
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining photochemistry and catalysis: rapid access to sp3 – rich polyheterocycles from simple pyrroles†
Emma E.
Blackham
,
Jonathan P.
Knowles
,
Jonathan
Burgess
and
Kevin I.
Booker-Milburn
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: k.booker-milburn@bristol.ac.uk; Web: http://www.chm.bris.ac.uk/org/bmilburn/index2.htm
First published on 4th January 2016
Abstract
Use of FEP flow reactor technology allows access to gram quantities of photochemically-generated tricyclic aziridines. These undergo a range of novel palladium-catalyzed ring-opening and cycloaddition reactions, likely driven by their inherent strain, allowing incorporation of further functionality by fusing additional heterocyclic rings onto these already complex polycyclic cores. This rapid, 2-step access to complex sp3 – rich heterocycles should be of interest to those in the fields of drug discovery and natural product synthesis.
Introduction
The strained 3-membered aziridine ring has long been exploited in a variety of ring-opening reactions, serving as a valuable strategy in the synthesis of amino compounds. Compared to epoxides, however, aziridines are generally less reactive due the lower electronegativity of N vs. O. Consequently the reactivity of aziridines is usually enhanced by carrying out reactions with nucleophiles in the presence of Brønsted or Lewis acids, or by the attachment of an electron withdrawing group to the nitrogen.1 The latter approach, however, restricts the type of aziridines that can be used and requires removal of the activating group in a subsequent step. We recently described a photochemical transformation of simple N-butenyl substituted pyrroles 1 into tricyclic fused aziridines 3, via cyclobutane 2 (Scheme 1a).2 However, we found the batch scale-up of this very powerful sequence to be somewhat limited. This is due to the low overall quantum yield of this 2-step, 2-photon sequence and the fact that, due to the high extinction coefficient (ε = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), the reaction has to be run at high dilution (<0.02 mM). This made further study of the chemistry of the resulting aziridines challenging.
000), the reaction has to be run at high dilution (<0.02 mM). This made further study of the chemistry of the resulting aziridines challenging.
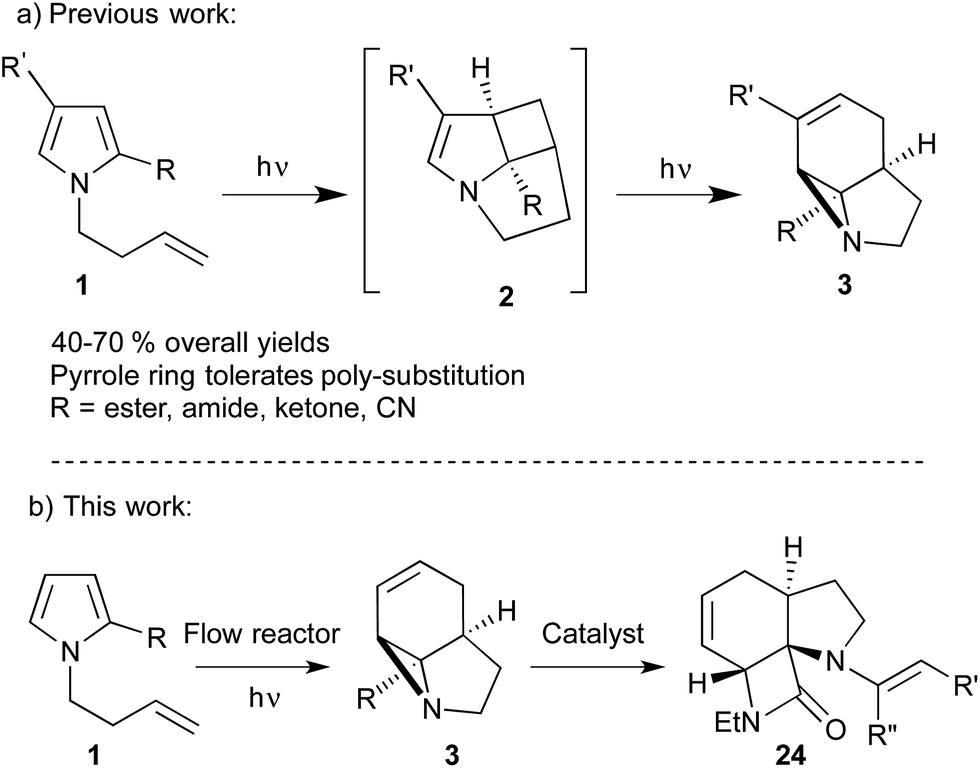 | ||
| Scheme 1 (a) Formation of aziridines by photocycloaddition/rearrangement of pyrrole derivatives. (b) Scale-up of photochemical aziridine synthesis and subsequent catalytic ring opening to generate polyheterocycles. | ||
Herein we describe the production of multigram quantities of aziridines using fluorinated ethylene propylene (FEP) flow reactor technology, a concept that has seen increasing use in recent years.3 The resulting structural complexity and close proximity of functional groups within these aziridines prompted us to explore their reactivity with various nucleophiles and ring-opening conditions. Their interesting, and sometimes unusual results are reported and include a novel β-lactam forming sequence (Scheme 1b).
