivatives that are essential motifs in natural products1 and valuable building blocks for various transformations.2 Chiral transition-metal-catalyzed carbene transfer, such as that using Cu(I)-, Rh(II)-, Ru(II)-, and Co(II)-complexes with diazo compounds, permits such cylcopropanations in high yields and selectivities.3 However, in view of the mild, selective and non-toxic characteristics of phenyliodonium ylides, they could provide an alternative to diazo precursors in the cyclopropanation.4 The asymmetric cyclopropanation of simple olefins using phenyliodonium ylide was successfully developed, mainly with chiral [Rh2{(S)-nttl}4]5 and Cu(I)-bis(oxazoline) complexes.6 The reaction mechanism generally involves a metallocarbene intermediate surrounded by chiral ligands (Scheme 1a). However, the study of DeLuca and coworkers shows that thermal decomposition of phenyliodonium ylide malonate under mild conditions appears to be an efficient source of free singlet carbene,7 which can undergo uncatalyzed cyclopropanation stereospecifically. This presents an opportunity for asymmetric cyclopropanation via enantiocontrol of an olefin substrate in the carbene transfer step, but the difficulty of gaining high stereoselectivity and yield lies in the background reaction and the readiness of carbene dimer formation.
C double bond provides efficient access to optically active cyclopropane derivatives that are essential motifs in natural products1 and valuable building blocks for various transformations.2 Chiral transition-metal-catalyzed carbene transfer, such as that using Cu(I)-, Rh(II)-, Ru(II)-, and Co(II)-complexes with diazo compounds, permits such cylcopropanations in high yields and selectivities.3 However, in view of the mild, selective and non-toxic characteristics of phenyliodonium ylides, they could provide an alternative to diazo precursors in the cyclopropanation.4 The asymmetric cyclopropanation of simple olefins using phenyliodonium ylide was successfully developed, mainly with chiral [Rh2{(S)-nttl}4]5 and Cu(I)-bis(oxazoline) complexes.6 The reaction mechanism generally involves a metallocarbene intermediate surrounded by chiral ligands (Scheme 1a). However, the study of DeLuca and coworkers shows that thermal decomposition of phenyliodonium ylide malonate under mild conditions appears to be an efficient source of free singlet carbene,7 which can undergo uncatalyzed cyclopropanation stereospecifically. This presents an opportunity for asymmetric cyclopropanation via enantiocontrol of an olefin substrate in the carbene transfer step, but the difficulty of gaining high stereoselectivity and yield lies in the background reaction and the readiness of carbene dimer formation.
 | ||
| Scheme 1 Catalytic enantioselective cyclopropanation of olefins with phenyliodonium ylide. | ||
Spirocyclopropane-oxindoles are versatile building blocks for the synthesis of natural products and pharmaceuticals.8 The asymmetric catalytic cyclopropanations of electron-deficient oxindolic olefins were generally realized with Michael-Initiated Ring-Closing sequence (MIRC) reaction.9 Moreover, olefin cyclopropanation with diazooxindoles via the carbene transfer could also give access to these targets.10 Based on our long-term endeavor in the development of chiral catalysts stemmed from metal/N,N′-dioxide complexes11 as well as our previous study of oxindole derivatives,12 we envision that a chiral Lewis acid catalyst of N,N′-dioxide could bind 3-alkenyl-oxindoles into a perfect chiral environment, benefiting the cyclopropanation of a free carbene generated from spontaneous decomposition of phenyliodonium ylide malonate (Scheme 1b). Herein, we reported a chiral N,N′-dioxide/Ni(OTf)2 complex catalyzed asymmetric cyclopropanation of 3-alkenyl-oxindoles with phenyliodonium ylide. Excellent diastereo and enantioselectivity were achieved for a variety of substituted spirocyclopropane-oxindoles under mild reaction conditions. Free carbene species formation was confirmed from EPR and HRMS analysis of the reaction system.
