
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective synthesis of protected L- and D-dideoxysugars and analogues via Prins cyclisations†
Ryan J.
Beattie
,
Thomas W.
Hornsby
,
Gemma
Craig
,
M. Carmen
Galan
* and
Christine L.
Willis
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: m.c.galan@bristol.ac.uk; chris.willis@bristol.ac.uk
First published on 11th January 2016
Abstract
A de novo approach for the rapid construction of orthogonally protected L- and D-deoxysugars and analogues is described. A novel and robust silicon-acetal undergoes Prins cyclisations with a series of homoallylic alcohols in high yield and excellent stereocontrol. Modified Tamao–Fleming oxidation of the resulting silyltetrahydropyrans gives direct access to deoxyglycoside analogues and the approach was showcased in the synthesis of protected L-oliose, a component of the anticancer agent aclacinomycin A.
Introduction
Deoxyglycosides are important components of a wide variety of natural products isolated from plants, fungi and bacteria including compounds exhibiting anticancer and antibiotic activities. Some have proved effective for use in the clinic (e.g. the antibiotic vancomycin and the anthracycline antibiotic altromycin B) or as lead compounds to pharmaceuticals.1 In addition, deoxyglycans are also prevalent in bacterial membrane glycoproteins, thus being a viable target for drug discovery and vaccine development.2The ability to fully understand and exploit the glycobiology of rare deoxysugars and analogues is hindered by the challenges of isolating pure materials in reasonable quantities from natural sources.1 In addition, synthetic approaches from naturally-abundant carbohydrates often require lengthy synthetic routes which make rare sugars very expensive.3 An alternative and potentially more versatile approach is the de novo asymmetric synthesis of deoxy sugars.4,5 An ideal synthetic strategy would be efficient, robust and readily adapted for the construction of a series of deoxysugars and derivatives. To this end, we have developed a new approach for the enantioselective synthesis of differentially-protected L- and D-deoxyglycosides and analogues via Prins cyclisations and its utility exemplified by the synthesis of 2,4- and 2,6-dideoxyglycosides including protected L-oliose.
Prins cyclisations involve acid-mediated reactions of homoallylic alcohols 1 (or derivatives thereof) to form an oxycarbenium ion I which cyclises via carbocation II and is subsequently trapped by a nucleophile, giving tetrahydropyrans 2 with excellent stereocontrol (Scheme 1).6 Reddy, Yadav and co-workers have used sugar derivatives as substrates in Prins cyclisations.7 Success of our proposed approach to deoxyglycosides relied upon use of a suitable electrophile bearing a hydroxyl surrogate (X in Scheme 1) which would need to be both stable to the acidic conditions required for the cyclisation and would readily be converted to a suitable functional group (e.g. acetate 3) for use in glycosylation reactions. An orthoformate was considered as the electrophile to directly introduce a 1-O-alkyl side-chain, but these have rarely been used in Prins cyclisations and are limited to substrates in which the reaction proceeds via a tertiary carbocation.8,9
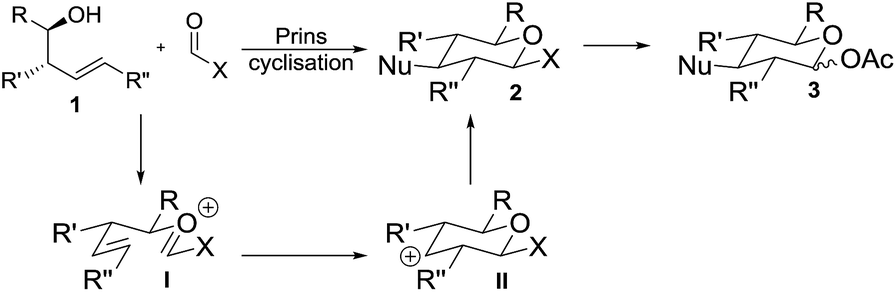 | ||
| Scheme 1 Proposed synthetic approach to deoxysugars. | ||
