
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCationic nickel porphyrinoids with unexpected reactivity†
Richard
Wicht
,
Stefanie
Bahnmüller
,
Kai
Brandhorst
,
Peter
Schweyen
and
Martin
Bröring
*
Institute for Inorganic and Analytical Chemistry, Technical University Braunschweig, Hagenring 30, 38106 Braunschweig, Germany. E-mail: m.broering@tu-bs.de
First published on 16th October 2015
Abstract
Cationic nickel(II) complexes of two ring-contracted porphyrinoid ligands distantly related to the corrins were prepared by metal templated macrocyclisation. The compounds show reversible electron transfer processes and were found to be the first porphyrinoid-based catalysts for C–C cross-coupling.
Introduction
The chemistry of ring-contracted porphyrinoids is currently one of the most intriguing topics in macrocycle research.1 Inspired by the cobalamins,2 an increasing number of novel porphyrinoid systems with small cavities and outstanding properties has been introduced to the field in recent years.3 Corroles have certainly taken the lead in this ongoing development.4 Artificial partially hydrogenated corroles, however, are hardly available by selective synthesis and have been reported on only in a very limited number.5 An inspiring idea for the synthesis of such a ligand comes from well-known porphyrin chemistry. As depicted in Fig. 1 the oxidative macrocyclisation of a methyl terminated linear tetrapyrrole 1 leads to metal chelates of the general form 2 if careful reaction conditions are ensured.6 Using 2,2′-bidipyrrins like 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 for an analogous transformation provides an option for the preparation of metal 9-methylisocorroles like 4. This ligand framework had been observed in serendipitously isolated rhodium and iridium complexes before,8 however, no logical synthesis nor any understanding of the formation of such chelates has so far been developed. A metal template assisted oxidative ring closure reaction is indeed successful with the precursors 5 and 6, and we report here about the synthesis and unexpected features of first cationic nickel(II) complexes 7 and 8 of such 9-methylisocorroles (Scheme 1).
7 for an analogous transformation provides an option for the preparation of metal 9-methylisocorroles like 4. This ligand framework had been observed in serendipitously isolated rhodium and iridium complexes before,8 however, no logical synthesis nor any understanding of the formation of such chelates has so far been developed. A metal template assisted oxidative ring closure reaction is indeed successful with the precursors 5 and 6, and we report here about the synthesis and unexpected features of first cationic nickel(II) complexes 7 and 8 of such 9-methylisocorroles (Scheme 1).
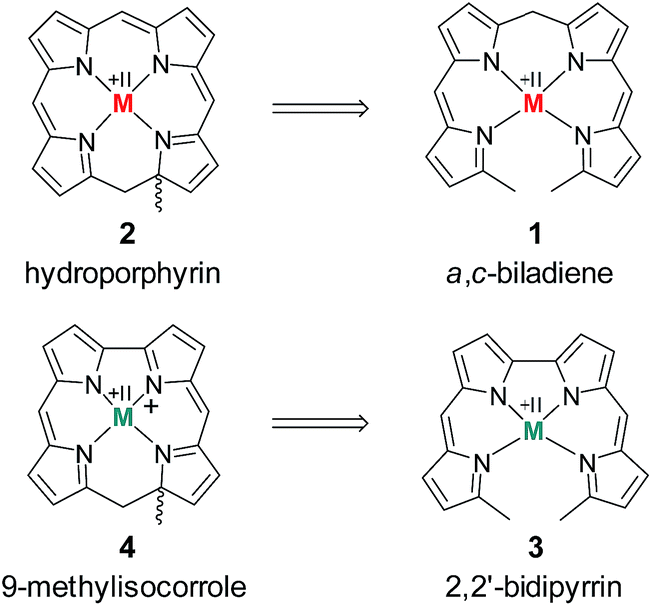 | ||
| Fig. 1 Strategies for hydroporphyrin and isocorrole syntheses. | ||
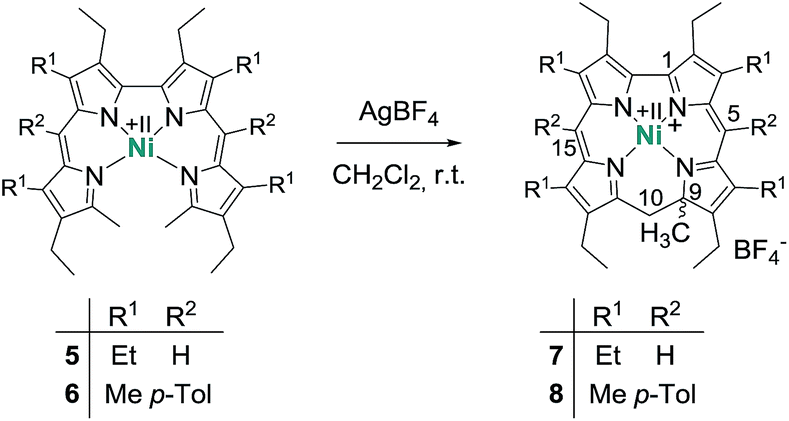 | ||
| Scheme 1 Preparation of nickel 9-methylisocorroles 7 and 8, with numbering scheme. | ||
Results and discussion
Treatment of nickel(II)-2,2′-bidipyrrin 57a or 6 with a slight excess of silver tetrafluoroborate in dichloromethane solution produces the desired complexes 7 and 8 in yields of 55% and 76%, respectively. This procedure contains several critical points which require a discussion. The choice of the anion of the silver salt is of uttermost importance for the successful synthesis. Weakly coordinating anions like BF4−, SbF6− or ClO4− lead to the desired complex cation, while more nucleophilic anions like NO3− or CH3CO2− result in the formation of complex mixtures of different unidentified and often oxygenated products.