Results and discussion
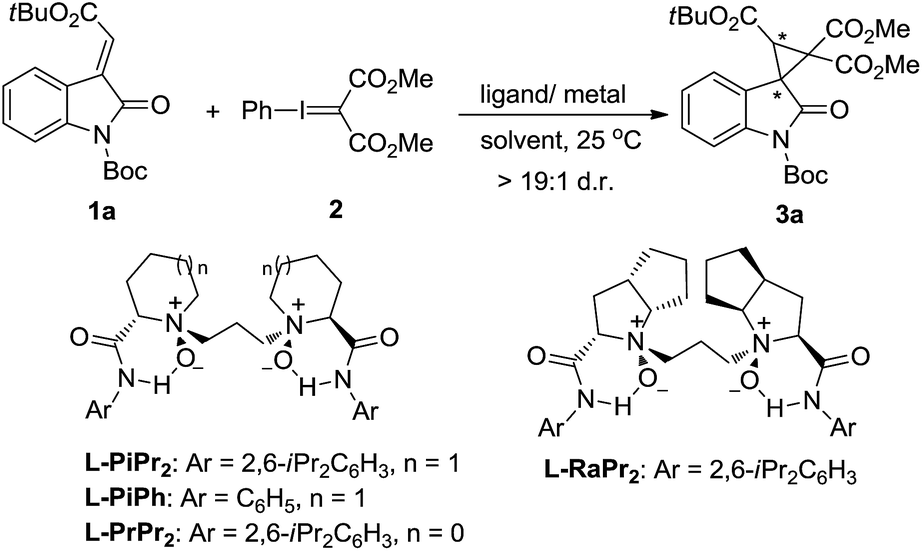
Our investigation commenced with the cyclopropanation of (E)-N-Boc-3-alkenyl-oxindole 1a as the model substrate with phenyliodonium ylide malonate 2 in CH2Cl2 at 25 °C. The known enantioselective activation of the substrate 1a by metal complexes of chiral N,N′-dioxide L-PiPr2 inspired us to examine it as the supporting ligand.11,12 CuBr only favored the carbene dimer of ethene-tetracarboxylate, and Cu(OTf)2 obtained a trace amount of the desired spirocyclopropane-oxindole 3a (Table 1, entries 1 and 2). This indicated that the prior formation of metal–carbene intermediate was detrimental in this system. However, chiral Lewis acid catalysts of both Ni(OTf)2 and Zn(OTf)2 enabled access to the product 3a in moderate yields and enantioselectivities yet high diastereoselectivities (>19:1 d.r.; entries 3 and 4). The results were appealingly consistent with our Lewis acid-activation pathway. The following survey of the ligands coordinated with Ni(OTf)2 showed that other N,N′-dioxides, including L-PiPh, L-PrPr2 and L-RaPr2, were less competent than L-PiPr2 in terms of the reactivities and enantioselectivities (entry 4 vs. entries 5–7). The reaction was further optimized through systematic study of several parameters. An improved enantioselectivity and moderate yield was observed in toluene (entry 8). An encouraging 85% yield with 98% ee of the product was given in Et2O (entries 9 and 10). It was suspected that Et2O may effectively prevent dimerization of the phenyliodonium ylide, but the poor solubility of the catalyst in Et2O led to the low performance of this catalytic system. Considering this question, a mixed solvent system was tested to improve the situation. A mixed solvent of CH2Cl2/Et2O (v/v = 1:4) was selected, and the asymmetric cyclopropanation gave a substantial improvement in catalytic yield with excellent diastereo and enantioselectivity (99% yield, >19:1 d.r., and 99% ee; entry 11).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Metal salt | Ligand | Solvent | Yieldb (%) | eec (%) |
|
a The reactions were carried out with 1a (0.1 mmol), metal/ligand (1:1, 5 mol%), and phenyliodonium ylide 2 (0.15 mmol) in a solvent (1.0 mL) at 25 °C for 24 h.
b Isolated yield.
c Determined by chiral HPLC analysis.
d Carbene dimer ethene-tetracarboxylate was the major product.
e The reverse of the enantioselectivity.