Results and discussion
Initially, we examined the scale-up irradiations of the three pyrroles 1a–c at 254 nm. This used the previously described trio of 1-layer FEP reactors with 36 W low-pressure Hg lamps.3b The three reactors were connected together in series for maximum productivity. Once optimized this gave gram quantities of the three aziridines 3a–c per 5–8 h run (Scheme 2). | ||
| Scheme 2 Scale up of photochemical aziridine synthesis using a trio of 1-layer FEP reactors with 36 W low-pressure Hg lamps. | ||
With these quantities in hand we first explored the ring opening of the amide-aziridine 3a with nucleophiles (Scheme 3). Using thiophenol the aziridine underwent SN2 ring-opening under mild conditions in THF in the presence of triethylamine. The high reactivity of the unactivated aziridine ring was highlighted by the clean ring opening with thiophenol in acetonitrile without added base to give 4. We considered that this ring-opening of an unactivated aziridine under such mild conditions pointed towards further strain imposed by the tricyclic system. Unfortunately ring opening could not be achieved using nucleophiles such as phenol or methanol under neutral or basic conditions. When the reaction was repeated with methanol under acid catalysis (p-TSA) an inseparable mixture of products was obtained which appeared to be a mixture of SN2′ ring-opened diastereomers. A screen of other catalysts was not carried out as the aziridine was prone to decomposition under acidic conditions.
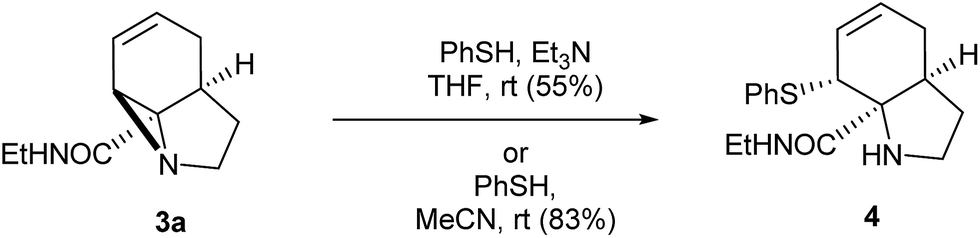 | ||
| Scheme 3 Initial ring opening reactions of aziridine 3a. | ||
Literature reports of Pd-catalysed Tsuji–Trost ring-opening reactions of vinyl aziridines are rare.4 Indeed there would appear to be only two reports involving the addition of carbon-nucleophile in an SN2′ mode.5 We were intrigued therefore to see if our highly strained systems would extend the scope of these reactions to a wider range of nucleophiles. Treatment of 3a with phenol and 5 mol% Pd(PPh3)4 in dioxane gave exclusively the anti product 5 (entry 1, Table 1). Despite the lack of literature precedent we then examined the reaction with a range of carbon-nucleophiles. Pleasingly the reaction of 3a and 3b also proceeded smoothly with a selection of soft carbon-nucleophiles (entries 3–10). It was also found that the anti/syn ratios could be switched on moving to more polar solvents.6 For example, reaction of 3a in DMF at 80 °C resulted in a 1:4 ratio of 5:6 (Nu = PhO, entry 2). Similarly reaction of 3a in MeCN with pentane-2,3-dione provided a switch in the anti/syn ratio to 1:9 (entry 6). This switch is likely to reflect a change from an outer sphere mechanism6a,b (direct attack of nucleophile on carbon – overall retention) to an inner sphere process6c (initial attack on metal – overall inversion) on moving to a more polar solvent. Such behaviour has been observed in Tsuji–Trost reactions of allylic acetates on switching from soft to hard nucleophiles,6d and appears to reflect the decrease in stability of the anion on moving to more polar solvents as judged from the pKa values of phenol in this case.7
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Solvent | NuH | T (°C) | Yield (%) |
5:6 |
| 1 | CONHEt | Dioxane | PhOH | rt | 49 | 1:0 |
| 2 | CONHEt | DMF | PhOH | 80 | 52 | 1:4 |
| 3 | CONHEt | Dioxane |
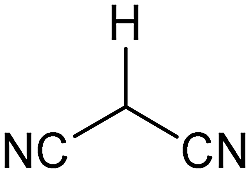
|
40 | 52 | 1:0 |
| 4 | CONHEt | MeCN |
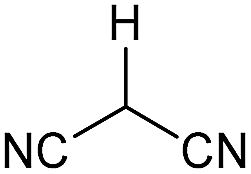
|
40 | 57 | 1:1 |
| 5 | CONHEt | Dioxane |
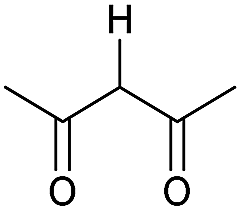
|
80 | 82 | 1:0 |
| 6 | CONHEt | MeCN |
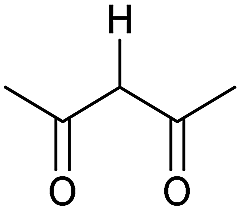
|
80 | 67 | 1:9 |
| 7 | CONHEt | Dioxane |