Secondly, the removal of excess silver ions in an early work-up step by precipitation with ammonium bromide is essential for the isolation of a pure product. Other halide salts lead to diminished yields or even decomposition of the nickel 9-methylisocorrole. During this process, no binding of the halide ion to the metal center is observed. This is remarkable as in all related cases, e.g. with the monoanionic N-methylporphyrins and analogues, neutral high spin (S = 1) halogenido nickel(II) complexes form immediately under such conditions, and are isolated as robust compounds.9 We assume that the small cavity sizes of the 9-methylisocorrole ligands of 7 and 8 inhibit a spin change from a small low spin (S = 0) to a large high spin (S = 1) nickel(II) ion, thus enforcing a square planar coordination mode of the 3d8 ion. The attempt to purify crude 7 or 8 by chromatography was unsuccessful due to the sensitivity of the products. Once purified by recrystallisation, however, the complexes are sufficiently stable for investigation under typical laboratory conditions, and could be stored as solids under nitrogen.
The cationic nickel 9-methylisocorroles 7 and 8 were identified by ESI-MS (m/z for [M − X]+: 593.3, and 717.3, respectively) and by NMR spectroscopy. At first glance, the complete loss of molecular symmetry is apparent in the 1H and 13C NMR spectra through the increased number of resonance lines. Particularly striking are the AB systems of the protons of the bridging CH2 groups, which appear well resolved in the spectrum of 7 (Fig. 2). Resonances for these protons occur at 3.49 ppm and 1.74 ppm, and are as characteristic as the singlet for the protons of the C9-bound methyl group at 1.13 ppm for the tetrapyrrole framework. In addition, the unusual quaternary carbon atom C9 and the methylene bridge carbon atom C10 appear as prominent signals at 77.9 ppm and 36.6 ppm, respectively, in the 13C NMR spectrum of 7. Similar data was obtained for 8, and the constitution of both compounds is further supported by 2D NMR spectra (see ESI†).
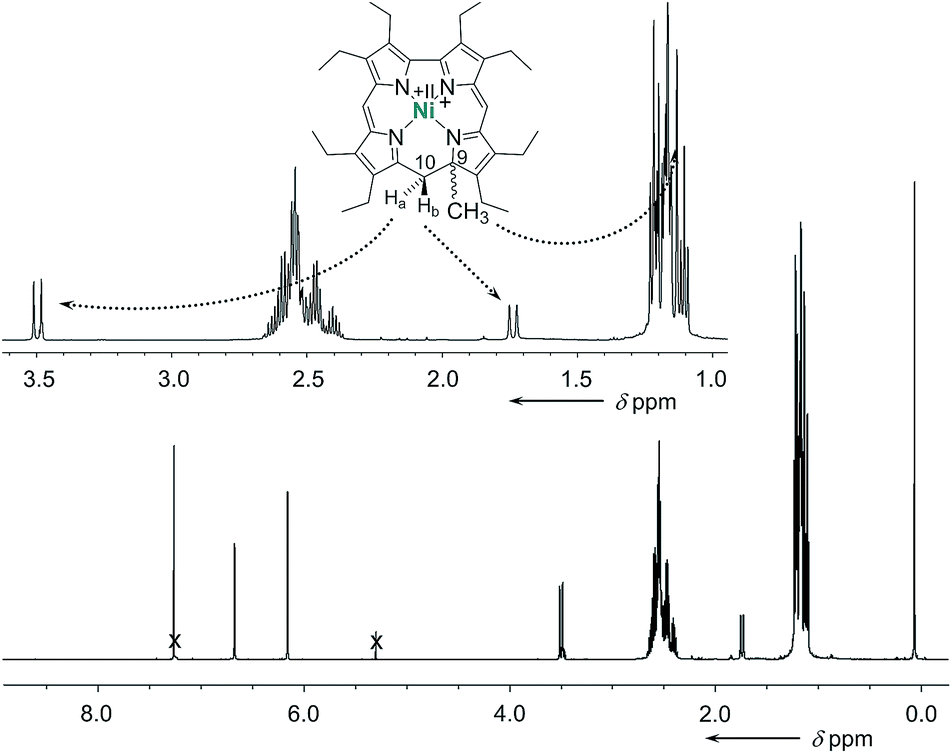 | ||
| Fig. 2 Details of the 1H NMR spectrum of the cationic nickel 9-methylisocorrole 7 (300 MHz, CDCl3). | ||
The redox behaviour of the new 9-methylisocorrole nickel complexes 7 and 8 was investigated in dichloromethane solution by cyclic voltammetry. Quasi-reversible reductions at E1/2 = −907/−920 mV and at −1515/−1548 mV, and quasi-reversible oxidations at E1/2 = +655/+603 mV vs. Fc/Fc+ are observed for 7 and 8, respectively.