|
|||||
| 1 | CuBr | L-PiPr2 | CH2Cl2 | NDd | |
| 2 | Cu(OTf)2 | L-PiPr2 | CH2Cl2 | Trace | — |
| 3 | Ni(OTf)2 | L-PiPr2 | CH2Cl2 | 65 | 65 |
| 4 | Zn(OTf)2 | L-PiPr2 | CH2Cl2 | 33 | 77 |
| 5 | Ni(OTf)2 | L-PiPh | CH2Cl2 | 37 | 7e |
| 6 | Ni(OTf)2 | L-PrPr2 | CH2Cl2 | 39 | 22e |
| 7 | Ni(OTf)2 | L-RaPr2 | CH2Cl2 | 50 | 15 |
| 8 | Ni(OTf)2 | L-PiPr2 | Toluene | 58 | 97 |
| 9 | Ni(OTf)2 | L-PiPr2 | THF | 81 | 89 |
| 10 | Ni(OTf)2 | L-PiPr2 | Et2O | 85 | 98 |
| 11 | Ni(OTf)2 | L-PiPr2 | CH2Cl2/Et2O (v/v = 1/4) | 99 | 99 |
The generality of the catalytic cyclopropanation with a range of (E)-3-ester-substituted methyleneindolinone derivatives was next surveyed under the optimized reaction conditions (Scheme 2). The Me-, Et-, iPr-, tBu-, and Bn-substituted ester groups had no influence on the yields or the stereoselectivities (3a–e). The electron-donating substituents at the C5-position of the oxindole ring were well-tolerated, giving slightly higher enantioselectivities than the electron-withdrawing ones (3f, gvs.3h–k). A 6-bromo-substituent resulted in a good yield (90% yield, 94% ee; 3l). 3-Acyl substituted methyleneindolinone derivatives produced the corresponding spirocyclopropane products 3m–p in excellent yields and enantioselectivities at lower reaction temperature. The alkyl-substituted alkenes, such as propyl, cyclohexyl and cyano, could also undergo this reaction smoothly, affording the desired adducts in moderate yields and good enantioselectivities (4a, 4a′ and 4a′′′). However, the reaction between (E)-1-Boc-3-tert-butylideneindolinone and phenyliodonium ylide 2 remained challenging due to the steric hindrance (4a′′).
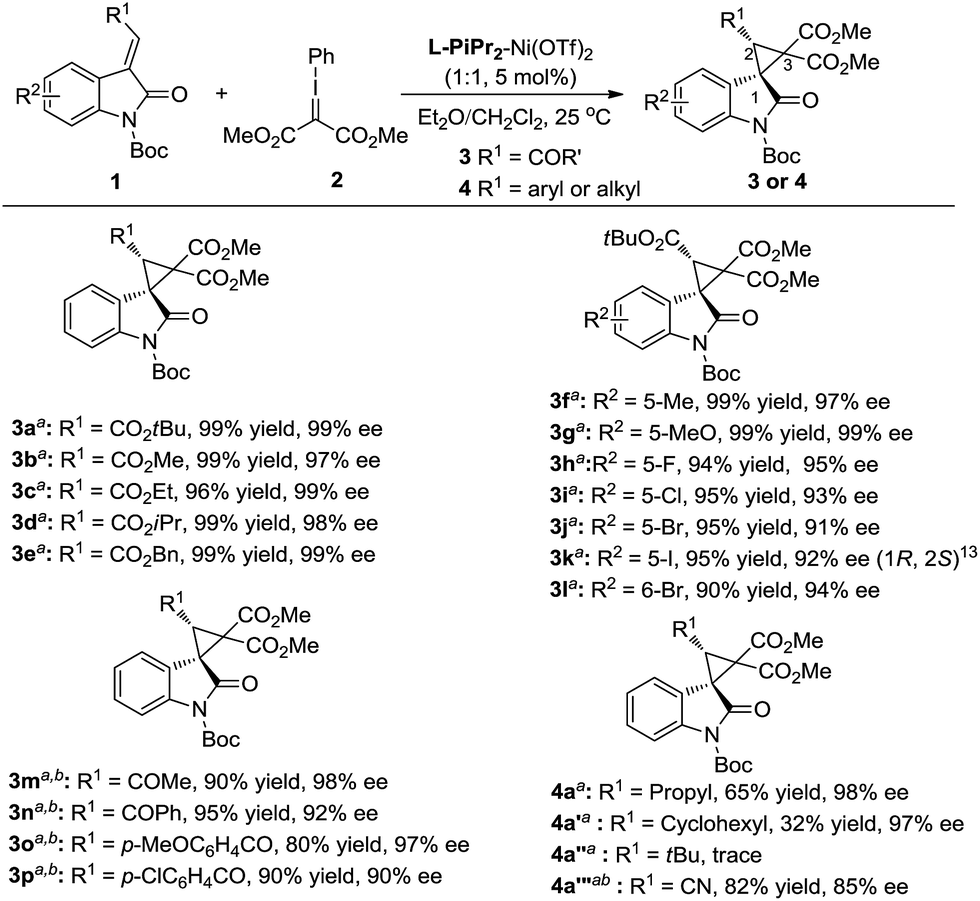 | ||
| Scheme 2 Substrate scope of the asymmetric cyclopropanation. a Reaction condition as in entry 11, Table 1.b Reaction performed at 0 °C. | ||