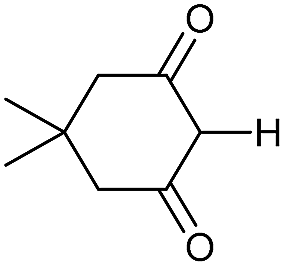
|
rt | 75 | 7:1 |
| 8 | CONHEt | Dioxane |
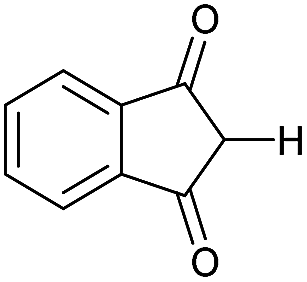
|
rt | 64 | 1:0 |
| 9 | COMe | Dioxane |
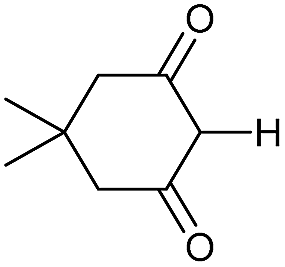
|
rt | 52 | 13:1 |
| 10 | COMe | Dioxane |
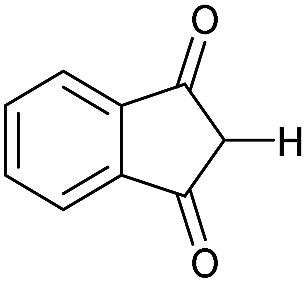
|
rt | 43 | 5:1 |
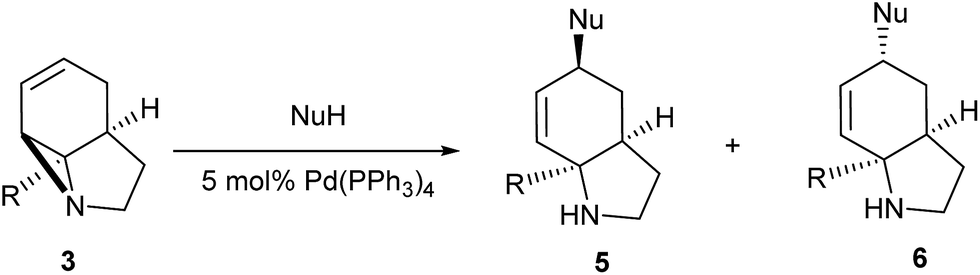
It has been previously demonstrated8 that N-alkyl substituted 2-vinyl aziridines reacted with isocyanates under Pd(0) catalysis to generate vinyl substituted cyclic ureas. If this could be applied to 3 then highly functionalised tricyclic ureas such as 7 would be accessible in just two steps from simple pyrroles. Table 2 describes the reaction between a wide range of aryl and sulfonyl isocyanates with tricyclic aziridines under Pd(0) catalysis. In general, the ureas 7 were formed in good yield using a 5 mol% loading of Pd(PPh3)4. It was found that the amide-aziridine 3a reacted more consistently than the COMe (3b) or CN (3c) substituted derivatives. A range of aryl isocyanates were tolerated, although 2-substituted examples tended to give lower yields and 2,6-substitution lead to mixed products from N/O cyclisation (entry 17). An interesting observation was noted with low (1 mol%) catalyst loadings (entries 19 & 20) and indeed no catalyst (entry 18), where exclusive formation of the cyclic imidates 8 was observed. In the case entry 19 increasing the quantity of Pd to 5 mol% results in rapid conversion of 8 to 7. These results are in good agreement with those observed by Alper for simpler vinyl aziridines.8a
|
|
||||
|---|---|---|---|---|
| Entry | R | R2 | Yield (%) |
7:8 |
| a Uncatalysed, dioxane, rt. b 1 mol% Pd(Ph3P)4, dioxane, rt. | ||||
| 1 | CONHEt | 4-MeC6H4SO2 | 83 | 1:0 |
| 2 | CN | 4-MeC6H4SO2 | 81 | 1:0 |
| 3 | COMe | 4-MeC6H4SO2 | 43 | 1:0 |
| 4 | CONHEt | 4-ClC6H4 | 58 | 1:0 |
| 5 | CONHEt | 2-ClC6H4 | 71 | 1:0 |
| 6 | CN | 2-ClC6H4 | 49 | 1:0 |
| 7 | CONHEt | 2-CF3C6H4 | 81 | 1:0 |
| 8 | CONHEt | 3-CF3C6H4 | 91 | 1:0 |
| 9 | CONHEt | 4-CF3C6H4 | 54 | 1:0 |
| 10 | COMe | 4-CF3C6H4 | 55 | 1:0 |
| 11 | CONHEt | 2-NO2C6H4 | 89 | 1:0 |
| 12 | CN | 2-NO2C6H4 | 54 | 1:0 |
| 13 | CONHEt | 4-NO2C6H4 | 89 | 1:0 |
| 14 | CONHEt | 4-MeCOC6H4 | 79 | 1:0 |
| 15 | CONHEt | 4-MeOC6H4 | 80 | 1:0 |
| 16 | CONHEt | 2-MeOC6H4 | 38 | 1:0 |
| 17 | CONHEt | 2,6-Cl2C6H3 | 80 | 10:1 |
| 18a | CONHEt | 4-MeC6H4SO2 | 51 | 0:1 |
| 19b | CONHEt | 4-MeC6H4SO2 | 85 | 0:1 |
| 20b | CONHEt | 2-ClC6H4 | 96 | 0:1 |
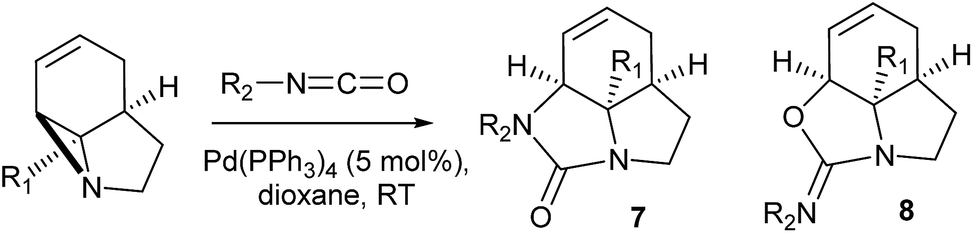
We then examined the potential [3 + 2] cycloaddition reactions of alkenes with the aziridines, as rapid access to the resulting tricyclic fused pyrrolidines would be attractive for alkaloid synthesis. Such Pd-catalysed [3 + 2] cycloadditions have been examined previously, but generally require activated aziridines.9 Initial results between 3a and simple alkenes such as methyl acrylate, acrylonitrile and methyl vinyl ketone showed no reactions. After much optimisation, benzylidine malononitrile was found to react under the specific conditions shown in Table 3 to give the pyrrolidine 10a as essentially one diastereomer. Higher yields were obtained by the use of the more reactive methylene diesters, which led to the formation of the tricyclic pyrrolidines 11a and 12a, b. Key to the success of these reactions was the use of triphenylphosphite as ligand;10 phosphine ligands gave lower yields and polymerisation of the reactive methylene diesters. However, use of nitrile aziridine 3c gave lower yields of the corresponding products (i.e.12c & 15c), with appreciable recovery of the starting material, due to the reduced reactivity of this substrate.