Results and discussion
A trialkylsilane was considered a suitable hydroxyl surrogate as, following cyclisation, a Tamao–Fleming oxidation would lead to the required acetal. Whilst dimethylphenylsilanes have been widely used,10 Hosomi and co-workers reported the benzyldimethylsilyl group (BDMS) as an attractive alternative that is readily oxidised to alcohols.11 An important criterion for our synthetic strategy was that the electrophile should be stable and readily prepared on a synthetically valuable scale. Thus, novel silyl acetal 5 was prepared in two steps and 76% overall yield via treatment of 2-lithio-1,3-dithiane12 with benzyldimethylsilyl chloride (BDMSCl) to give dithiane 4 followed by mercuric-mediated deprotection in ethanol (Scheme 2).13 The reaction was conducted on a multigram scale and the acetal is stable with no apparent decomposition after 6 months on the bench.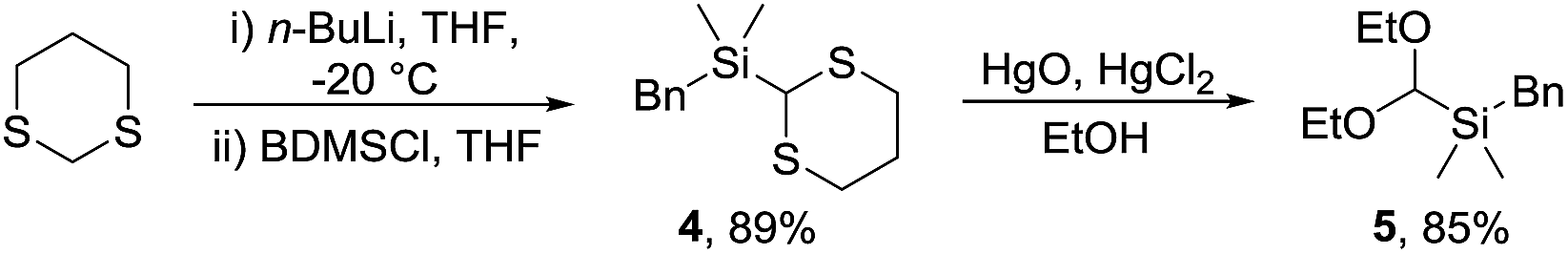 | ||
| Scheme 2 Synthesis of acetal 5. | ||
Initially, the key Prins cyclisation was optimised using the known (R)-homoallylic alcohol 6 prepared from dihydrocinnamaldehyde via a Nokami crotyl transfer reaction.14 Several methods have been reported for the introduction of oxygen nucleophiles,15 and in this case treatment of alcohol 6 and acetal 5 with trifluoroacetic acid (TFA) at 0 °C,16 then hydrolysis of the resultant ester gave alcohol 7 in 97% yield (Scheme 3). A single diastereomer was isolated in which all four substituents were equatorial.
 | ||
| Scheme 3 Cyclisation of homoallylic alcohols 6 and 8. | ||
Our ultimate targets, 2,6- and 2,4-dideoxysugar analogues, lack a substituent at C-2 and their synthesis requires a substrate with a terminal alkene. Hence (R)-homoallylic alcohol 8 (prepared via a Brown allylation17) was treated with acetal 5 under the optimised reaction conditions to give alcohol 9 in 93% yield. It is known that the mechanism of Prins cyclisations is not simple and, depending on the nature of the substrate and reaction conditions, competing processes may occur.18 To ensure that there was no loss of stereochemical integrity during the cyclisation, the enantiopurity (97.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5 e.r.) of 9 was confirmed by chiral SFC.
:2.5 e.r.) of 9 was confirmed by chiral SFC.
An alternative synthetic approach to the silyltetrahydropyrans was to incorporate the silyl moiety into the alkene coupling partner which on reaction with an aldehyde would enable the facile introduction of a range of side-chains at C-5 of the target deoxysugars (Scheme 4). Several methods were investigated for the synthesis of α-silyl-homoallylic alcohol 10via acid-mediated allylation of acetal 5 (e.g. in the presence of InCl3, AgNO3, SnCl2) but none of the required product was isolated. In contrast, when silyl acetal 5 was treated with allyltributylstannane and LiBF4 in wet acetonitrile, alcohol 10 was isolated in 61% yield.19 The TFA mediated reaction of 10 with either acetaldehyde or 3-benzyloxypropanal followed by hydrolysis of the resultant ester gave silyltetrahydropyrans 11 and 12 in 89% and 82% yields, respectively, from acetal 5. By varying the reaction conditions the analogous acetates 13 and 14 were readily prepared. Further studies are ongoing to investigate the enantioselective allylation of acetal 5.
