
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The design of a readily attachable and cleavable molecular scaffold for ortho-selective C–H alkenylation of arene alcohols†
Brian J.
Knight
,
Jacob O.
Rothbaum
and
Eric M.
Ferreira
*
Department of Chemistry, University of Georgia, Athens, GA 30602, USA. E-mail: emferr@uga.edu
First published on 14th December 2015
Abstract
We describe herein the design of a novel molecular scaffold that can induce facile oxidative olefinations when attached to alcohols. Benzylic, homo-, and bishomobenzylic alcohols are utilized. The scaffold can act as a protecting group for the alcohol in other transformations, and it is recoverable in excellent yield. The overall sequence can also be telescoped without purifications of intermediates, representing a net alcohol-based directed ortho-alkenylation.
Introduction
The ability to forge an array of bond types via metal-catalyzed selective C–H functionalization is well appreciated.1 A substantial percentage of these transformations rely on the directing capability of the substrate. That is, the appropriate positioning of a Lewis basic functional group will present a metal catalyst center to a specific site, enabling the precise activation of the commonly unreactive C–H bond.2 Several commonly encountered functional groups can impart this directing capability. Alcohols can conceivably be one such group, but there are noted restrictions.3 Almost all cases of directed catalytic C–H functionalization employ tertiary alcohols;4 there are, to our knowledge, very few outliers using primary and secondary alcohols, with functionalizations in comparatively diminished yields.4a–c Implicit hypotheses suggest that this general substrate class is prone to oxidation, fragmentation, and other decomposition pathways.An alternative strategy is the utilization of alcohol surrogates, species that can be directly attached to and later removed from the alcohol functional group, which will permit metal-catalyzed functionalizations when connected.5 In addition to a few cases where this approach has been employed in sp3 C–H functionalization,6 this strategy has been applied toward arene sp2 C–H reactions. Interestingly, the large majority of examples have been based on phenolic precursors (Fig. 1a), including both Pd-catalyzed7–11 and non-Pd-catalyzed examples.12,13 In contrast, few catalytic sp2 C–H functionalizations have been based on aliphatic alcohol surrogates. Hartwig's iridium-catalyzed dehydrogenative functionalization with a silane is a landmark example (Fig. 1b).14 Tan15 and Yu16 have both developed attachable and cleavable groups for alcohols that induce