However, the deprotection of the Boc-group of the 3-aryl-substituted methyleneindolinone derivatives occurred, which prevented the cyclopropanation process. Changing the ratio of the substrates 1 and phenyliodonium ylide 2 from 1:1.5 to 1.5:1 made the reaction more applicable. Therefore, a wide range of aryl substituted 3-alkenyl-oxindoles 1 was investigated (Scheme 3). The reactions went well, obtaining the corresponding products 4 in 60–93% yields with 94–99% ee. Both the electronic nature and the position of the substituents on the 3-aryl group of 1 had obvious influence on the yield but not the enantioselectivity (4b–4n). It is noteworthy that no diastereomers were detected in most cases (>19:1 d.r.), except for benzo[d][1,3]dioxole substituted 4o and 2-naphthyl substituted 4p. The sense of diastereoselectivity in the latter two cases was appreciably decreased, and a trace amount of the diastereomer was confirmed by 1H NMR spectroscopy (19:1 d.r.), whereas the enantioselectivity was unaffected. As a representative substituted, 3-benzylidene-indolinone underwent efficient cyclopropanation, giving 4q in excellent yields. The absolute configuration of the product 3k and 4b was determined to be (1R, 2S) by X-ray analysis.13 When the catalytic system was applied to other non-oxindolic olefins, we found that coumarins were also able to provide the bridge ring derivatives with good yields and enantioselectivities (4r–4s). Benzofuran-2(3H)-one enabled access to the desired product with an excellent yield; however, the outcome of enantiocontrol was disappointing (4t). Compared with the N-Boc oxindoles, the loss of the necessary bidentate manner of two carbonyl groups might have led to poor chiral induction.