 | ||
| Scheme 4 Alternative cyclisation strategy to silyltetrahydropyrans. | ||
The second stage of our synthetic strategy required oxidation of the benzyldimethylsilyl group; Trost and Donohoe have reported the use of tetrabutylammonium fluoride (TBAF) followed by hydrogen peroxide for similar transformations.20 Following detailed investigations we established suitable conditions for the successful oxidation of silane 7 (Scheme 5). It was evident that two steps are involved. First addition of TBAF converted silane 7 to silanol 15 which could be isolated and characterised.21
 | ||
| Scheme 5 Oxidation of silane 7. | ||
However, it was not necessary to isolate 15 as it was converted in situ to hemiacetal 16via a urea hydrogen peroxide oxidation and then directly acetylated to give 17 in 73% yield from silane 7. It proved vital to keep the temperature of the fluoride activation step in the range 0–5 °C, as at higher temperatures disiloxanes were formed from the condensation of two silanols, which were only slowly oxidised under the reaction conditions.22
To confirm that the oxidation/acetylation protocol was compatible with different protecting groups commonly used in carbohydrate chemistry, the secondary alcohols in 7 and 9 were converted to acetates (18 and 19) and benzyl ethers (20 and 21) in high yields using standard reaction conditions (Scheme 6). Oxidation of each silane gave the corresponding anomeric acetates (17, 22–24) in 64–80% isolated yield.
 | ||
| Scheme 6 Oxidation of acetoxy and benzyloxy derivatives. | ||
Next the optimised cyclisation/oxidation/acetylation strategy was applied to the preparation of protected 2,4-dideoxyglycosides (Scheme 7). Homoallylic alcohol 26 was prepared in 88% overall yield from (S)-glycidol via protection of the alcohol as silyl ether 25 and ring opening of the oxirane with vinylmagnesium bromide and CuCN. Treatment of 26 with silyl acetal 5 and TESOTf in acetic acid23 gave silyltetrahydropyran 27 in 65% yield which was readily converted to triacetate 28 as a 1:2 mixture of anomers using the oxidation/acetylation protocol. Interestingly, treatment of the mixture of homoallylic alcohol 26 and acetal 5 with TFA, our standard cyclisation conditions, gave none of the expected product, instead the analogous ethyl ether 29 was isolated.24 Some deoxysugars indeed have ethers at C-3, for example, D-oleandrose is a component of the highly potent and selective anticancer agent apoptolidin25 and L-cymarose, found in the DNA-helicase inhibitor, heliquinomycin26 and so this unexpected result has potential significant synthetic value.
 | ||
| Scheme 7 Preparation of 2,4-dideoxysugar 28. | ||
To access orthogonally protected 2,4-dideoxyglycosides, benzyl-protected tetrahydropyran 30 was prepared via a similar protection/vinyl addition/cyclisation strategy from (S)-glycidol benzyl ether in 71% overall yield (Scheme 8). Oxidation of 30 gave diacetate 31 which subsequently was used to glycosylate cyclohexanol in the presence of BF3·OEt2 giving 32 as exclusively the α-anomer in 72% yield. The synthetic approach was extended to 2,4-dideoxyglycosides with an axial C-3 oxygenated substituent via hydrolysis of acetate 30 and Mitsunobu inversion to give 33 which was oxidised to protected glycoside 34 in 75% yield.
 | ||
| Scheme 8 Synthesis of protected 2,4-dideoxyglycosides 31 and 34. | ||
Next we turned out attention to the synthesis of protected 2,6-dideoxysugars as all diastereoisomers of D- and L-2,6-dideoxyhexoses have been found in biologically active natural products e.g.D-olivose is a component of angucylcline antibiotic landomycin A27 whilst D-digitoxose is present in the steroidal glycoside digitoxin (Fig. 1).