meta-selective alkenylations. Recently, oximes have been demonstrated by both Zhao17 and Dong18 to be competent functional groups for Pd-catalyzed ortho-selective functionalizations of benzylic and homobenzylic alcohol precursors (Fig. 1c). In these latter oxime cases, they exploit the “exo-directing mode” of the functional group for cyclometalation to afford alkenylated, arylated, and acetoxylated products. Although the oxime has proven viable in this capacity, its synthetic manipulation does have its requisite demands (e.g., generally 3 steps for the transformation of alcohol → oxime,19 lack of recoverability, reductive oxime removal), and thus surrogates of different design may offer unique advantages. Herein, we disclose the development of a novel molecular scaffold based on a heterocycle-incorporated acetal, delineating the structural features required to impart this desired type of reactivity. Additionally, we illustrate how the scaffold is readily attached and cleaved, highlighting the lability of this specific system and its translation to an overall streamlined process.
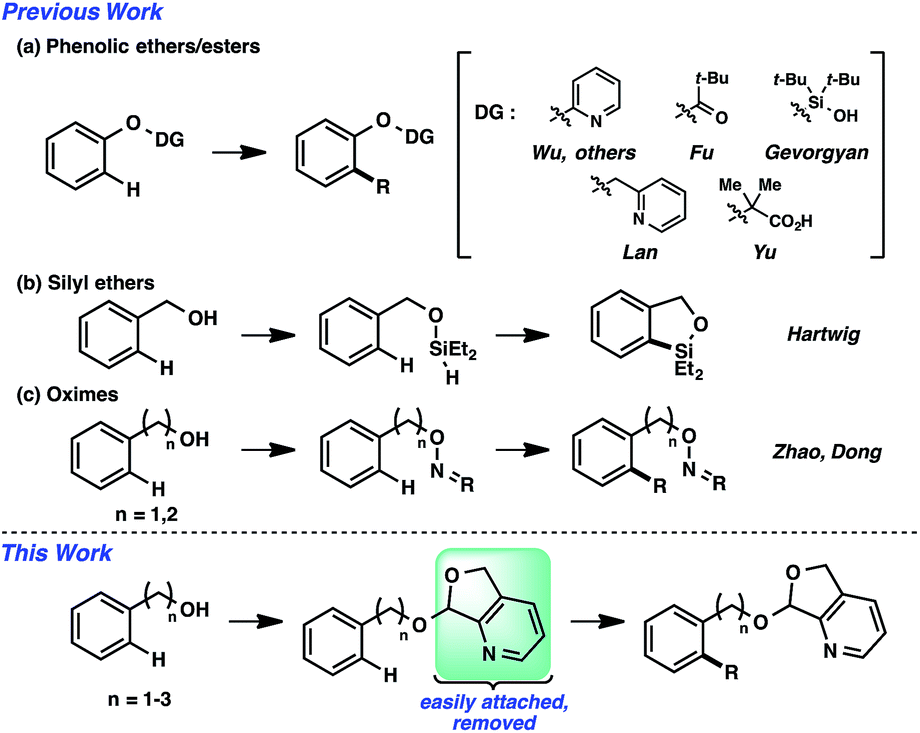 | ||
| Fig. 1 Surrogate approach to hydroxyl-directed C–H functionalization. | ||
Results and discussion
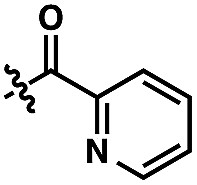
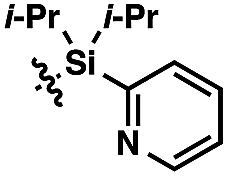
We anticipated at the onset that certain common functional groups may serve as straightforward solutions for our goal. It was expected that known protecting groups may exploit the potential Lewis basicity of the alcohol oxygen, or more likely, attach a functional group that endows its own directing capacity to induce the metalation. Our analysis of select groups in oxidative olefinations, using ligand-accelerated conditions inspired by the developments of Yu and coworkers20 (ethyl acrylate, 10 mol% Pd(OAc)2, 20 mol% Ac–Gly–OH, 3 equiv. AgOAc, HFIP, 90 °C), is depicted in Table 1. As can be seen, standard protecting groups were uniformly ineffectual. Ethers, acetals, esters, and carbamates all gave little to no reactivity. The di-tert-butyl-silanol group, which had been applied for phenol-based functionalization,7 also did not afford appreciable reactivity (entry 8). Pyridyl-incorporated esters and ethers also failed to induce directed olefination (entries 9, 10).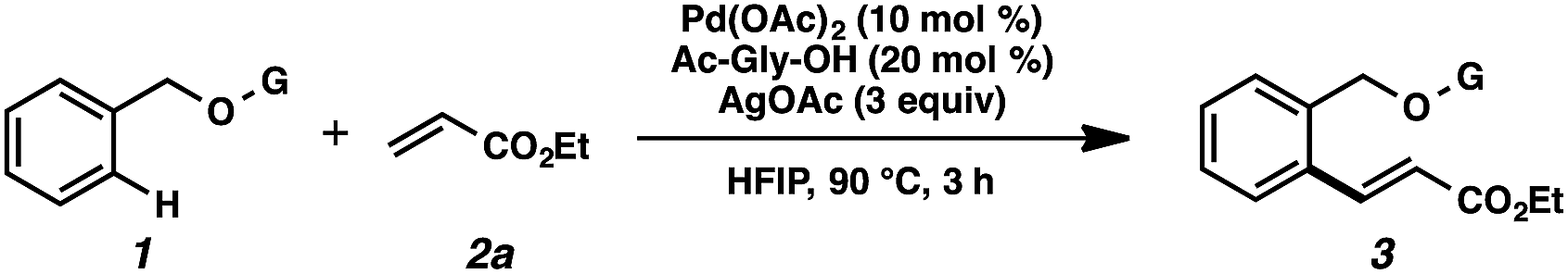
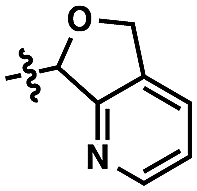