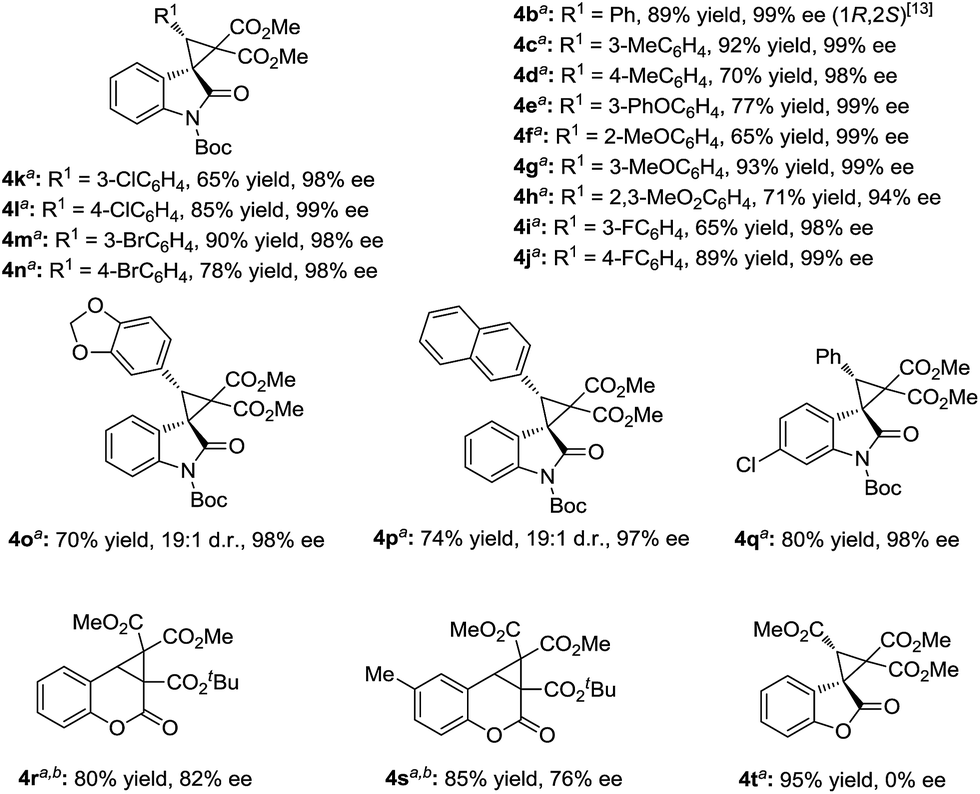 | ||
| Scheme 3 Substrate scope of aryl substituted olefins. a The reactions were carried out with 1a (0.15 mmol), metal/ligand (1:1, 5 mol%), and phenyliodonium ylide 2 (0.1 mmol) in solvent (1.0 mL) at 25 °C for 48 h. b The solvent was changed from CH2Cl2/Et2O (v/v = 1:4) to MTBE. | ||
Furthermore, the synthetic value of the reaction was investigated. The cyclopropanation of oxindole 1a with phenyliodonium ylide malonate 2 was carried out on a gram scale. The desired product 3a was generated in 99% yield, >19:1 d.r. and 99% ee (Scheme 4a). On the other hand, the product 3a could be easily transformed into the amino-functionalized acyclic β-amino acid derivative 5 (90% yield, 99% ee, and >19:1 d.r.) through a Lewis acid catalyzed nucleophilic ring-opening reaction using aniline as the nucleophile (Scheme 4b).
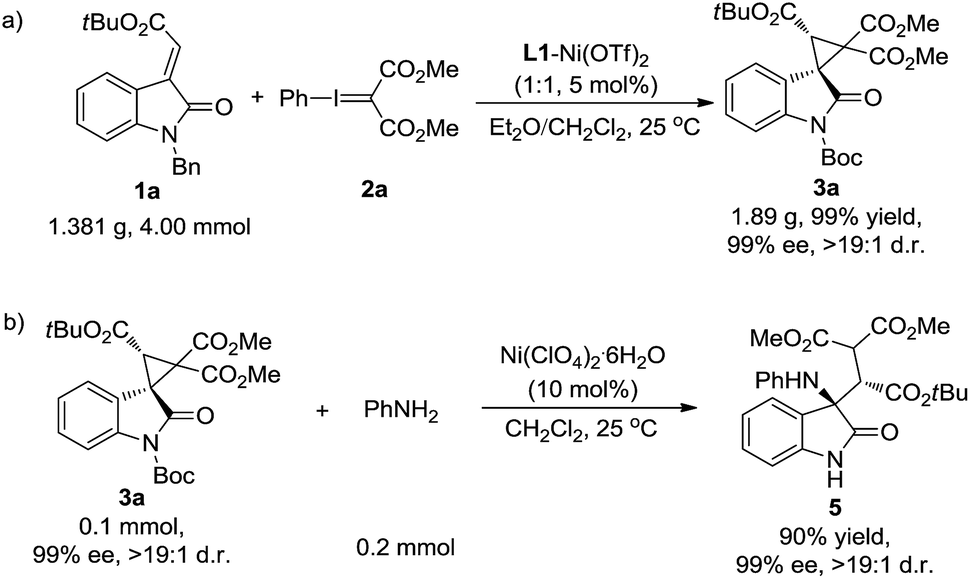 | ||
| Scheme 4 (a) Scaled-up version of the reaction and (b) the ring-open reaction of 3a with aniline. | ||
A series of experiments were conducted to probe into the reaction mechanism. The reaction between (E)-N-Boc-3-alkenyl-oxindole 1a and phenyliodonium ylide malonate 2 proceeded smoothly in the absence of the catalyst, giving the desired product in 40% yield; neither the N,N′-dioxide nor Ni(OTf)2 could substantially enhance the reaction. The outcomes of the reaction were unaffected when carried out in the dark. Clearly, the catalytic system of N,N′-dioxide–Ni(OTf)2 is a ligand-accelerated process in view of the excellent yields previously discussed. The Ni(II)-complex of L-PiPr2 has been confirmed by X-ray analysis in our early study.11a–c The bonding of oxindole