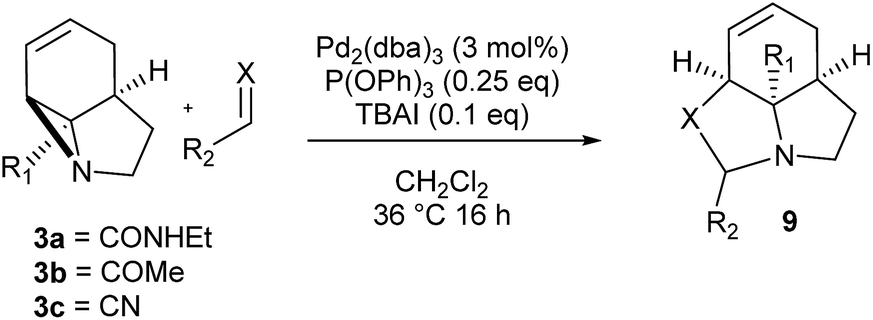
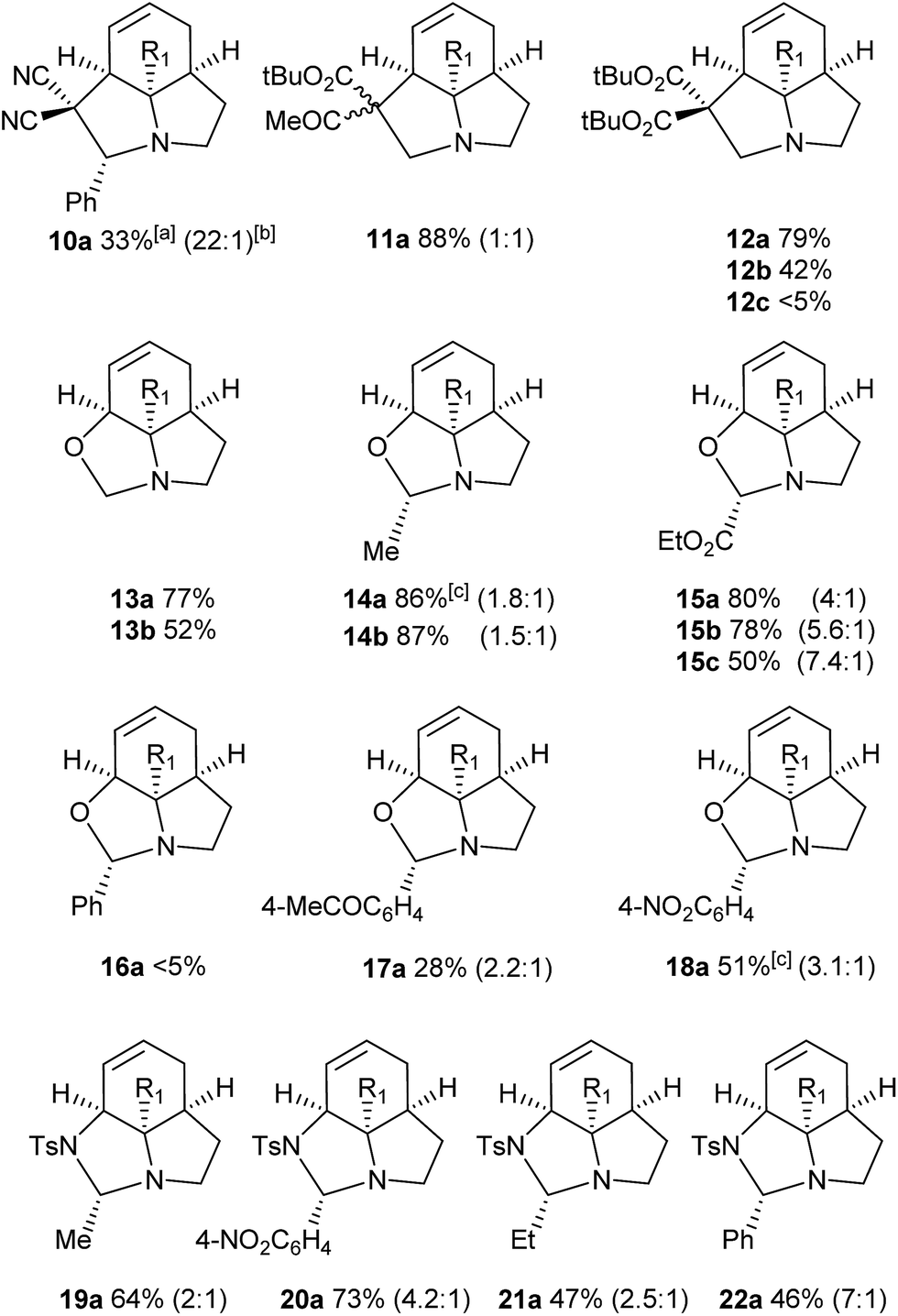
There have been a number of reports of the reaction of aldehydes with aziridines in a [3 + 2] manner, but with few exceptions11 these all involved activated aziridines under Lewis acid catalysis.12 We were intrigued to discover whether our strained aziridines would undergo [3 + 2] cycloadditions with aldehydes under the aforementioned optimised Pd-catalysed conditions. With the exception of benzaldehyde, a range of aldehydes reacted well with the aziridines to give a variety of functionalised oxazolidines with good diastereoselection (13–18). These results highlight the exceptional reactivity of these tricyclic aziridines compared to the more conventionally activated documented examples. It should be noted that none of these reactions gave oxazolidines in the absence of palladium (vide infra). Following on from this success we were keen to explore the use of imines in this reaction. Gratifyingly, it was found that a selection N-Ts imines reacted with 3a to give the tricyclic fused aminals 19–22 with reasonable to good levels of diastereoselection. This appears to be a previously unreported mode of addition for aziridines, allowing novel access to highly functionalised aminals.13
Finally, we decided to examine whether the limited scope of [3 + 2] cycloaddition reactions of alkenes (3 examples, Table 3) could be extended using alkyne based Michael acceptors. It was envisioned that the aziridines would react with alkynes under Pd-catalysis to generate tricyclic systems containing a partially