For our goal to ultimately be realized, it was clear that we needed to devise a more unique solution. The design of an original scaffold to address this challenge required particular components: namely (1) a robust directing group, (2) a functional group to allow straightforward attachment and removal, and (3) structural features that would ideally induce proximal direction. Hybridizing these requirements led us to compound 6 (Scheme 1). Pyridines as intramolecular directing groups are well-established, the acetal linkage would enable simple connectivity, and the fused ring system may substantially lower the entropic barriers for the desired cyclometalation. To that end, we synthesized the PyA (![[P with combining low line]](https://www.rsc.org/images/entities/b_char_0050_0332.gif)
![[y with combining low line]](https://www.rsc.org/images/entities/b_char_0079_0332.gif) ridyl–
ridyl–![[A with combining low line]](https://www.rsc.org/images/entities/b_char_0041_0332.gif) cetal) scaffold, available in two steps from commercial dimethyl pyridine-2,3-dicarboxylate.21
cetal) scaffold, available in two steps from commercial dimethyl pyridine-2,3-dicarboxylate.21
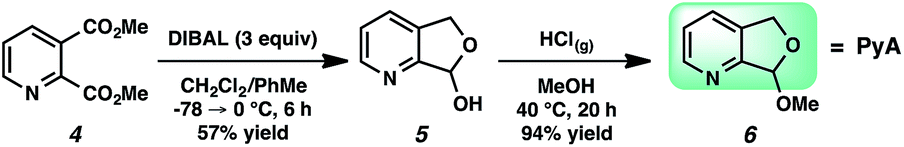 | ||
| Scheme 1 Pyridyl–acetal (PyA) synthesis. | ||
To our delight, we found that scaffold attachment to benzyl alcohol was straightforward using 5 Å molecular sieves, and subsequent Pd-catalyzed functionalization proceeded with marked improvement over the other cases (Table 1, entry 11 and Scheme 2). We were able to observe notably high levels of alkenylation, isolating monoolefin 8aamono and diolefin 8aadi in 51% and 31% yield, respectively.22
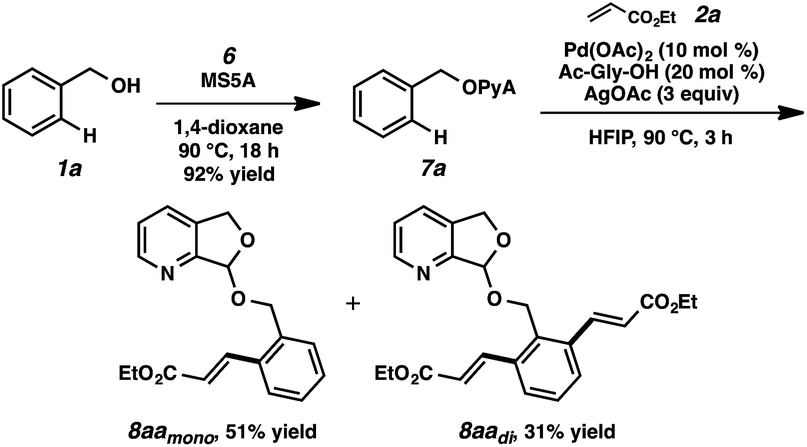 | ||
| Scheme 2 Initial test of PyA-based attachment and C–H olefination. | ||
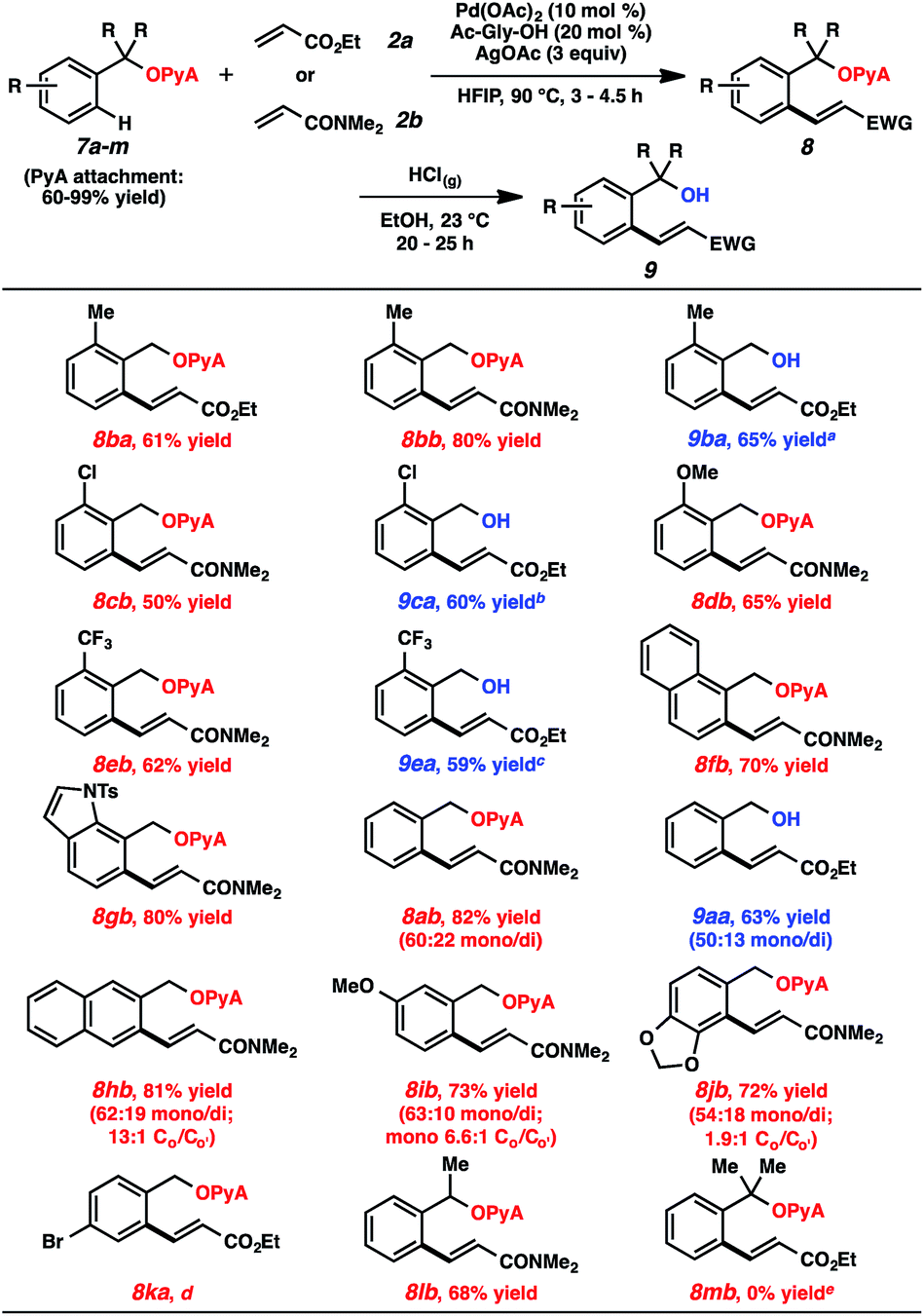
A range of scaffold-attached benzylic alcohol substrates were evaluated in the oxidative olefination using either ethyl acrylate or N,N-dimethylacrylamide (Table 2). In cases where only monoolefination can occur (i.e., ortho-substituted arenes), moderate to very good yields of alkene products were observed. In several of these cases, alcoholysis was also performed immediately following olefination (HCl, EtOH, 23 °C),23 and the resulting alcohols were formed in synthetically useful levels for the two-step sequence.24 Heterocycles are compatible with this reaction (e.g., compound 8gb). For cases where mono- and diolefination were possible, approximately 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures were generally produced, with monoolefination products being formed with ranging regioselectivities.25 Aryl bromides were incompatible with this process, likely due to competitive oxidative addition pathways. A secondary alcohol-based substrate was reactive and remarkably selective for monoalkenylation (8lb); a tertiary alcohol-based substrate, in contrast, was unproductive under these conditions.