substrate 1a or phenyl substituted 3-alkenyl-oxindole 1b′ to the metal cation of the chiral catalyst was detected from ESI-MS spectra. Peaks at m/z 1200.4629 and 1176.4937 were assigned to [Ni2+ + L-PiPr2 + 1a + TfO−]+ and [Ni2+ + L-PiPr2 + 1b′ + TfO−]+, respectively (see ESI† for details).
To determine the carbene intermediate, the reaction system was further characterized by EPR spectroscopy. The EPR X-band spectrum of N,N′-dioxide–Ni(OTf)2 showed no signals, indicating that there is no unpaired electron on the nickel(II) center due to the strong coordination of the supporting ligands. Interestingly, the EPR spectrum of the mixture of oxindole 1a and phenyliodonium ylide 2 with or without the catalyst exhibits a similar rhombic band and is centered around g = 2.003 (Fig. 1b–d). The intensity of the band is stronger when the chiral catalyst is added (Fig. 1dvs.1b). The time profile of the reaction showed that the EPR band disappeared gradually. These results suggest the presence of unpaired electrons on the carbene intermediate. The reaction proceeded more likely via a free carbene intermediate than a metallocarbene, as in the studies of the Müller's5 and the Tang's.6c The intermediacy of such a free carbene upon thermal decomposition of phenyliodonium ylide 2 may be a singlet one 1:C(CO2Me)2 initially. The cyclopropanation was slower than intersystem crossing to the more stable triplet carbene 3:C(CO2Me)2, which exhibits two unpaired electrons. In this circumstance, the cyclopropanation occurs through a stepwise mechanism involving an analogous biradical intermediate. The unresolved hyperfine structure implied the interaction of 3:C(CO2Me)2 with the substrate.