unsaturated pyrrolidine ring. This proved successful in the reaction of 3c with a range of alkynes and gave rapid access to the highly functionalised dihydropyrrole 23. Interestingly, these cycloadditions proceeded equally well with or without catalyst (Table 4) suggesting, in this case, a purely thermal process. Moving to amide 3a a quite different outcome was observed, and remarkably the tricyclic fused β-lactams 24 were obtained. This sequence produces a single diastereomeric product (Table 5). One notable feature here is that although Pd-catalysis generally gave the highest yields and shortest reaction times, the reactions were also found to proceed without catalyst at room temperature.
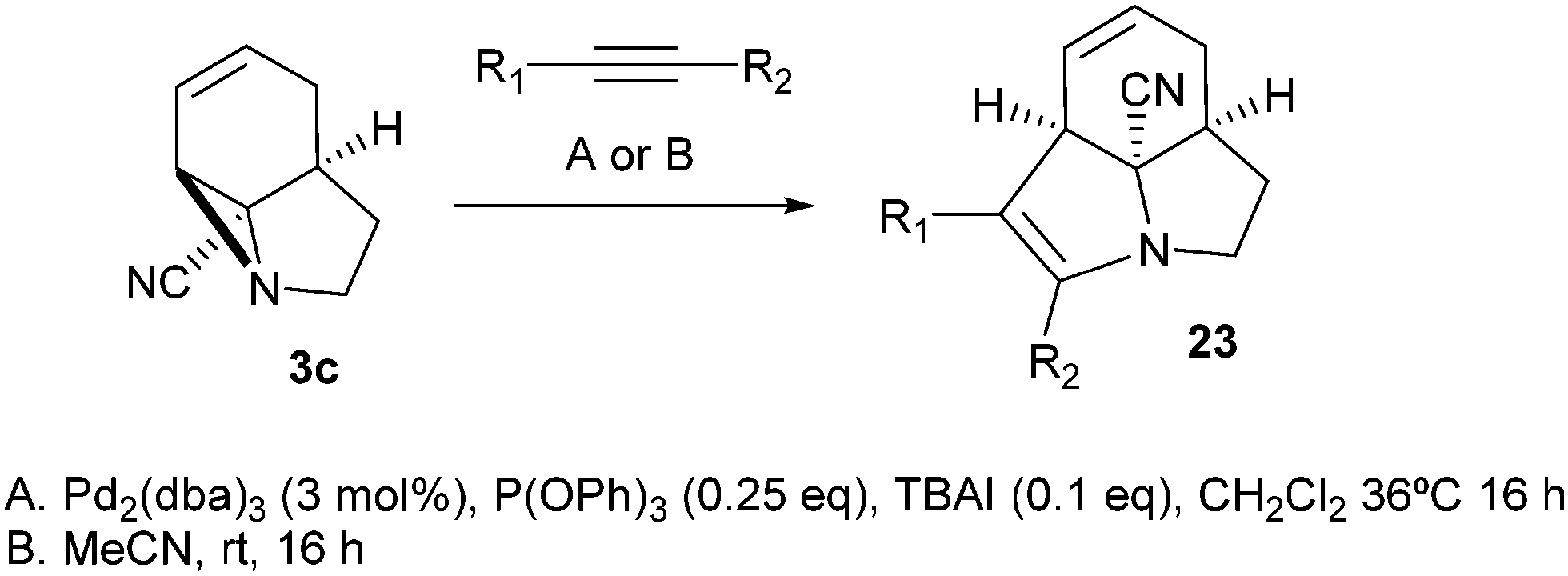
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | t (h) | Conditions A yield (%) | t (h) | Conditions B yield (%) |
| a Desilylated product obtained on work-up (R1 = CHO; R2 = H). b 15% of an oxazolidinone by-product was also isolated – see ESI for details. c Includes 9% of the imidate isomer – see ESI for details. | ||||||
| 1 | COMe | H | 16 | 98 | 16 | 82 |
| 2 | CO2Me | H | 16 | 83 | 24 | 54 |
| 3 | CO2Et | H | 16 | 78 | 47 | 61 |
| 4 | CONH2 | H | 16 | 0 | 16 | 0 |
| 5 | CHO | TMSa | 16 | 46b | 20 | 60c |
| 6 | CO2Me | CO2Me | 16 | 32 | 6.5 | 48 |
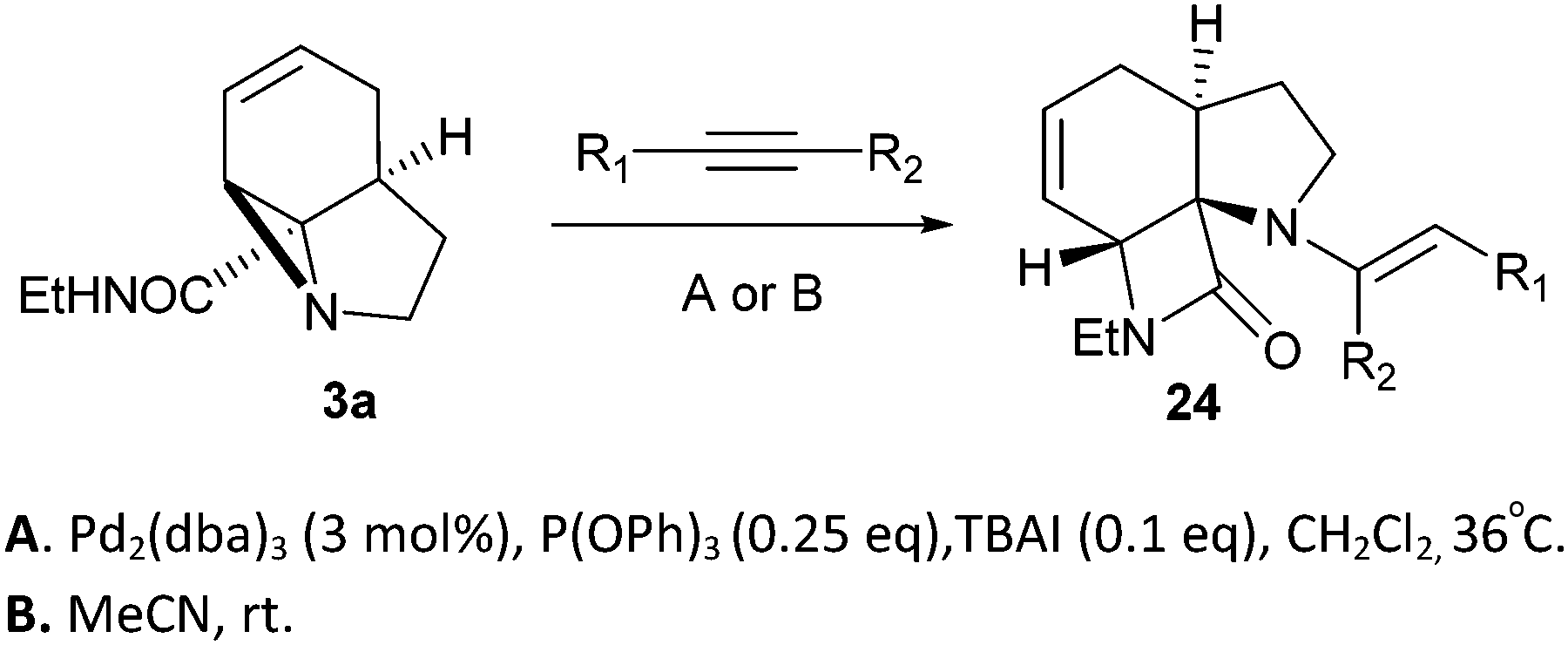
Although these reactions may seem diverse, mechanistically they are all likely to be related, with subtle differences depending on the substrates and conditions used. The unusual Tsuji–Trost type ring opening reactions described in Table 1 are likely to