:1 mixtures were generally produced, with monoolefination products being formed with ranging regioselectivities.25 Aryl bromides were incompatible with this process, likely due to competitive oxidative addition pathways. A secondary alcohol-based substrate was reactive and remarkably selective for monoalkenylation (8lb); a tertiary alcohol-based substrate, in contrast, was unproductive under these conditions.
Alkene partners were also evaluated (Table 3). In addition to ethyl acrylate and N,N-dimethylacrylamide, other acrylates and acrylamides were effective reactants (8bc–g). Electron-deficient styrenes were competent participants (8bh, 8bi), while more electron-neutral ones afforded the products in diminished yields (8bj, 8bk). Oxazolidinones and phosphine oxides were compatible (8bl, 8bm), but acrylonitrile was only marginally reactive (8bn).26
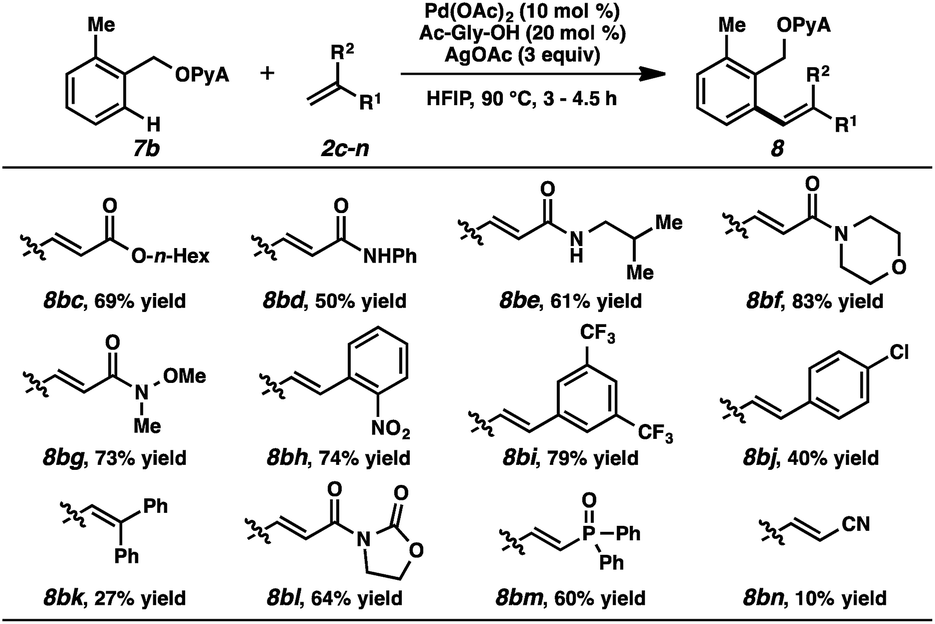
|
It merits mention that the PyA scaffold can be employed as a protecting group akin to conventional acetal species. Scheme 3 is illustrative; desilylation, oxidation, etherification, and Cu-catalyzed amidation27 were all effective in the presence of this moiety. A key attribute of this scaffold is its ability to be leveraged in protecting group strategies. Alkenylations of the respective products proceeded smoothly to generate diversely functionalized olefinic compounds. Of note, under these alkenylation conditions, the PyA scaffold outcompetes the ketone, ether, and acetamide for reactivity, despite these latter functional groups' directing capacity for Pd-catalyzed functionalization.28
 | ||
| Scheme 3 PyA scaffold as a protecting and directing group. (a) TBAF, THF, 23 °C, 3 h, 87% yield. (b) Dess–Martin periodinane, CH2Cl2, 23 °C, 1.5 h, 97% yield. (c) N,N-Dimethylacrylamide, Pd(OAc)2 (10 mol%), Ac-Gly-OH (20 mol%), AgOAc (3 equiv.), HFIP, 4.5 h, 90 °C. (d) NaH, MeI, THF, 0 °C, 2 h, 85% yield. (e) AcNH2, CuI (30 mol%), N,N,N′,N′-tetramethylethylenediamine (90 mol%), K3PO4, KI, DMF, 110 °C, 15 h, 95% yield. | ||
Toward increasing the breadth of utility, we sought to expand the transformations to more remote cases (i.e., beyond benzylic alcohols). Examples of these types of remote bond-forming events have been more rare, presumably due to the challenges of forming larger cyclometalated species. The substrates derived from both homobenzylic and bishomobenzylic alcohols can be effectively olefinated using this approach (Scheme 4).29 In comparison, when the parent alcohols (12, 16) were subjected to the alkenylation conditions, little to no reactivity was observed. Preliminary attempts to functionalize the further-extended acetal (20) were also unsuccessful.