Fig. 3 illustrates the results from cyclic voltammetry and square wave voltammetry measurements on 7. Comparing these data with the potentials found for the linear tetrapyrrole complexes 5 and 6 under the same conditions (E1/2 = −129/−161 mV; E1/2 = +475/+382 mV) reveals unusually large increases of both, oxidation and first reduction potentials, upon ring closure. With respect to other nickel porphyrinoids, hydroporphyrinoids, and corrinoids the potentials of the isocorrole complexes 7 and 8 are shifted by about 0.9 V for the first reduction step, and by about 0.4 V for the first oxidation.10 DFT calculations on the one-electron reduced 7′ suggest a ligand centered reduction process. The calculations are in agreement with the results from an EPR study (Fig. 4). Here, chemical reduction of 7 was performed by Na/Hg treatment in dry tetrahydrofurane, and the one-electron reduced product was shown to be a purely organic radical without apparent Ni(I) contributions.
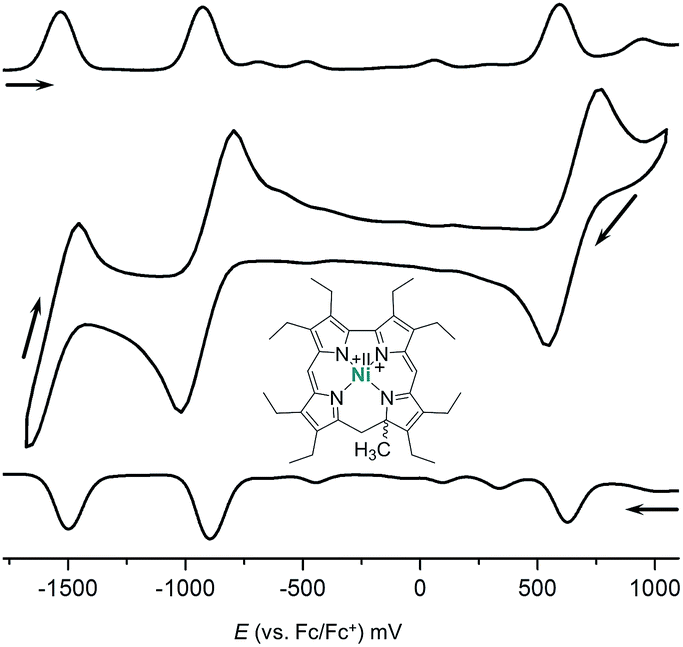 | ||
| Fig. 3 Cyclic voltammetry and square wave voltammetry results for nickel 9-methylisocorrole 7 (dichloromethane, 0.1 mol L−1nBu4NPF6; scan rate 200 mV s−1). | ||
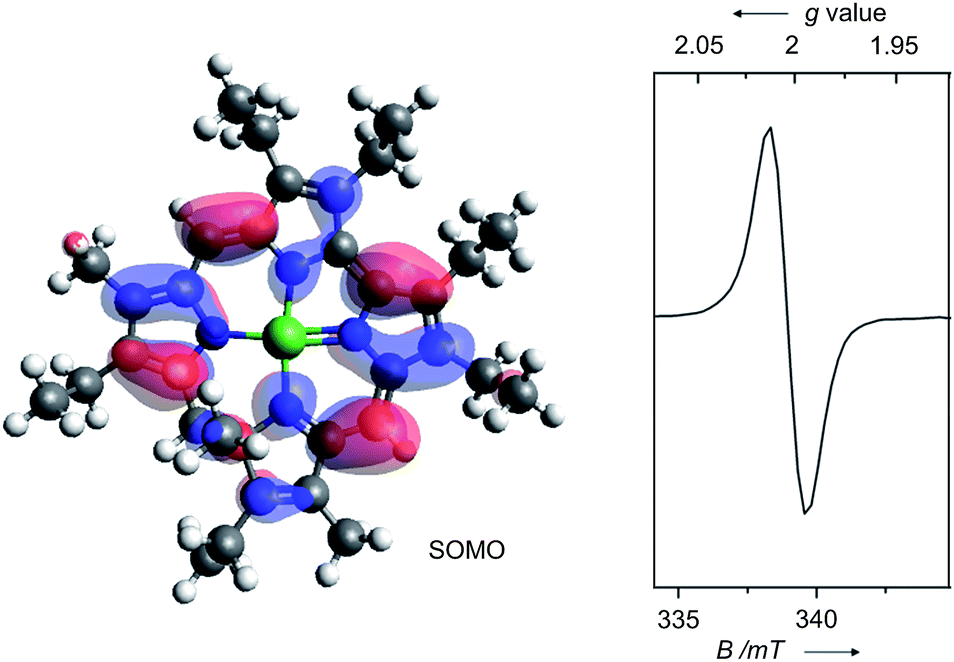 | ||
| Fig. 4 Graphical representation of the singly occupied molecular orbital (SOMO) from DFT calculations (B3LYP/TZVP, WTBS)11 and X-band EPR spectrum (tetrahydrofurane, 28 K) of the one-electron reduced nickel 9-methylisocorrole 7′. | ||
Other than the reduction, chemical oxidation of 7 by dioxygen from air occurs over a day in solution, and results in the formation of a mixture of unassigned species. In one case, a single crystal grew from this complex mixture and allowed the crystallographic identification of an oxidation product. Quite surprisingly, this analysis showed a species very similar to 7 but dimerised through the bridging methylene unit C10.3b,12Fig. 5 shows the molecular structure and provides details of the coordination environment of the nickel ion as well as of the conformational state of the macrocycle.
