
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric [3 + 2] cycloaddition of donor–acceptor aziridines with aldehydes via carbon–carbon bond cleavage†
Yuting
Liao
a,
Xiaohua
Liu
*a,
Yu
Zhang
a,
Yali
Xu
a,
Yong
Xia
a,
Lili
Lin
a and
Xiaoming
Feng
*ab
aKey Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: liuxh@scu.edu.cn; xmfeng@scu.edu.cn; Fax: +86 28 85418249; Tel: +86 28 85418249
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), China
First published on 23rd February 2016
Abstract
An enantioselective [3 + 2] annulation of donor–acceptor aziridines with aldehydes has been realized using a Nd(OTf)3/N,N′-dioxide/LiNTf2 catalyst system, providing various chiral cis-1,3-oxazolidines in moderate to good yields with high levels of stereocontrol. A relay catalytic process is proposed where LiNTf2 promotes the formation of azomethine ylide intermediates, and a chiral Nd(III)–N,N′-dioxide complex accelerates the asymmetric cycloaddition.
Introduction
The cycloaddition of aziridines is an attractive method for obtaining various nitrogen-containing heterocycles.1 Among them, highly enantioselective [3 + 2] cycloadditions via C–N bond cleavage of racemic aziridines have been realized. For example, the asymmetric cycloadditions of racemic vinyl aziridines with isocyanates and α,β-unsaturated ketones have been reported by Trost and Hou, respectively (Scheme 1a).2 Very recently, Wang realized the chiral copper(I) complex catalyzed [3 + 2] annulation of racemic 2-aryl-N-tosylaziridines with indoles (Scheme 1b).3 As a type of donor–acceptor (DA) variation, 2,3-diester aziridines favor C–C bond heterolytic cleavage, and the formed transient azomethine ylide intermediates can undergo cycloadditions with various dipolarophiles (Scheme 1c). These transformations can be promoted by Lewis acids under mild conditions in comparison to photochemically or thermally induced conditions.4 A pioneering example is the ZnCl2 catalyzed cycloaddition between N-aryl-2,3-diester aziridines and electron-rich alkenes developed by Johnson.5a The Zhang group and others have further explored Lewis acid accelerated cycloadditions of N-tosylaziridinedicarboxylates with aldehydes,5b,c imines,5c,d electron-rich alkenes,5e alkynes,5f 2,3-disubstituted indoles,5g,h donor–acceptor cyclopropanes,5i isocyanides5j and heterocumulenes.5k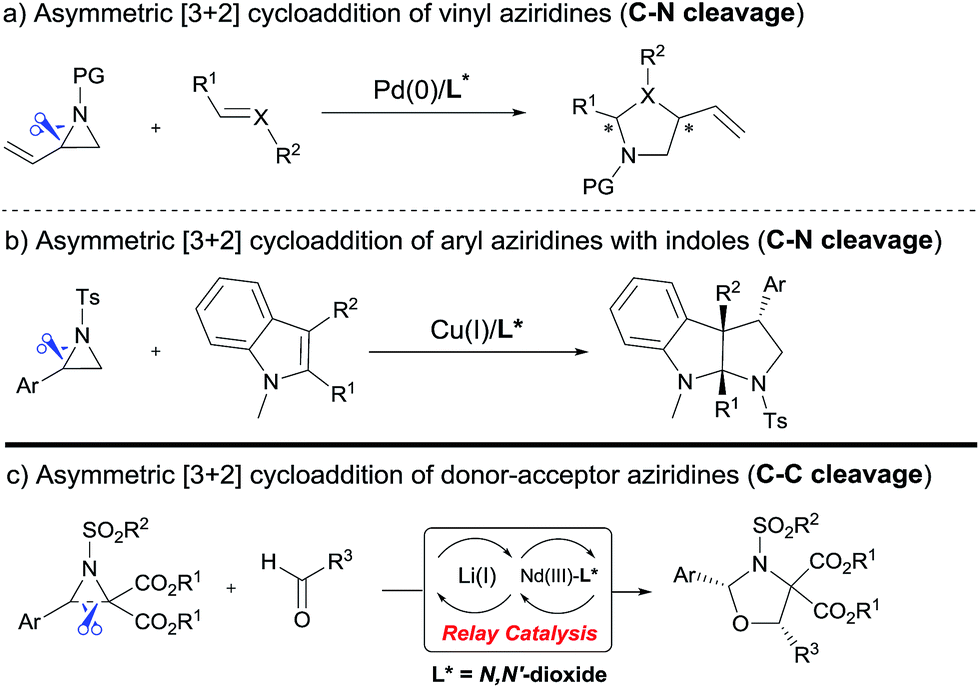 | ||
| Scheme 1 Asymmetric [3 + 2] cycloadditions of racemic aziridines. | ||
Despite these important racemic examples, catalytic asymmetric [3 + 2] cycloadditions of DA aziridines are rare, with the results yielding no more than 70% ee.5b,d–f Compared with their cousins, DA cyclopropanes6 and DA oxiranes,7 enantioselective [3 + 2] cycloadditions of aziridines are more difficult with considerable challenges: (1) the azomethine ylide via C–C bond cleavage is transient, being further converted to 2-amino malonate and aldehyde in the presence of unavoidable trace amounts of water that prevent cycloadditions.5a,e (2) DA aziridine has a relatively congested structure tethering four substituents on a three-membered ring, which hampers its interaction with a chiral catalyst. Additionally, the competitive coordination of aldehydes to the