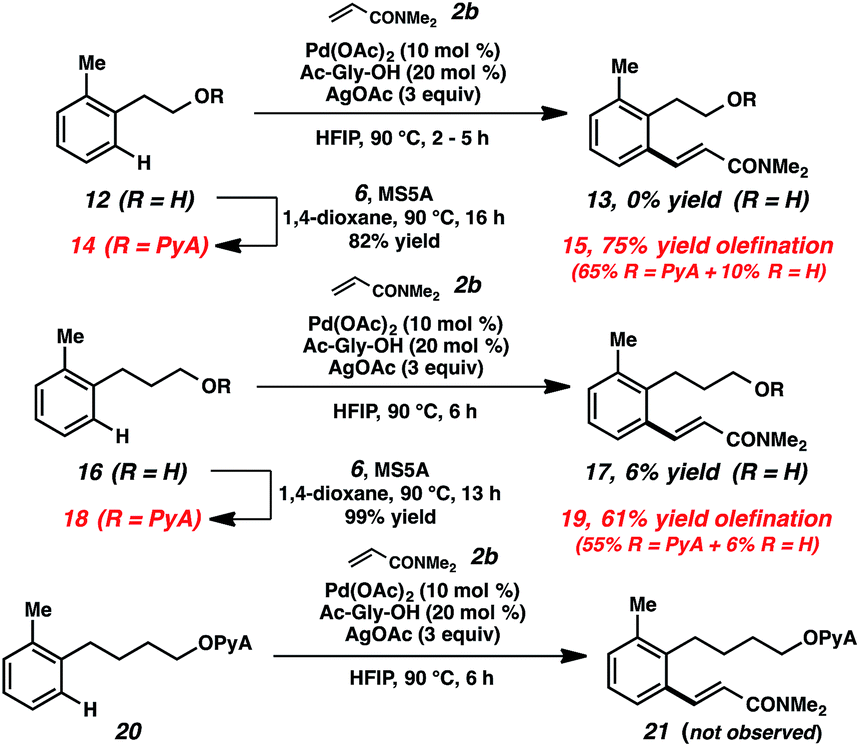 | ||
| Scheme 4 Remote functionalization using PyA scaffold. | ||
We encountered a limitation of this system when we attempted attaching particularly electron deficient alcohol substrates. For example, the use of alcohol 1q, bearing an electron-withdrawing ester moiety, was ineffective in attachment using acetal 6 (Scheme 5).30 Gratifyingly, however, the application of hemiacetal 5 under dehydrative conditions (10 mol% TsOH·H2O, 3 equiv. MgSO4) circumvented this problem, and attachment and alkenylation were both possible.31 The ease of this hemiacetal approach, where residual hemiacetal 5 was readily removed in the reaction workup, also established a path for the execution of a telescoping sequence.32 As illustrated in Scheme 5, benzylic alcohol 1b could be converted to the olefinic product without any intermediary purifications, representing a net alcohol-based directed ortho-functionalization.33 The scaffold can also be recovered in the alcoholysis, further highlighting the utility of this scaffold.
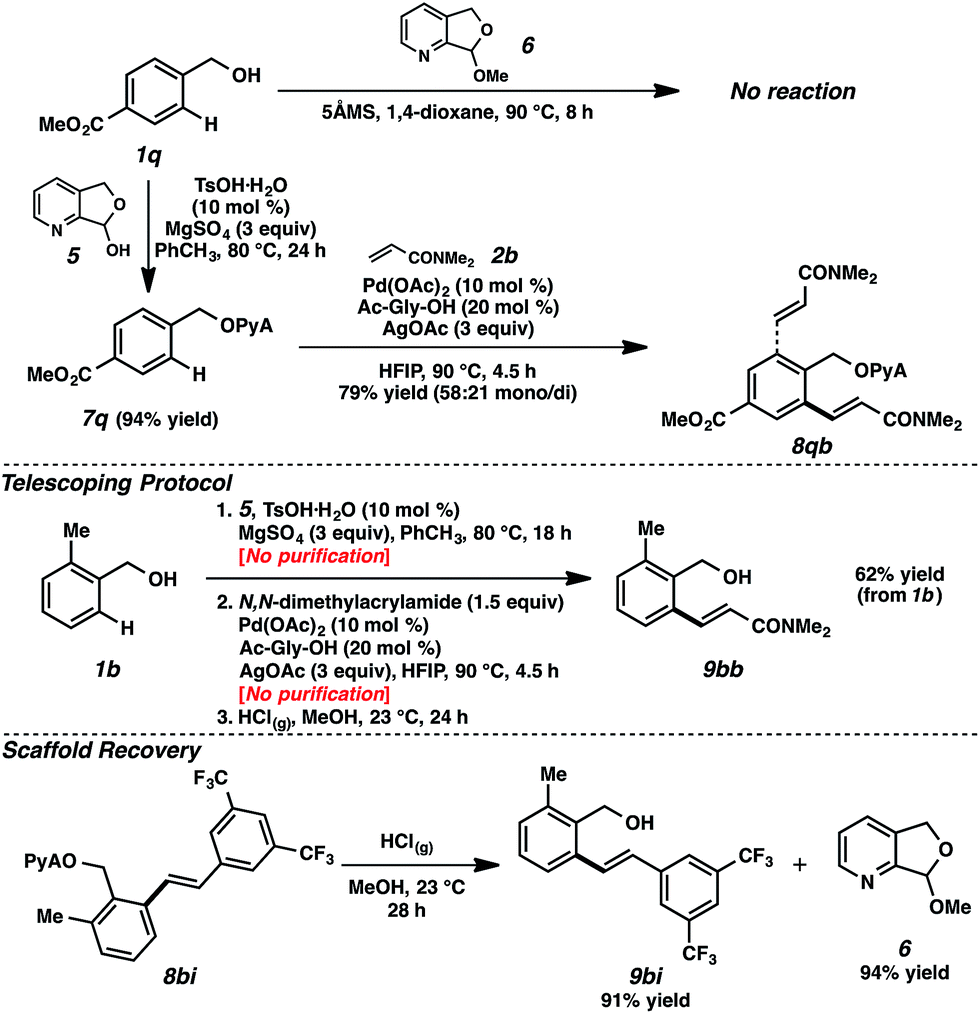 | ||
| Scheme 5 Alternative scaffold attachment method, telescoping, and recovery. | ||
The nature of the directing group merits further discussion. As mentioned above, we anticipated that this particular scaffold possessed specific features that rendered it uniquely effective for this function. Structural variants were revealing (Scheme 6). No reactivity was observed in the cases of acetal 22 or pyridine 23.34 When pyridyl ether 24 was subjected to the alkenylation conditions, minimal reactivity was observed.35 When compound 26 was tested, however, high reactivity was restored. From this set of experiments, a few conclusions can be drawn. First, the bidentate capability of the nitrogen and oxygen atoms is likely important at some stage of the overall reaction.36 Second, the fused ring system decreases the conformational degrees of freedom, presumably encouraging a proximal relationship between the metal center and the C–H bond. Lastly, the acetal functional group is not important for catalytic functionalization and only facilitates attachment and cleavage. Based on this data, we believe cyclometalated species 28, with scaffold bidentate coordination, is a probable intermediate in the overall alkenylation.37