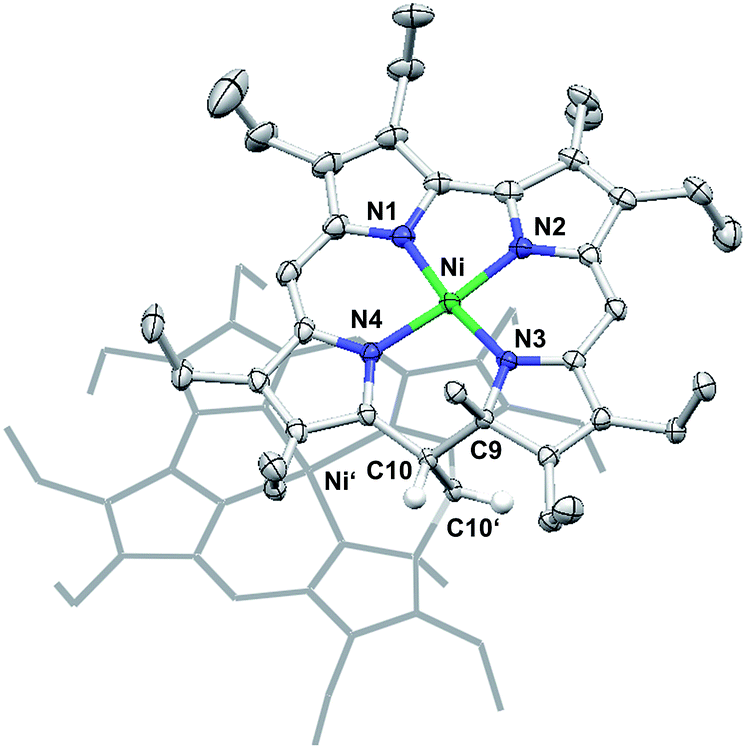 | ||
| Fig. 5 Molecular structure of the dicationic dimer of 7. Selected bond lengths [Å]: Ni–N1 1.846(3), Ni–N2 1.832(3), Ni–N3 1.868(3), Ni–N4 1.873(3), C9–C10 1.565(4), C10–C10′ 1.586(4). Thermal ellipsoids are set at 50% probability. The second subunit is shown as stick representation only, and most hydrogen atoms are removed for clarity. | ||
The dimer crystallises as a chloroform solvate in the space group P21/c. Two nickel atoms in distorted square-planar coordination geometry and with Ni–N distances ranging from 1.832(3) Å to 1.873(3) Å are found in the central N4 cavities of the macrocyclic subunits and support the expected low spin-d8 configuration of the metal ions. As a single positive charge is calculated per subunit, two BF4− anions are found for each dimer. These anions are isolated from the dication and do not bind to the nickel(II) atoms. The conformation of the macrocyclic 9-methylisocorrole is mainly governed by the steric requirements of the C9 bound methyl group and the methylene bridge C10 involved in the dimer formation. At this position of the macrocycle an offset is observed, resulting in a weakly helical conformation. Additional characteristic non-planar distortions, which may be expected from the peripheral substitution, are not detected.13
The persistent low spin configuration of the cationic complexes 7 and 8, and the unusual potential of the first reduction step tempted us to investigate whether organometallic reactions could be conducted with this compound. Indeed, catalytic C–C cross-coupling reactions were observed upon treatment of the nickel 9-methylisocorroles 7 and 8 with Grignard reagents in the presence of arylhalides (Scheme 2). Best results are obtained if the Grignard reagent is added in small aliquots over the course of 2 h to a solution of the nickel complex 7 or 8 (5 mol%) and the arylhalide in dry THF at ambient temperature, with the two complexes behaving basically identical. These conditions generally give high to complete conversion of the halide component, with high selectivity for the cross-coupling product. The reaction can be run in dichloromethane or toluene solution, too, but with reduced yield of the desired heterodiaryl. A reduced catalyst loading elongates the reaction time as expected. However, excess Grignard reagent (2.5–4 equiv.) is required in all cases, and significant amounts of the homocoupling product from the organometallic component are present throughout. As judged by the stoichiometry, another and as yet unassigned by-reaction of the Grignard reagent must occur, presumably by solvolysis. These by-products form preferentially upon long reaction times, e.g. upon attempts to reduce the catalyst loading. In the absence of a nickel 9-methylisocorrole 7 or 8, or in the presence of nickel tetraphenylporphyrin, no cross-coupling reaction and no homocoupling of the Grignard component occurs, and upon using simple nickel(II) acetate as catalyst only the unwanted symmetric biaryl and the very slow formation of the cross-coupling product could be detected (<9% after 300 h).