 | ||
| Fig. 1 Examples of 2,6-dideoxyhexoses. | ||
Initial studies revealed that whilst homoallylic alcohol 35 was readily prepared via Brown allylation of allyl ethyl ether,28 reaction of 35 with silyl acetal 5 under our standard TFA conditions gave the 5-membered ring aldehyde 36 in 46% yield (Scheme 9). Aldehyde 36 is likely to be formed via a Prins-pinacol reaction29 involving oxonia-Cope rearrangement of oxycarbenium ion III to enol ether IV followed by cyclisation to tetrahydrofuran V and finally O-alkyl cleavage to generate the carbonyl group.30 Hence to favour formation of a tetrahydropyran over a tetrahydrofuran we reasoned that an electron withdrawing group rather than an ether was required and a carbamate protecting group was selected.31
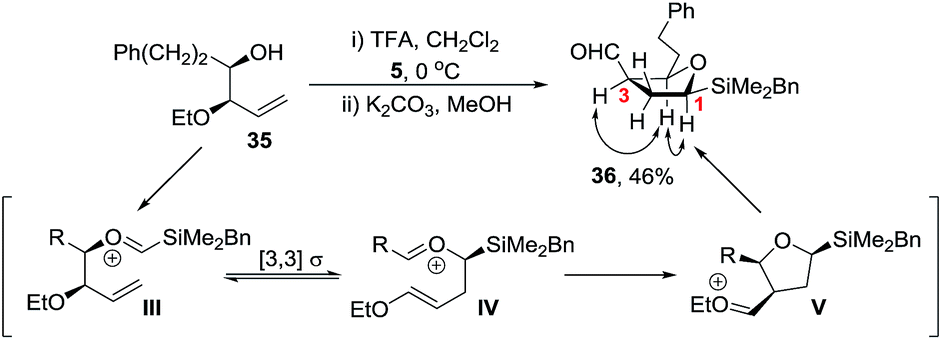 | ||
| Scheme 9 Cyclisation to tetrahydrofuran 36 and nOe correlations. | ||
Thus an asymmetric synthesis of homoallylic alcohol 40 was required which could be readily adapted for both the L- or D-protected 2,6-dideoxysugars since, for example, L-oliose is a component of aclarubicin, clinically used for the treatment of acute leukaemias,32 whilst D-oliose is present in the antitumour drugs mithramycin and chromocyclomycin,33 as well as the HIV-inhibitor durhamycin A.34
Singh and Guiry reported that Sharpless asymmetric epoxidation (SAE) of divinylcarbinol, followed by Mitsunobu inversion of the resulting alcohol gives epoxide 37 (Scheme 10).35 Importantly, choice of (−)-DIPT or (+)-DIPT in the SAE step allows access to the D- and L-series, respectively. Protection of alcohol 37 with N,N-diisopropylcarbamoyl chloride gave a mixture of chlorohydrin 38 and the required epoxide 39. Chlorohydrin 38 was readily converted to 39 by treatment with NaOH in THF at room temperature within a matter of minutes. Reductive ring opening of the oxirane with DIBALH gave mono-protected syn allylic diol 40 which cyclised with acetal 5 to give the required tetrahydropyran 41 with an equatorial C-3 alcohol and the axial C-4 protected hydroxyl. Finally oxidation of silane 41 gave protected L-oliose 42 in 77% yield as a 1:1 mixture of anomers. The methodology could be extended to the synthesis of L-olivose via protected alcohol 44 which was prepared from epoxide 43 using the same conditions as for assembly of the diastereomer 40 (Scheme 11). Interestingly, reaction of 44 with acetal 5 under the standard conditions gave a mixture of products due to migration of the carbamoyl group but on reduction of the mixture with LiAlH4, diol 45 was isolated in 57% yield. It is possible that neighbouring group participation by the carbamoyl group traps the intermediate carbocation I in the Prins cyclisation giving II and resulting in migration of the carbomyl group but this has not been proven.