proceed via a conventional pathway where strain induces a higher reactivity compared to standard non-activated aziridines. It is possible, however, the aziridine is further activated by initial protonation from the carbon-nucleophiles used, which would not be possible for more conventionally activated aziridines (e.g. NTs). The reactions in Tables 2 and 3 can be described by the mechanisms in Scheme 4. Firstly the aziridine 3 reacts by nucleophilic attack on the isocyanate/ketone/aldehyde/imine to generate the zwitterion 25. This is in contrast to initial attack of Pd to give a π-allyl Pd complex as previously described by Alper.8a We propose this based on the absence of any Pd-induced reactions (e.g. ring opening/β-hydride elimination) when the electrophile is either not present or insufficiently reactive. Palladium then reacts with the zwitterion 25 to give π-allyl Pd complex 26 (Path A), which after cyclisation forms the various heterocycles 27 and regenerates the Pd(0) catalyst. Where the reaction proceeds without catalyst (e.g.Table 2, entry 18) it is possible that the zwitterion 25 is in equilibrium with the ring-opened form 28, which then undergoes cyclisation to 27 (Path B). In the case of β-lactam formation it is likely that after the aziridine adds to the alkyne and undergoes ring opening with Pd(0), the resulting vinyl anion 29 deprotonates the secondary amide to give 30 which then cyclises to the 4-membered lactam (Path C). The higher pKa of the vinyl anion 29 compared to other species (i.e.26 & 28) explains why β-lactam formation was not observed in other cases.