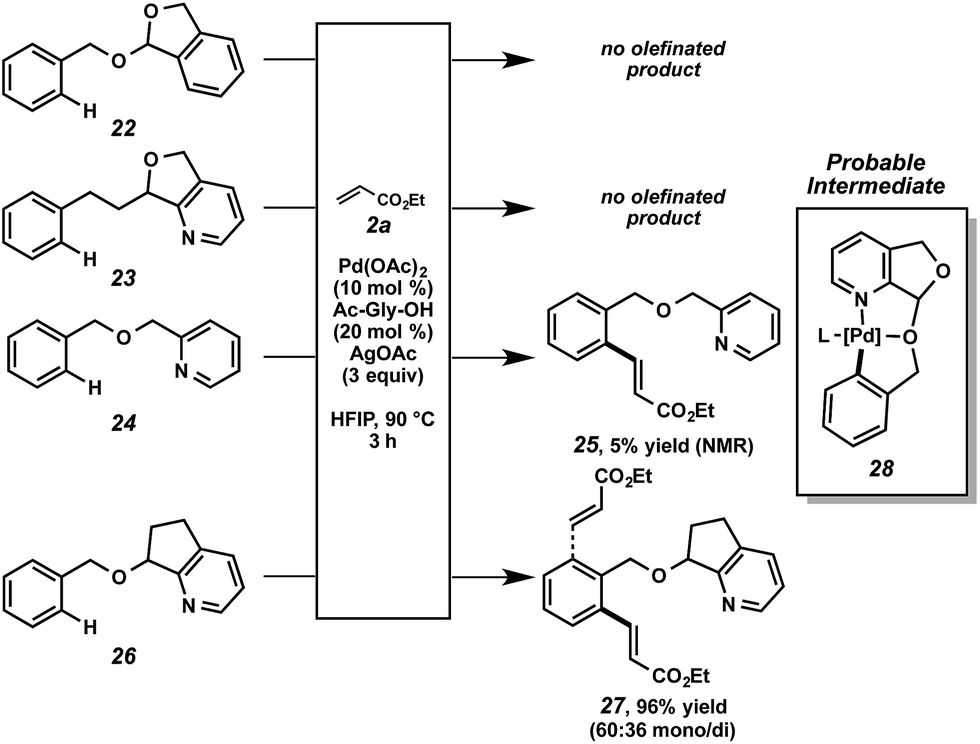 | ||
| Scheme 6 Reaction analysis of PyA scaffold variants. | ||
Conclusions
New strategies in catalytic C–H functionalizations that improve utility hold potential for achieving broad synthetic applicability. Our efforts herein outline a convenient and straightforward functionalization of alcohol-based substrates based on a simple molecular scaffolding approach. The scaffold is easily synthesized38 and is attachable and removable with high-yielding recovery. It has the capacity to serve as an alcohol protecting group, it can be incorporated into a telescoping process, and it can induce olefinations from notably remote positions on a molecule. We anticipate that these scaffold attributes will provide distinct advantages in catalytic functionalization chemistry.39 Further developments and applications in catalysis are underway and will be reported in due course.Acknowledgements
The University of Georgia is acknowledged for support of this research. The Summer Undergraduate Research Opportunity program at UGA is acknowledged for support of JOR.Notes and references
- (a) K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS PubMed; (b) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (c) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (d) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (e) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed.
- (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (b) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed; (c) Y.-H. Zhang, G.-F. Shi and J.-Q. Yu, in Comprehensive Organic Synthesis, ed. P. Knochel and G. A. Molander, Elsevier, Amsterdam, 2nd edn, 2014, vol. 3, pp. 1101–1209 Search PubMed.
- For a review on relevant strategies toward C–H functionalization of alcohol substrates, see, F. Mo, J. R. Tabor and G. Dong, Chem. Lett., 2014, 43, 264–271 CrossRef CAS.