 | ||
| Scheme 10 Enantioselective synthesis of protected L-oliose 42. | ||
 | ||
| Scheme 11 Enantioselective synthesis of L-olivose precursor 45. | ||
Conclusions
In conclusion, a de novo approach for the rapid construction of a series of orthogonally protected L- and D-dideoxyglycosides and analogues is described from simple starting materials. A stable acetal 5 was prepared in two high yielding steps and used in a series of acid-mediated Prins cyclisations with different homoallylic alcohols to give the corresponding tetrahydropyrans in good yield and excellent diastereoselectivity. These reactions are readily performed on gram scales. A modified Tamao–Fleming oxidation/acetylation protocol gave the target 2,4-dideoxysugars with an acetyl group at the anomeric position. Extending the utility of the new methodology to the synthesis of 2,6-dideoxysugars revealed the importance of the choice of protecting group to avoid formation of tetrahydrofuranals. The enantioselective synthesis of protected L-oliose is described using N,N-diisopropylcarbamoyl as a protecting group. Silane 41 has potential value for the synthesis of other 2,6-dideoxyhexoses for example methylation of the free hydroxyl group will lead to protected L-diginose while Mitsunobu inversion will give L-boivinose derivatives and subsequent methylation to L-sarmentose and these investigations are ongoing in our laboratories.Acknowledgements
We thank the Bristol Chemical Synthesis Centre for Doctoral Training for the provision of PhD studentships EP/G0367641/1 (RJB, TWH) and EPSRC EP/L001926/1 and EP/J002542/1(MCG).Notes and references
- For reviews see: (a) A. C. Weymouth-Wilson, Nat. Prod. Rep., 1997, 14, 99–110 RSC; (b) V. Křen and T. Řezanka, FEMS Microbiol. Rev., 2008, 32, 858 CrossRef PubMed; (c) A. Kirschning, A. F. W. Bechthold and J. Rohr, Bioorganic Chemistry Deoxysugars, Polyketides and Related Classes: Synthesis, Biosynthesis, Enzymes, in Topics in Current Chemistry, Springer Berlin/Heidelberg, 1997, vol. 188, pp. 1–84 Search PubMed.
- For recent reviews on the importance of oligosaccharides see: (a) P. Stallforth, B. Lepenies, A. Adibekian and P. H. Seeberger, J. Med. Chem., 2009, 52, 5561–5577 CrossRef CAS PubMed; (b) M. C. Galan, D. Benito-Alifonso and G. M. Watt, Org. Biomol. Chem., 2011, 9, 3598–3610 RSC; (c) M. C. Galan, P. Dumy and O. Renaudet, Chem. Soc. Rev., 2013, 42, 4599–4612 RSC.
- Recently some elegant strategies have been reported for the synthesis of D-deoxyglycosides starting from readily available carbohydrates, e.g. M. Emmadi and S. S. Kulkarni, Org. Biomol. Chem., 2013, 11, 3098–3102 CAS.
- For selected reviews see: (a) A. Kirschning, M. Jesberger and K. U. Schoning, Synthesis, 2001, 507–540 CrossRef CAS; (b) R. M. De Lederkremer and C. Marino, in Advances in Carbohydrate Chemistry and Biochemistry, vol. 188, ed. D. Horton, 2008, vol. 61, pp. 143–216 Search PubMed; (c) A. Z. Aljahdali, P. Shi, Y. Zhong and G. A. O'Doherty, in Advances in Carbohydrate Chemistry and Biochemistry, ed. D. Horton, 2013, vol. 69, pp. 55–123 Search PubMed.
- For selected examples see: (a) H. Guo and G. A. O'Doherty, Angew. Chem., Int. Ed., 2007, 46, 5206–5208 CrossRef CAS PubMed; (b) R. S. Babu, Q. Chen, S.-W. Kang, M. Zhou and G. A. O'Doherty, J. Am. Chem. Soc., 2012, 134, 11952–11955 CrossRef CAS PubMed; (c) D. Leonori and P. H. Seeberger, Beilstein J. Org. Chem., 2013, 9, 332–341 CrossRef CAS PubMed; (d) R. Pragani, P. Stallforth and P. H. Seeberger, Org. Lett., 2010, 12, 1624–1627 CrossRef CAS PubMed.
- For reviews see: (a) I. M. Pastor and M. Yus, Curr. Org. Chem., 2007, 11, 925–957 CrossRef CAS; (b) C. Olier, M. Kaafarani, S. Gastaldi and M. P. Bertrand, Tetrahedron, 2010, 66, 413–445 CrossRef CAS.
- (a) B. V. S. Reddy, A. V. Ganesh, A. S. Krisna, G. G. K. S. N. Kumar and J. S. Yadav, Tetrahedron Lett., 2011, 52, 3342–3344 CrossRef CAS; (b) J. S. Yadav, B. V. S. Reddy, A. P. Singh, D. N. Chaya and D. Chatterjee, Tetrahedron Lett., 2010, 51, 1475–1478 CrossRef CAS.