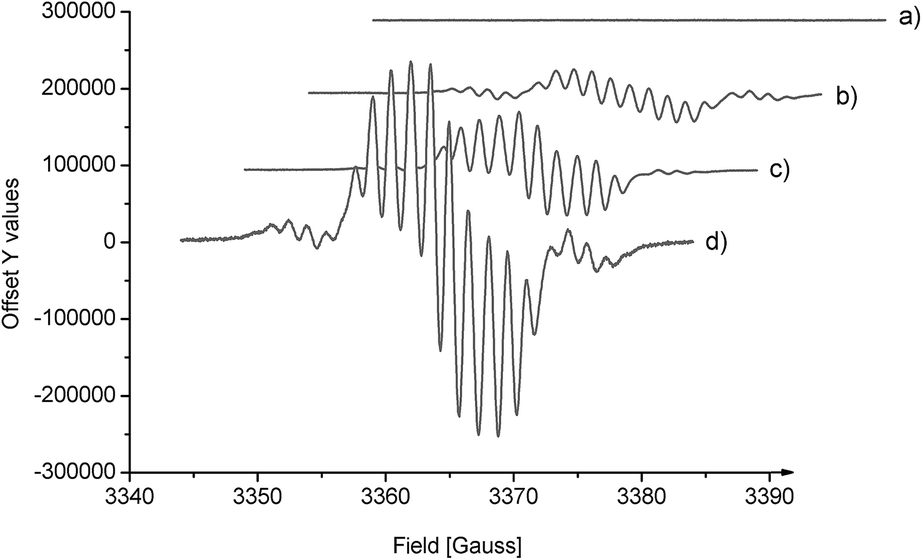 | ||
| Fig. 1 The electroparamagnetic resonance (EPR) spectra (X band, 9.43 GHz, RT, in Et2O/CH2Cl2 = 4/1). (a) 2; (b) 2 and 1a (1:1); (c) Ni(OTf)2 (20 mol%), 2 and 1a (1:1); (d) Ni(OTf)2/L-PiPr2 (20 mol%), 2 and 1a (1:1). | ||
Therefore, in view of the aforementioned consequences as well as the structures of the catalyst12 and the products, we proposed a chiral Lewis acid-promoted asymmetric cyclopropanation mechanism via a free carbene intermediate (Scheme 5). Substrate 1 coordinates to the chiral N,N′-dioxide–Ni(II) center in a bidentate manner with two carbonyl groups. The facial-control of the carbene addition was directed by the blocking of the amide unit underneath the ligand. Initially, the decomposition of the phenyliodonium ylide generated a more stable triplet carbene. It would prefer electronic addition to the outer CC bond because of the low steric hindrance and the stability of the triplet biradical intermediate. Due to the steric hindrance of the substituents on the biradical intermediates, the C–C bond rotation is slower than spin flip of the intermediate. Therefore, high diastereo and enantioselectivity of the products were given.
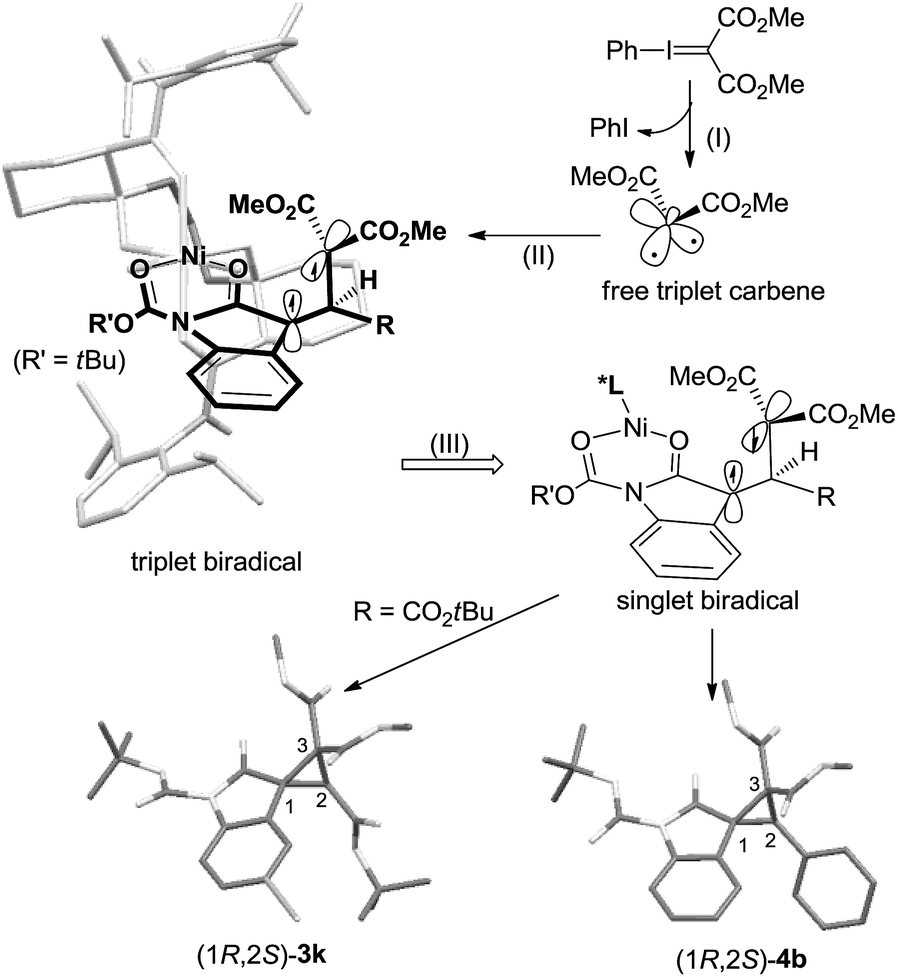 | ||
| Scheme 5 Proposed stereochemical model. | ||
Conclusions
In conclusion, we have developed a new asymmetric catalytic strategy for cyclopropanation of olefins. The chiral N,N′-dioxide/Ni(OTf)2 complex exhibited excellent performance in the reaction of 3-alkenyl-oxindoles with phenyliodonium ylide malonate under mild reaction conditions. The desired spirocyclopropane-oxindoles with contiguous tertiary and all carbon quaternary centers were attained in high yields and stereoselectivities (up to 99% yield, >19:1 d.r., and 99% ee). At the same time, when the catalytic system was applied to other non-oxindolic olefins, coumarins were also able to provide the bridge ring derivatives with good yields and enantioselectivities. A stepwise biradical process is suggested based on EPR spectroscopy. Further application of iodonium ylides and chiral N,N′-dioxide–metal complexes in asymmetric transformations are underway.