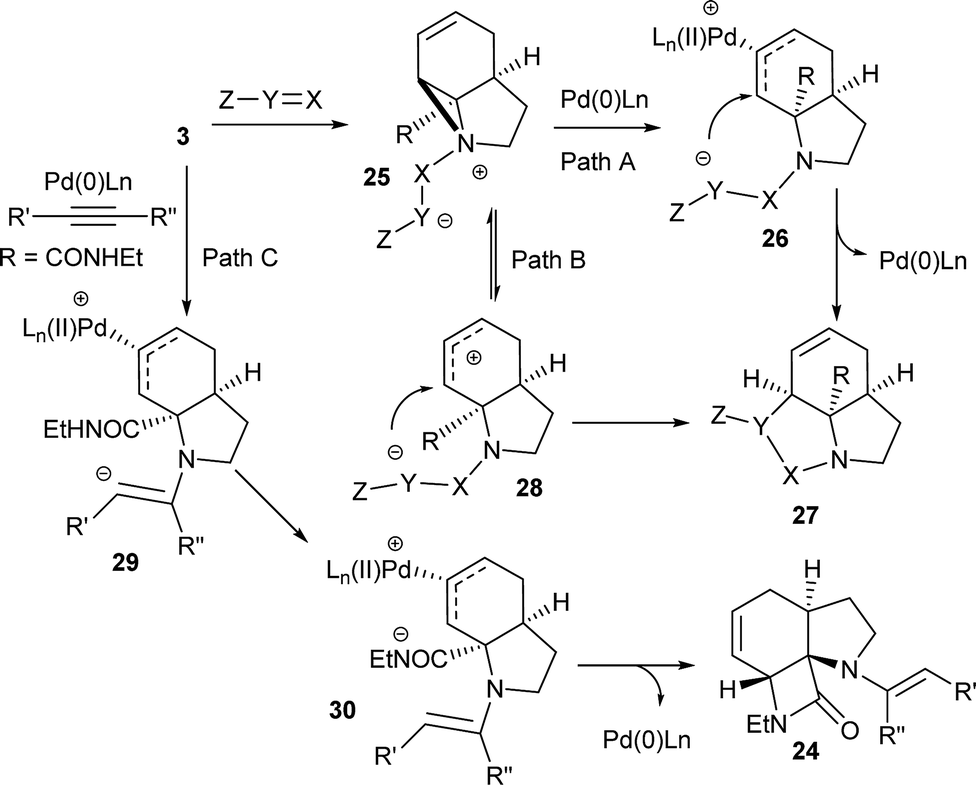 | ||
| Scheme 4 Plausible mechanistic rationale for formation of all products. | ||
Conclusions
In summary, highly strained and compact fused tricyclic aziridines have been shown to undergo a range of ring-opening and cycloaddition reactions under mild conditions. This is in contrast with the usual behaviour of aziridines, which normally require an activating electron-withdrawing group to be placed on the nitrogen. Notable features include: (a) a very rare and extremely mild Tsuji–Trost type aziridine ring-opening using carbon-nucleophiles; (b) a Pd-catalysed [3 + 2] cycloaddition of aldehydes and imines, which has hitherto not been reported; (c) a novel, mild and effective method for the stereo-controlled formation of tricyclic fused β-lactams. Together these diverse reactions highlight the power of combining an initial photochemical step with a secondary catalytic process, in this case resulting in highly complex products in just two steps from simple pyrroles. We have also shown that the photochemical step to give these complex aziridines intermediates is readily scalable using the FEP flow reactors we have previously described,3a–c allowing access to gram quantities of the substrates for these catalytic processes. As such these results may prove useful to chemists looking to exploit short routes to complex, sp3-rich compounds as potential scaffolds in drug discovery and alkaloid synthesis.Acknowledgements
We thank the EPSRC Bristol Chemical Synthesis Doctoral Training Centre (EP/G036764/1) and the University of Bristol for PhD studentship funding (E.E.B). We thank Dr Hazel A. Sparkes for X-ray crystallographic analysis.Notes and references
- (a) J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247–258 RSC; (b) D. Tanner, Angew. Chem., Int. Ed. Engl., 1994, 33, 599–619 CrossRef; (c) For a review dealing specifically with vinyl aziridines see: H. Ohno, Chem. Rev., 2014, 114, 7784–7814 CrossRef CAS PubMed.
- K. G. Maskill, J. P. Knowles, L. D. Elliott, R. W. Alder and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2013, 52, 1499–1502 CrossRef CAS PubMed.
- (a) B. D. A. Hook, W. Dohle, P. R. Hirst, M. Pickworth, M. Berry and K. I. Booker-Milburn, J. Org. Chem., 2005, 70, 7558–7564 CrossRef CAS PubMed; (b) L. D. Elliott, J. P. Knowles, P. J. Koovits, K. G. Maskill, M. J. Ralph, G. Lejeune, L. J. Edwards, R. I. Robinson, I. R. Clemens, B. Cox, D. D. Pascoe, G. Koch, M. Eberle, M. B. Berry and K. I. Booker-Milburn, Chem.–Eur. J., 2014, 20, 15226–15232 CrossRef CAS PubMed; (c) J. P. Knowles, L. D. Elliott and K. I. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025–2052 CrossRef CAS PubMed; (d) S. Yuanha, K. Kuijpersa, V. Hessela and T. Noël, Reaction Chemistry & Engineering, 2016 10.1039/c5re00021a; (e) A. C. Gutierrez and T. Jamison, Org. Lett., 2011, 13, 6414–6417 CrossRef CAS PubMed; (f) A. Yavorskyy, O. Shvydkiv, N. Hoffmann, K. Nolan and M. Oelgemoller, Org. Lett., 2012, 14, 4343–4345 CrossRef PubMed.
- (a) B. M. Trost, M. Osipov and G. Dong, J. Am. Chem. Soc., 2010, 132, 15800–15807 CrossRef CAS PubMed; (b) S. Sebelius, V. J. Olsson and K. J. Szabó, J. Am. Chem. Soc., 2005, 127, 10478–10479 CrossRef CAS PubMed.