- (a) F. Perron-Sierra, M. A. Promo, V. A. Martin and K. F. Albizati, J. Org. Chem., 1991, 56, 6188–6199 CrossRef CAS; (b) G. E. Keck, J. A. Covel, T. Schiff and T. Yu, Org. Lett., 2002, 4, 1189–1192 CrossRef CAS PubMed.
- Prins cyclisations have been used to assemble tetrahydropyrans with a hydroxymethyl side-chain which then can be oxidatively cleaved to give lactones, see for example, J. S. Yadav, M. S. Reddy and A. R. Prasad, Tetrahedron Lett., 2005, 46, 2133–2136 CrossRef CAS.
- K. Tamao, Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci., 2008, 84, 123–133 CrossRef CAS PubMed.
- K. Miura, T. Hondo, T. Takahashi and A. Hosomi, Tetrahedron Lett., 2000, 41, 2129–2132 CrossRef CAS.
- (a) E. J. Corey, D. Seebach and R. Freedman, J. Am. Chem. Soc., 1967, 89, 434–436 CrossRef CAS; (b) A. G. Brook, J. M. Duff, P. F. Jones and N. R. Davis, J. Am. Chem. Soc., 1967, 89, 431–434 CrossRef CAS.
- A. Degl'Innocenti, P. Scafato, A. Capperucci, L. Bartoletti, C. Spezzacatena and R. Ruzziconi, Synlett, 1997, 361, 362 Search PubMed.
- (a) J. Nokami, M. Ohga, H. Nakamoto, T. Matsubara, I. Hussain and K. Kataoka, J. Am. Chem. Soc., 2001, 123, 9168–9169 CrossRef CAS PubMed; (b) C. S. Barry, N. Bushby, J. R. Harding and C. L. Willis, Org. Lett., 2005, 7, 2683–2686 CrossRef CAS PubMed.
- (a) J. J. Jaber, K. Mitsui and S. D. Rychnovsky, J. Org. Chem., 2001, 66, 4679–4686 CrossRef CAS PubMed; (b) E. H. Al-Mutairi, S. R. Crosby, J. Darzi, J. R. Harding, R. A. Hughes, C. D. King, T. J. Simpson, R. W. Smith and C. L. Willis, Chem. Commun., 2001, 835–836 RSC; (c) K. Tadpetch and S. D. Rychnovsky, Org. Lett., 2008, 10, 4839–4842 CrossRef CAS PubMed; (d) J. S. Yadav, B. V. Subba Reddy, G. G. K. S. Narayana Kumar and S. Aravind, Synthesis, 2008, 395–400 CrossRef CAS; (e) J. S. Yadav, B. V. S. Reddy, A. V. Ganesh and G. G. K. S. Narayana Kumar, Tetrahedron Lett., 2010, 51, 2963–2966 CrossRef CAS.
- (a) C. S. J. Barry, S. R. Crosby, J. R. Harding, R. A. Hughes, C. D. King, G. D. Parker and C. L. Willis, Org. Lett., 2003, 5, 2429–2432 CrossRef CAS PubMed; (b) C. S. Barry, J. D. Elsworth, P. T. Seden, N. Bushby, J. R. Harding, R. W. Alder and C. L. Willis, Org. Lett., 2006, 8, 3319–3322 CrossRef CAS PubMed; (c) P. T. Seden, J. P. H. Charmant and C. L. Willis, Org. Lett., 2008, 10, 1637–1640 CrossRef CAS PubMed.
- H. C. Brown and P. K. Jadhav, J. Am. Chem. Soc., 1983, 105, 2092–2093 CrossRef CAS.
- (a) S. Marumoto, J. J. Jaber, J. P. Vitale and S. D. Rychnovsky, Org. Lett., 2002, 4, 3919–3922 CrossRef CAS PubMed; (b) R. Jasti and S. D. Rychnovsky, J. Am. Chem. Soc., 2006, 128, 13640–13648 CrossRef CAS PubMed; (c) C. S. Barry, N. Bushby, J. R. Harding, R. A. Hughes, G. D. Gregory, R. Roe and C. L. Willis, Chem. Commun., 2005, 3727–3729 RSC.
- B. H. Lipshutz and D. F. Harvey, Synth. Commun., 1982, 12, 267 CrossRef CAS.