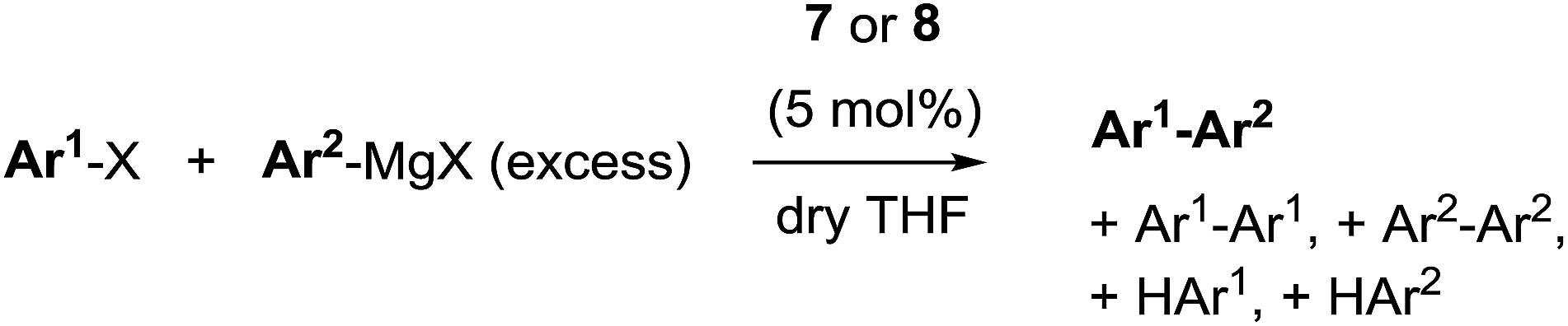 | ||
| Scheme 2 Catalytic Caryl–Caryl cross-coupling with 7 and 8 (X = Cl, Br, I). | ||
Table 1 summarises the results using different catalysts, reaction conditions, and starting materials. Both components, the aryl halide as well as the Grignard reagent, are variable within successful protocols. Beside the solvent, the influence of the size of the arylhalide is remarkable as the larger anthracenyl bromide shows the fastest reaction, followed by the naphthyl and phenyl systems. The selectivity of the formation of the cross-coupling product is higher for electron poor substrates, accompanied with a significant decrease of the reaction rate. The expected influence was observed for different halides at the aryl halide component where the reaction rate increases while the selectivity decreases in the order chlorine, bromine, iodine. Sterically hindered mesityl derivatives could not be coupled, and the reaction stops at −78 °C. Beside the homocoupling product from the Grignard component, which seems to be formed during the activation of the nickel 9-methylisocorrole cations 7 and 8, the major by-products stem from dehalogenation and from homocoupling of the aryl halide. These by-products point to a radical mechanism of the catalytic reaction. Addition of TEMPO indeed quenches the cross-coupling reaction. The reaction is initiated upon the addition of the Grignard component to 7 and 8, and UV/Vis and EPR spectroscopy indicate the formation of the one-electron reduced neutral radicals 7′ and 8′, respectively, upon this treatment (see ESI† for reference). All further steps are speculative at this point. However, as the nickel 9-methylisocorrole scaffold most probably does not allow the binding of two organic residues to the nickel at the same side of the macrocycle, a step-wise mechanism as depicted in Scheme 3 is proposed. As the rate determining step the reduced catalyst 7′ or 8′ activates the aryl halide by one-electron transfer to the radical [Ar1X]˙− and is itself reactivated by the rapid reaction with the Grignard component, producing the radical [Ar2]˙ to close the catalytic cycle. Product and by-product formation may be rationalised as the possible radical combination processes. Other than for the known nickel catalysed C–C coupling reactions like the Kumada coupling14 or the example recently described by Tilley et al.,15 this new process takes place without any redox change at the metal atom and can be related to the non-innocent behaviour of the 9-methylisocorrole ligand. The selectivity for the cross-coupling may be understood kinetically if the activation of the nickel complex by the Grignard reagent is faster than the diffusion of the aryl halide radical. However, the origin of the H atom necessary for the dehalogenation products remains unclear, and further studies will be necessary to provide solid grounds for the proposed mechanism.
| Cat. (mol%) | Ar1X | Ar2MgY (equiv.) | Time (h) | Conv.a (%) | Yielda,b (%) | |||
|---|---|---|---|---|---|---|---|---|
| a Determined by GC, rel. to the arylhalide component Ar1X. b Ar1Ar2 Ar1Ar1 HAr1 Ar2Ar2. Anth = anthracene; ani = anisole; PhF = 4-CF3C6H4; PhF2 = 3,5-(CF3)2C6H3. | ||||||||