Acknowledgements
The study was funded by the National Natural Science Foundation of China (No. 21432006, 21290182, and 21321061).Notes and references
- For selected reviews, see
(a)
J. Salaun, in Cyclopropane derivatives and their diverse biological activities, ed. A. Meijere, vol. 207, Springer, Berlin, 2000 Search PubMed
; (b) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed
-
(a) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051 RSC
- For selected reviews and recent examples, see:
(a) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS PubMed
- For selected reviews on phenyliodonium ylides, see:
(a) R. M. Moriarty and R. K. Vaid, Synthesis, 1990, 431 CrossRef CAS
-
(a) P. Müller, Y. Allenbach and E. Robert, Tetrahedron: Asymmetry, 2003, 14, 779 CrossRef
-
(a) B. Moreau and A. B. Charette, J. Am. Chem. Soc., 2005, 127, 18014 CrossRef CAS PubMed
- M. B. Camacho, A. E. Clark, T. A. Liebrecht and J. P. DeLuca, J. Am. Chem. Soc., 2000, 122, 5210 CrossRef CAS
-
(a)
Y. He, T. Jiang, K. L. Kuhen, Y.-H. Ellis, B. Wu, T. Y.-H. Wu and B. Bursulaya, Oxindoles with Anti-HIV Activity, US Patent, WO 2004/037247 A1, IRM LLC, Scripps Research Institute, 2004
-
(a) A. Noole, N. S. Sucman, M. A. Kabeshov, T. Kanger, F. Z. Macaev and A. V. Malkov, Chem.–Eur. J., 2012, 18, 14929 CrossRef CAS PubMed
- Z.-Y. Cao, X. M. Wang, C. Tan, X.-L. Zhao, J. Zhou and K. L. Ding, J. Am. Chem. Soc., 2013, 135, 8197 CrossRef CAS PubMed
- For recent examples of N,N′-dioxide–metal complexes, see:
(a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS PubMed
-
(a) G. Wang, X. H. Liu, T. Y. Huang, Y. L. Kuang, L. L. Lin and X. M. Feng, Org. Lett., 2013, 15, 76 CrossRef CAS PubMed
- ESI†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1038443 and 1038445. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03658e |
| This journal is © The Royal Society of Chemistry 2016 |