- (a) Y. Lu, D.-H. Wang, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 5916–5921 CrossRef CAS PubMed; (b) X. Wang, Y. Lu, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 12203–12205 CrossRef CAS PubMed; (c) Y. Lu, D. Leow, X. Wang, K. M. Engle and J.-Q. Yu, Chem. Sci., 2011, 2, 967–971 RSC; (d) K. Morimoto, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2011, 76, 9548–9551 CrossRef CAS PubMed; (e) S. R. Kandukuri, L.-Y. Jiao, A. B. Machotta and M. Oestreich, Adv. Synth. Catal., 2014, 356, 1597–1609 CrossRef CAS; (f) S. Nakanowatari and L. Ackermann, Chem.–Eur. J., 2014, 20, 5409–5413 CrossRef CAS PubMed.
- For recent reviews on functional group surrogates for C–H functionalization, see: (a) G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450–2494 CrossRef CAS PubMed; (b) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906–6919 RSC; (c) C. Wang and Y. Huang, Synlett, 2013, 24, 145–149 CAS; (d) E. M. Ferreira, Nat. Chem., 2014, 6, 94–96 CrossRef CAS PubMed; (e) R. K. Rit, M. R. Yadav, K. Ghosh and A. K. Sahoo, Tetrahedron, 2015, 71, 4450–4459 CrossRef CAS.
- For select examples of this strategy in sp3 C–H functionalization using Pd catalysis, see: (a) Z. Ren, F. Mo and G. Dong, J. Am. Chem. Soc., 2012, 134, 16991–16994 CrossRef CAS PubMed; (b) P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2011, 133, 12584–12589 CrossRef CAS PubMed; (c) K. J. Fraunhoffer and M. C. White, J. Am. Chem. Soc., 2007, 129, 7274–7276 CrossRef CAS PubMed; (d) G. T. Rice and M. C. White, J. Am. Chem. Soc., 2009, 131, 11707–11711 CrossRef CAS PubMed.
- Di-tert-butyl-silanol: (a) C. Huang, B. Chattopadhyay and V. Gevorgyan, J. Am. Chem. Soc., 2011, 133, 12406–12409 CrossRef CAS PubMed; (b) C. Huang, N. Ghavtadze, B. Chattopadhyay and V. Gevorgyan, J. Am. Chem. Soc., 2011, 133, 17630–17633 CrossRef CAS PubMed; (c) Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2015, 54, 2255–2259 CrossRef CAS PubMed.
- 2-Pyridyl: (a) J.-H. Chu, P.-S. Lin and M.-J. Wu, Organometallics, 2010, 29, 4058–4065 CrossRef CAS. For recent examples of the use of this group in C–H functionalization, see: (b) M. Kim, S. Sharma, J. Park, M. Kim, Y. Choi, Y. Jeon, J. H. Kwak and I. S. Kim, Tetrahedron, 2013, 69, 6552–6559 CrossRef CAS; (c) B. Liu, H.-Z. Jiang and B.-F. Shi, J. Org. Chem., 2014, 79, 1521–1526 CrossRef CAS PubMed; (d) W. Zhang, J. Zhang, S. Ren and Y. Liu, J. Org. Chem., 2014, 79, 11508–11516 CrossRef CAS PubMed.
- 2-Pyridinylmethyl: X. Cong, J. You, G. Gao and J. Lan, Chem. Commun., 2013, 49, 662–664 RSC.
- Pivaloyl: B. Xiao, Y. Fu, J. Xu, T.-J. Gong, J.-J. Dai, J. Yi and L. Liu, J. Am. Chem. Soc., 2010, 132, 468–469 CrossRef CAS PubMed.
- 2-Carboxypropan-2-yl: H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567–7571 CrossRef CAS PubMed.
- (a) L. Ackermann, E. Diers and A. Manvar, Org. Lett., 2012, 14, 1154–1157 CrossRef CAS PubMed; (b) W. Ma and L. Ackermann, Chem.–Eur. J., 2013, 19, 13925–13928 CrossRef CAS PubMed.
- Also noteworthy is the use of Rh catalysis with phosphinite species that dynamically exchange with phenols to provide in situ directing groups. For key examples, see: (a) L. N. Lewis and J. F. Smith, J. Am. Chem. Soc., 1986, 108, 2728–2735 CrossRef CAS; (b) S. Oi, S.-i. Watanabe, S. Fukita and Y. Inoue, Tetrahedron Lett., 2003, 44, 8665–8668 CrossRef CAS; (c) R. B. Bedford, S. J. Coles, M. B. Hursthouse and M. E. Limmert, Angew. Chem., Int. Ed., 2003, 42, 112–114 CrossRef CAS; (d) J. C. Lewis, J. Wu, R. G. Bergman and J. A. Ellman, Organometallics, 2005, 24, 5737–5746 CrossRef CAS; (e) M. C. Carrión and D. J. Cole-Hamilton, Chem. Commun., 2006, 4527–4529 RSC.
- (a) E. M. Simmons and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 17092–17095 CrossRef CAS PubMed; (b) For a related case in the functionalization of sp3 C–H bonds, see: E. M. Simmons and J. F. Hartwig, Nature, 2012, 483, 70–73 CrossRef CAS PubMed.
- S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778–18781 CrossRef CAS PubMed.
- L. Chu, M. Shang, K. Tanaka, Q. Chen, N. Pissarnitski, E. Streckfuss and J.-Q. Yu, ACS Cent. Sci., 2015, 1, 394–399 CrossRef CAS.
- (a) K. Guo, X. Chen, M. Guan and Y. Zhao, Org. Lett., 2015, 17, 1802–1805 CrossRef CAS PubMed; (b) K. Guo, X. Chen, J. Zhang and Y. Zhao, Chem.–Eur. J., 2015, 21, 17474–17478 CAS.