| 7 (5) | 1-BrNaph | PhMgCl (1.5) | 0.5 | 46 | 41 | 3 | 2 | 20 |
| 7 (5) | 1-BrNaph | PhMgCl (5) | 0.5 | 100 | 71 | 8 | 20 | 70 |
| 7 (5) | 1-BrNaph | PhMgCl (6 × 0.5) | 1.25 | 100 | 86 | 8 | 5 | 43 |
| 7 (5) | 1-BrNaph | PhMgBr (6 × 0.5) | 1.25 | 100 | 88 | 9 | 2 | 35 |
| 7 (1) | 1-BrNaph | PhMgCl (5 × 1) | 23.33 | 93 | 77 | 9 | 7 | 182 |
| 7 (5) | 1-ClNaph | PhMgCl (1.5) | 1.33 | 44 | 42 | 2 | 0 | 21 |
| 7 (5) | 1-ClNaph | PhMgCl (6 × 0.5) | 23.33 | 81 | 76 | 5 | 0 | 52 |
| 7 (5) | 1-INaph | PhMgCl (1.5) | 0.33 | 45 | 34 | 6 | 5 | 19 |
| 8 (5) | PhBr | AniMgBr (3 × 2) | 40 | 100 | 95 | 5 | 0 | 163 |
| 8 (5) | 4-BrAni | PhMgBr (4 × 1) | 40 | 38 | 15 | 4 | 19 | 111 |
| 8 (5) | 1-BrPhF | AniMgBr (4 × 1) | 23.33 | 70 | 61 | 9 | 0 | 108 |
| 8 (5) | 1-BrNaph | PhMgBr (1.5) | 0.5 | 51 | 44 | 6 | 1 | 11 |
| 8 (1) | 1-BrNaph | PhMgCl(1.5) | 0.5 | 10 | 9 | <1 | <1 | 4 |
| 8 (5) | 1-BrNaph | PhMgBr (15) | 0.5 | 100 | 81 | 12 | 7 | 35 |
| 8 (5) | 1-BrNaph | AniMgBr (4 × 1) | 1 | 91 | 64 | 7 | 20 | 67 |
| 8 (5) | 1-BrNaph | PhF2MgBr (4 × 1) | 23.33 | 69 | 69 | 0 | 0 | 107 |
| 8 (5) | 5-BrAnth | AniMgBr (4 × 1) | 1 | 100 | 100 | 0 | 0 | 27 |
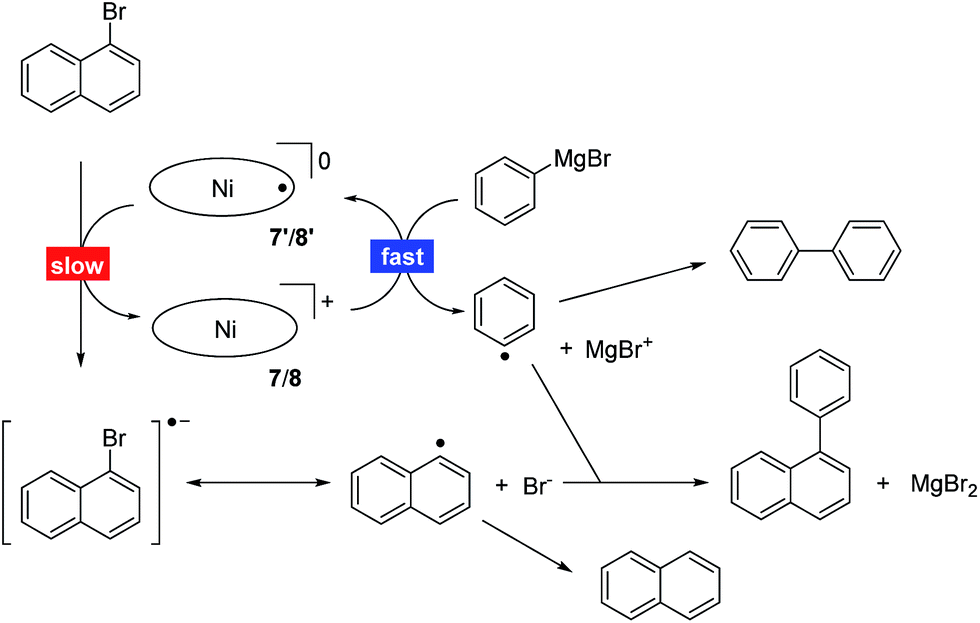 | ||
| Scheme 3 Proposed radical mechanism for the nickel 9-methylisocorrole catalysed Caryl–Caryl cross-coupling of PhMgBr with 1-bromonaphthalene. | ||
Conclusions
The successful synthesis of cationic nickel(II) 9-methylisocorroles from 2,2′-bidipyrrin-type tetrapyrroles by a templated oxidative macrocyclisation reaction using silver salts of weakly coordinating anions as oxidants paves the road to the investigation of a group of new contracted metal porphyrinoids. Our first investigations have revealed interesting redox and ligand exchange behaviour for the nickel complexes which differ largely from what is known for nickel porphyrins. Indeed, no change in the oxidation state of the nickel atom is observed upon oxidation or reduction, and the low spin character of the Ni(II) ion in the 9-methylisocorrole complexes remains persistent even in the presence of additional donors like halogenido or phenyl ligands. These features, which are unprecedented in the chemistry of nickel porphyrins, apparently lead to catalytic activity in C–C cross-coupling reactions. Such organometallic reactivity is typically unexpected for metal porphyrins due to the lack of two adjacent coordination sites at the metal atom.16 In this case, however, the catalytic transformation seems to proceed via kinetically advantageous multi-step radical reactions in very good selectivity, with the non-innocent 9-methylisocorrole ligand playing the role of an electron relay. This example shows that nickel porphyrinoids can be very attractive compounds and may well become useful in fields like catalysis and sensors,17 and that nickel can do more than stabilising macrocycles in porphyrin research.Notes and references
- Expanded, Contracted and Isomeric Porphyrins, ed. J. L. Sessler and S. J. Weghorn, Elsevier, Oxford, 1997 Search PubMed.
- K. Gruber, B. Puffer and B. Kräutler, Chem. Soc. Rev., 2011, 40, 4346–4363 RSC.