- (a) B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2005, 127, 17644–17655 CrossRef CAS PubMed; (b) B. M. Trost, M. R. Machacek and Z. T. Ball, Org. Lett., 2003, 5, 1895–1898 CrossRef CAS PubMed; (c) T. J. Donohoe, P. C. M. Winship, B. S. Pilgrim, D. S. Walter and C. K. A. Callens, Chem. Commun., 2010, 46, 7310–7312 RSC.
- (a) S. E. Denmark, R. F. Sweis and D. Wehrli, J. Am. Chem. Soc., 2004, 126, 4865–4875 CrossRef CAS PubMed; (b) S. E. Denmark, D. Wehrli and J. Y. Choi, Org. Lett., 2000, 2, 2491–2494 CrossRef CAS PubMed.
- E. J. Rayment, N. Summerhill and E. A. Anderson, J. Org. Chem., 2012, 77, 7052–7060 CrossRef CAS PubMed.
- S. K. Woo, M. S. Kwon and E. Lee, Angew. Chem., Int. Ed., 2008, 47, 3242–3244 CrossRef CAS PubMed.
- For selected examples where the source of nucleophilic trapping originates from the electrophile, see: (a) K. Zheng, X. Liu, S. Qin, M. Xie, L. Lin, C. Hu and X. Feng, J. Am. Chem. Soc., 2012, 134, 17564–17573 CrossRef CAS PubMed; (b) J. S. Yadav, B. V. Subba Reddy, G. Mahesh Kumar and C. V. S. R. Murthy, Tetrahedron Lett., 2001, 42, 89–91 CrossRef CAS.
- Y. Hayakawa, J. W. Kim, H. Adachi, K. Shin-ya, K.-I. Fujita and H. Seto, J. Am. Chem. Soc., 1998, 120, 3524–3525 CrossRef CAS.
- M. Brasholz, S. Sörgel, C. Azap and H.-U. Reißig, Eur. J. Org. Chem., 2007, 3801–3814 CrossRef CAS.
- R. T. Crow, B. Rosenbaum, R. Smith III, Y. Guo, K. S. Ramos and G. A. Sulikowski, Bioorg. Med. Chem. Lett., 1999, 9, 1663–1666 CrossRef CAS PubMed.
- H. C. Brown, P. K. Jadhav and K. S. Bhat, J. Am. Chem. Soc., 1988, 110, 1535–1538 CrossRef CAS.
- Prins-pinacol reactions have been used in the synthesis of tetrahydrofurans, (a) N. Hanaki, J. T. Link, D. W. C. MacMillan, L. E. Overman, W. G. Trankle and J. A. Wurster, Org. Lett., 2000, 2, 223–226 CrossRef CAS PubMed; (b) L. E. Overman and L. D. Pennington, J. Org. Chem., 2003, 68, 7143–7157 CrossRef CAS PubMed.
- (a) L. D. M. Lolkema, H. Hiemstra, C. Semeyn and W. N. Speckamp, Tetrahedron, 1994, 50, 7115–7128 CrossRef CAS; (b) L. D. M. Lolkema, C. Semeyn, L. Ashek, H. Hiemstra and W. N. Speckamp, Tetrahedron, 1994, 50, 7129–7140 CrossRef CAS.
- Carbamates have been used to good effect in intramolecular Samurai cyclisations, B. Leroy and I. E. Markó, Tetrahedron Lett., 2001, 42, 8685–8688 CrossRef CAS.
- W. Kersten, H. Kersten, W. Szybalski and M. Fiandt, Biochemistry, 1966, 5, 236–244 CrossRef CAS PubMed.
- H. Jayasuriya, R. B. Lingham, P. Graham, D. Quamina, L. Herranz, O. Genilloud, M. Gagliardi, R. Danzeisen, J. E. Tomassini, D. L. Zink, Z. Q. Guan and S. B. Singh, J. Nat. Prod., 2002, 65, 1091–1095 CrossRef CAS.
- W. Lu, C. Leimkuhler, M. Oberthür, D. Kahne and C. T. Walsh, Biochemistry, 2004, 43, 4548–4558 CrossRef CAS PubMed.
- S. Singh and P. J. Guiry, J. Org. Chem., 2009, 74, 5758–5761 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04144a |
| This journal is © The Royal Society of Chemistry 2016 |