- (a) A. A. Cantrill, A. N. Jarvis, H. M. I. Osborn, A. Ouadi and J. B. Sweeney, Synlett, 1996, 847–848 CrossRef CAS; (b) T. Kawamura, N. Matsuo, D. Yamauchi, Y. Tanabe and H. Nemoto, Tetrahedron, 2013, 69, 5331–5341 CrossRef CAS.
- (a) B. M. Trost and T. R. Verhoeven, J. Org. Chem., 1976, 41, 3215–3216 CrossRef CAS; (b) B. M. Trost and P. E. Strege, J. Am. Chem. Soc., 1977, 99, 1649–1651 CrossRef CAS; (c) H. Matsushita and E. Negishi, J. Chem. Soc., Chem. Commun., 1982, 160–161 RSC; (d) J. Fiaud and J. Legros, J. Org. Chem., 1987, 52, 1907–1911 CrossRef CAS; (e) M. Luparia, M. T. Oliveira, D. Audisio, F. Frébault, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2011, 50, 12631–12635 CrossRef CAS PubMed.
- (a) K. Evstigneev and P. Maiorova, Russ. J. Gen. Chem., 1997, 67, 1475–1477 Search PubMed; (b) K. Araki, K. Iwamoto, S. Shinkai and T. Matsuda, Bull. Chem. Soc. Jpn., 1990, 63, 3480–3485 CrossRef CAS; (c) C. J. A. McCaw, A. O'Byrne and W. J. Spillane, Eur. J. Org. Chem., 2008, 24, 4200–4205 Search PubMed; (d) D. Celadon, F. Maran, M. G. Severin and E. Vianello, J. Am. Chem. Soc., 1991, 113, 9320–9329 CrossRef.
- (a) D. C. D. Butler, G. A. Inman and H. Alper, J. Org. Chem., 2000, 65, 5887–5890 CrossRef CAS PubMed; (b) B. M. Trost and D. R. Fandrick, J. Am. Chem. Soc., 2003, 125, 11836–11837 CrossRef CAS PubMed.
- (a) K. Aoyagi, H. Nakamura and Y. Yamamoto, J. Org. Chem., 2002, 67, 5977–5980 CrossRef CAS PubMed; (b) M. A. Lowe, M. Ostovar, S. Ferrini, C. C. Chen, P. G. Lawrence, F. Fontana, A. A. Calabrese and V. K. Aggarwal, Angew. Chem., Int. Ed., 2011, 50, 6370–6374 CrossRef CAS PubMed; (c) K. Fugami, Y. Morizawa, K. Ishima and H. Nozaki, Tetrahedron Lett., 1985, 26, 857–860 CrossRef CAS; (d) N. T. Patil and Y. Yamamoto, Synlett, 2007, 1994–2005 CAS.
- B. M. Trost and S. R. Angle, J. Am. Chem. Soc., 1985, 107, 6123–6124 CrossRef CAS.
- (a) M. Hanaoka, A. Ashimori, H. Yamagishi and S. Yasuda, Chem. Pharm. Bull., 1983, 31, 2172–2175 CrossRef CAS; (b) N. Murugesan, G. Blaskó, R. D. Minard and M. Shamma, Tetrahedron Lett., 1981, 22, 3131–3134 CrossRef CAS; (c) M. Hanaoka, M. Kohzu and S. Yasuda, Chem. Pharm. Bull., 1985, 33, 4113–4115 CrossRef CAS; (d) J. B. Doughty, C. L. Lazzell and A. R. Collett, J. Am. Chem. Soc., 1950, 72, 2866–2867 CrossRef CAS.
- (a) B. Kang, A. W. Miller, S. Goyal and S. T. Nguyen, Chem. Commun., 2009, 3928–3930 RSC; (b) V. K. Yadav and V. Sriramurthy, J. Am. Chem. Soc., 2005, 127, 16366–16367 CrossRef CAS PubMed; (c) S. Gandhi, A. Bisai, B. A. B. Prasad and V. K. Singh, J. Org. Chem., 2007, 72, 2133–2142 CrossRef CAS PubMed; (d) M. K. Ghorai and K. Ghosh, Tetrahedron Lett., 2007, 48, 3191–3195 CrossRef CAS; (e) R. Maeda, R. Ishibashi, R. Kamaishi, K. Hirotaki, H. Furuno and T. Hanamoto, Org. Lett., 2011, 13, 6240–6243 CrossRef CAS PubMed; (f) H.-T. Yang, M.-L. Xing, Y.-F. Zhu, X.-Q. Sun, J. Cheng, C.-B. Miao and F.-B. Li, J. Org. Chem., 2014, 79, 1487–1492 CrossRef CAS PubMed; (g) K. Okawa, T. Kinutani and K. Sakai, Bull. Chem. Soc. Jpn., 1968, 41, 1353–1355 CrossRef CAS.
- C–C bond thermolysis of aziridines generates azomethine ylides which undergo [3 + 2] cycloaddition reactions with imines: (a) A. Boruah, B. Baruah, D. Prajapati, J. S. Sandhu and A. C. Ghosh, Tetrahedron Lett., 1996, 37, 4203–4204 CrossRef CAS; (b) M. T. Hancock and A. R. Pinhas, Tetrahedron Lett., 2003, 44, 7125–7128 CrossRef CAS; (c) X. Wua and J. Zhang, Synthesis, 2012, 44, 2147–2154 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectroscopic data for all new compounds. CCDC 1441499–1441501. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04062k |
| This journal is © The Royal Society of Chemistry 2016 |