- Norcorroles: (a) A. Ghosh, I. H. Wasbotten, W. Davis and J. C. Swarts, Eur. J. Inorg. Chem., 2005, 4479–4485 CrossRef CAS; (b) M. Bröring, S. Köhler and C. Kleeberg, Angew. Chem., Int. Ed., 2008, 47, 5658–5660 CrossRef PubMed; (c) T. Ito, Y. Hayashi, S. Shimizu, J.-Y. Shin, N. Kobayashi and H. Shinokubo, Angew. Chem., Int. Ed., 2012, 51, 8542–8545 CrossRef CAS PubMed; Isocorroles: (d) E. Vogel, B. Binsack, Y. Hellwig, C. Erben, A. Heger, J. Lex and Y.-D. Wu, Angew. Chem., Int. Ed. Engl., 1997, 36, 2612–2615 CrossRef CAS; (e) H.-J. Kim and J. S. Lindsey, J. Org. Chem., 2005, 70, 5475–5486 CrossRef CAS PubMed; (f) J.-I. Setsune, A. Tsukajima and J. Watanabe, Tetrahedron Lett., 2006, 47, 1817–1820 CrossRef CAS; Heterocorroles: (g) A. W. Johnson, I. T. Kay and R. Rodrigo, J. Chem. Soc., 1963, 2336–2342 RSC; (h) M. J. Broadhurst, R. Grigg and A. W. Johnson, J. Chem. Soc., Perkin Trans. 1, 1972, 1124–1135 RSC; (i) A. Ghosh, T. Chatterjee, W.-Z. Lee and M. Ravikanth, Org. Lett., 2013, 15, 1040–1043 CrossRef CAS PubMed; (j) M. Bröring, F. Brégier, E. Cónsul Tejero, C. Hell and M. C. Holthausen, Angew. Chem., Int. Ed., 2007, 46, 445–448 CrossRef PubMed; (k) M. Horie, Y. Hayashi, S. Yamaguchi and H. Shinokubo, Chem.–Eur. J., 2012, 18, 5919–5923 CrossRef CAS PubMed; (l) H. Kamiya, T. Kondo, T. Sakida, S. Yamaguchi and H. Shinokubo, Chem.–Eur. J., 2012, 18, 16129–16135 CrossRef CAS PubMed; (m) D. Sakow, B. Böker, K. Brandhorst, O. Burghaus and M. Bröring, Angew. Chem., Int. Ed., 2013, 52, 4912–4915 CrossRef CAS PubMed; (n) D. Sakow, D. Baabe, B. Böker, O. Burghaus, M. Funk, C. Kleeberg, D. Menzel, C. Pietzonka and M. Bröring, Chem.–Eur. J., 2014, 20, 2913–2924 CrossRef CAS PubMed; Corrolazine: (o) B. Ramdhanie, C. L. Stern and D. P. Goldberg, J. Am. Chem. Soc., 2001, 123, 9447–9448 CrossRef CAS PubMed; N-confused Corroles: (p) K. Fujino, Y. Hirata, Y. Kawabe, T. Morimoto, A. Srinivasan, M. Toganoh, Y. Miseki, A. Kudo and H. Furuta, Angew. Chem., Int. Ed., 2011, 50, 6855–6859 CrossRef CAS PubMed; (q) H. Furuta, H. Maeda and A. Osuka, J. Am. Chem. Soc., 2001, 123, 6435–6436 CrossRef CAS PubMed; Subporphyrins and Subphthalocyanines: (r) T. Torres, Angew. Chem., Int. Ed., 2006, 45, 2834–2837 CrossRef CAS PubMed; (s) Y. Inokuma, Z. S. Yoon, D. Kim and A. Osuka, J. Am. Chem. Soc., 2007, 129, 4747–4761 CrossRef CAS PubMed; (t) Y. Takeuchi, A. Matsuda and N. Kobayashi, J. Am. Chem. Soc., 2007, 129, 8271–8281 CrossRef CAS PubMed.
- Overviews are provided by: (a) D. T. Gryko, Eur. J. Org. Chem., 2002, 1735–1742 CrossRef CAS; (b) S. Nardis, D. Monti and R. Paolesse, Mini-Rev. Org. Chem., 2005, 2, 355–372 CrossRef CAS; (c) I. Aviv and Z. Gross, Chem. Commun., 2007, 1987–1999 RSC; (d) K. E. Thomas, A. B. Alemayehu, J. Conradie, C. M. Beavers and A. Ghosh, Acc. Chem. Res., 2012, 45, 1203–1214 CrossRef CAS PubMed.
- (a) T. Hayashi, Y. Morita, E. Mizuhata, K. Oohora, J. Ohbayashi, T. Inoue and Y. Hisaeda, Chem. Commun., 2014, 50, 12560–12563 RSC; (b) K. Aravindu, M. Krayer, H.-J. Kim and J. S. Lindsey, New J. Chem., 2011, 35, 1376–1384 RSC; (c) H.-J. Kim and J. S. Lindsey, J. Org. Chem., 2005, 70, 5475–5486 CrossRef CAS PubMed; (d) C.-J. Liu, A. Thompson and D. Dolphin, J. Inorg. Biochem., 2001, 83, 133–138 CrossRef CAS PubMed; (e) F.-P. Montforts, Angew. Chem., Int. Ed. Engl., 1982, 21, 214–215 CrossRef; (f) C. Angst, C. Kratky and A. Eschenmoser, Angew. Chem., Int. Ed. Engl., 1981, 20, 263–265 CrossRef; (g) V. Rasetti, B. Kräutler, A. Pfaltz and A. Eschenmoser, Angew. Chem., Int. Ed. Engl., 1977, 16, 459–461 CrossRef; (h) D. Dolphin, R. L. N. Harris, J. L. Huppatz, A. W. Johnson and I. T. Kay, J. Chem. Soc. C, 1966, 30–40 RSC.
- K. M. Smith, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic, San Diego, CA, 2000, vol. 1, pp. 119–148 Search PubMed.