- Z. Ren, J. E. Schulz and G. Dong, Org. Lett., 2015, 17, 2696–2699 CrossRef CAS PubMed.
- The three steps involve Mitsunobu reaction with N-hydroxyphthalimide, cleavage of the imide, and carbonyl condensation. The overall transformation can be conveniently accomplished in one pot; see ref. 17. Alternatively, this net transformation can be accomplished in two steps using Ellman's oxaziridine for direct O-amination, but the yields are generally lower and this option is less frequently employed.
- (a) K. M. Engle, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137–14151 CrossRef CAS PubMed; (b) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed.
- We found it more cost-effective to synthesize diester 4 from the dicarboxylic acid via Fischer esterification. See the ESI for details.†.
- Optimization of the olefination conditions is described in the ESI.† We found that after functionalization, treatment with celite and ethylenediamine overnight gave a notable improvement in isolated yield, most likely the result of decomplexation of the metal (Pd and/or Ag) to the pyridine-containing product. The treatment could be performed for shorter periods of time (<1 h), but we opted for the longer time to ensure completion across all products formed.
- (a) The decision to cleave or retain the PyA group was largely predicated on ease of chromatographic purification, as the alkenylation products using 2a tended to co-elute with the starting materials. As can be seen with products 8ba and 9ba, the alcoholysis was not imperative, and the yields were comparable with or without cleavage. The slightly lower yield of 8ba compared to 9ba reflects the small amounts of acetal cleavage that would occur over the course of the alkenylation; (b) The PyA group could also be cleaved using 1 M aq. HCl at 23 °C. See the ESI for details.†.
- The yields for the alcohol products are reported for over the two step procedue (olefination and alcoholysis).
- It is unclear what factors, sterics and/or electronics, are governing the overall regioselectivities for monoolefination. The selectivity difference between products 8ib and 8jb, for example, is rather striking and suggest subtle influences are at play.
- Enals, enones, and vinylsulfones were not successful coupling partners in this transformation.
- (a) A. Klapars, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421–7428 CrossRef CAS PubMed; (b) A. W. Mitra, M. M. Hansen, M. E. Laurila, S. P. Kolis and J. R. Martinelli, Tetrahedron Lett., 2013, 54, 6580–6583 CrossRef CAS.
- (a) For ketones, see: G. Li, L. Wan, G. Zhang, D. Leow, J. Spangler and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 4391–4397 CrossRef CAS PubMed, and references therein; (b) For ethers, see: G. Li, D. Leow, L. Wan and J.-Q. Yu, Angew. Chem., Int. Ed., 2012, 52, 1245–1247 CrossRef PubMed; (c) For acetamides, see: M. D. K. Boele, G. P. F. van Strijdonck, A. H. M. de Vries, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2002, 124, 1586–1587 CrossRef CAS PubMed.
- Zhao (oximes, ref. 17) and Yu (ethers, ref. 28b) have demonstrated and the functionalization of homobenzylic alcohol-based compounds.
- As a reference point, the o-CF3 benzylic alcohol was operative (i.e., the precursor to compound 8eb) in scaffold attachment, albeit in a lower yield on the spectrum of alcohol substrates (72%).
- In general, scaffolds 5 and 6 were similarly efficient for the attachment of electron-rich alcohol substrates. There does not appear to be a clear advantage of using 6vs.5 in these cases, although the conditions for attachment are different and thus one may be more appropriate for certain systems.
- Hemiacetal 5 is highly water-soluble and is easily removed in an aqueous wash. In contrast, residual methyl acetal (6) was not selectively removable by this protocol and complicated the subsequent functionalization reaction.
- As a comparison, the three-step procedure with intermediate purifications is higher yielding (74% overall yield). See the ESI.†.
- For compound 22, substantial decomposition was observed. Compared to other ethers and acetals (Table 1, entries 2, 3 and 5), we suspect that the decomposition arises from the greater lability of the benzylic acetal.
- This specific lack of reactivity was surprising, given this ether had been employed in phenol-based functionalizations. See ref. 9.
- For select reviews on bidentate coordination in C–H functionalizations, see: (a) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (b) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS PubMed.
- L2-type (neutral) bidentate ligands in catalytic C–H functionalization are notably more rare than LX-type (monoanionic) bidentate ligands. For select examples implicating the former, see: (a) P. L. Alsters, P. F. Engel, M. P. Hogerheide, M. Copijn, A. L. Spek and G. van Koten, Organometallics, 1993, 12, 1831–1844 CrossRef CAS; (b) R. K. Rit, M. R. Yadav and A. K. Sahoo, Org. Lett., 2014, 16, 968 CrossRef CAS PubMed; (c) Y. Xu, G. Yan, Z. Ren and G. Dong, Nat. Chem., 2015, 7, 829–834 CrossRef CAS PubMed; (d) Ref. 18 .
- Both 5 and 6 can be synthesized in multigram quantities, and they are both bench-stable compounds that have been stored for several months with no apparent degradation.
- Preliminary experiments toward catalytic scaffold have not been successful. Our current hypothesis is that under these specific oxidative conditions, free alcohols are not sufficiently tolerated, consisent with the low yields Yu had observed in alcohol-based C–H functionalizations. See ref. 4a–c.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc03948g |
| This journal is © The Royal Society of Chemistry 2016 |