- (a) M. Bröring, C. D. Brandt, J. Lex, H.-U. Humpf, J. Bley-Escrich and J.-P. Gisselbrecht, Eur. J. Inorg. Chem., 2001, 2549–2556 CrossRef; (b) M. Bröring, D. Griebel, C. Hell and A. Pfister, J. Porphyrins Phthalocyanines, 2001, 5, 708–714 CrossRef; (c) M. Bröring, Synthesis, 2000, 1291–1294 CrossRef.
- (a) M. Bröring, E. Cónsul Tejero, A. Pfister, C. D. Brandt and J. J. Pérez Torrente, Chem. Commun., 2002, 3058–3059 RSC; (b) M. Bröring, C. D. Brandt and E. Cónsul-Tejero, Z. Anorg. Allg. Chem., 2005, 631, 1793–1798 CrossRef; (c) M. Bröring and E. Cónsul-Tejero, J. Organomet. Chem., 2005, 690, 5290–5299 CrossRef.
- (a) P. J. Chmielewski, L. Latos-Grażyński, M. M. Olmstead and A. L. Balch, Chem.–Eur. J., 1997, 3, 268–278 CrossRef CAS PubMed; (b) P. J. Chmielewski and L. Latos-Grażyński, Inorg. Chem., 1992, 31, 5231–5235 CrossRef CAS; (c) J. Lisowski, L. Latos-Grażyński and L. Szterenberg, Inorg. Chem., 1992, 31, 1933–1940 CrossRef CAS; (d) L. Latos-Grażyński, M. M. Olmstead and A. L. Balch, Inorg. Chem., 1989, 28, 4066–4068 CrossRef; (e) L. Latos-Grażyński, J. Lisowski, M. M. Olmstead and A. L. Balch, Inorg. Chem., 1989, 28, 1183–1188 CrossRef; (f) D. K. Lavallee, Inorg. Chem., 1977, 16, 955–958 CrossRef CAS.
- (a) G. Pomarico, X. Xiao, S. Nardis, R. Paolesse, F. R. Fronczek, K. M. Smith, Y. Fang, Z. Ou and K. M. Kadish, Inorg. Chem., 2010, 49, 5766–5774 CrossRef CAS PubMed; (b) A. M. Stolzenberg and M. T. Stershic, Inorg. Chem., 1988, 27, 1614–1620 CrossRef CAS; (c) A. M. Stolzenberg and M. T. Stershic, J. Am. Chem. Soc., 1988, 110, 6391–6402 CrossRef CAS; (d) D. Chang, T. Malinski, A. Ulman and K. M. Kadish, Inorg. Chem., 1984, 23, 817–824 CrossRef CAS; (e) C. Kratky, A. Fässler, A. Pfaltz, B. Kräutler, B. Jaun and A. Eschenmoser, J. Chem. Soc., Chem. Commun., 1984, 1368–1371 RSC.
- (a) A. Schaefer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef CAS; (b) S. Huzinaga and B. Miguel, Chem. Phys. Lett., 1990, 175, 289–291 CrossRef CAS; (c) S. Huzinaga and M. Klobukowski, Chem. Phys. Lett., 1993, 212, 260–264 CrossRef CAS.
- A similar dimerization has occasionally been observed for other tetrapyrroles: (a) H. Scheer and C. Krauss, Photochem. Photobiol., 1977, 25, 311–314 CrossRef CAS PubMed; (b) E. Vogel, M. Bröring, S. J. Weghorn, P. Scholz, R. Deponte, J. Lex, H. Schmickler, K. Schaffner, S. E. Braslavsky, M. Müller, S. Pörting, C. J. Fowler and J. L. Sessler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1651–1654 CrossRef CAS.
- (a) M. O. Senge, Chem. Commun., 2006, 243–256 RSC; (b) M. Nakamura, Coord. Chem. Rev., 2006, 250, 2271–2294 CrossRef CAS; (c) A. Ikezaki, M. Takahashi and M. Nakamura, Angew. Chem., Int. Ed., 2009, 48, 6300–6303 CrossRef CAS PubMed.
- Selected reviews: (a) K. Tamao, J. Organomet. Chem., 2002, 653, 23–26 CrossRef CAS; (b) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed; (c) P. Kumar and J. Louie, Angew. Chem., Int. Ed., 2011, 50, 10768–10769 CrossRef CAS PubMed.
- M. I. Lipschutz and T. D. Tilley, Angew. Chem., Int. Ed., 2014, 53, 7290–7294 CrossRef CAS PubMed.
- (a) J. V. Ruppel, K. B. Fields, N. L. Snyder and X. P. Zhang, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2010, vol. 10, pp. 1–84 Search PubMed; (b) Y. Hisaeda and H. Shimakoshi, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2010, vol. 10, pp. 313–370 Search PubMed.
- (a) S. Thies, C. Bornholdt, F. Köhler, F. D. Sönnichsen, C. Näther, F. Tuczek and R. Herges, Chem.–Eur. J., 2010, 16, 10074–10083 CrossRef CAS PubMed; (b) M. Dommaschk, M. Peters, F. Gutzeit, C. Schütt, C. Näther, F. D. Sännichsen, S. Tiwari, C. Riedel, S. Boretius and R. Herges, J. Am. Chem. Soc., 2015, 137, 7552–7555 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation and analytical data of 7 and 8; details of DFT calculations and catalytic investigations. CCDC 1057061. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03663a |
| This journal is © The Royal Society of Chemistry 2016 |