chiral catalyst is also disadvantageous to the yield and stereocontrol. Therefore, asymmetric cycloadditions of DA aziridines require a powerful catalytic system enabling both C–C bond cleavage and enantiocontrol. Here we report a relay catalyst system of Nd(OTf)3/N,N′-dioxide/LiNTf2 for the asymmetric cycloaddition of DA aziridines with aldehydes (Scheme 1c).8 The relay approach is to use an achiral metal salt to accelerate the formation of the azomethine ylide intermediate, which is then transformed into a chiral catalytic environment to undergo asymmetric cycloaddition. Chiral cis-1,3-oxazolidines, which have emerged as crucial structural units in chiral catalysts and biologically active compounds,9 could be formed in moderate to high yield with good enantioselectivity.
Results and discussion
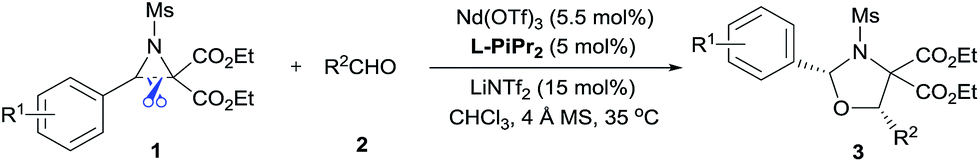
We selected the model [3 + 2] annulation of DA aziridine 1a with benzaldehyde 2a as our starting point (see Table 1). In the presence of 10 mol% N,N′-dioxide L-PiPr2 and 4 Å MS, metal salts that have previously been established as efficient Lewis acids for catalytic racemic transformations were tested (entries 1–4). Disappointingly, low yields and enantioselectivities were obtained with Sc(OTf)3, Ni(ClO4)2, Zn(OTf)2 and La(OTf)3 due to the decomposition of aziridine 1a. La(OTf)3/L-PiPr2 provided the cis-1,3-oxazolidines 3aa in 14% yield with 36% ee in toluene at 35 °C. We surmised whether extra Lewis acid could assist in furnishing the desired transformation.7 Encouragingly, the addition of LiNTf2 (10 mol%) was indeed beneficial for both the reactivity and enantioselectivity (entry 5). After examining a series of lanthanide metal salts (see the ESI†), Nd(OTf)3 was selected as the best one, resulting in 30% yield and 71% ee (entry 6). Next, the systematic modification of N,N′-dioxides through alteration of their amino acid backbones and amide moieties, as well as the length of the linkage, showed L-PiPr2 was the optimal ligand (see the ESI†). Empirically changing the solvent to CHCl3 instead of toluene improved the enantioselectivity to 85% ee (entry 7). Optimization of other reaction conditions by adjusting the ratio of Nd(OTf)3 to L-PiPr2 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and increasing the amount of LiNTf2, 4 Å MS and aldehyde to speed up the reaction provided the product 3aa in a good yield of 65% with 87% ee (entry 8). It should be considered that the azomethine ylide intermediate derived from DA aziridine was very unstable, thereby a 2-fold excess of benzaldehyde 2a was necessary to achieve a satisfying outcome. Furthermore, chromatography purification using basic Al2O3 as the stationary phase instead of silica gel, led to an encouraging outcome with 68% yield and 91% ee (entry 9). Basic Al2O3 might prevent the partial racemization of chiral cis-1,3-oxazolidines that occurs in silica gel. Notably, L-PiPr2 at a loading of 2.5 mol% was able to catalyse the reaction efficiently without any changes to the outcome (entry 10).
:1) and increasing the amount of LiNTf2, 4 Å MS and aldehyde to speed up the reaction provided the product 3aa in a good yield of 65% with 87% ee (entry 8). It should be considered that the azomethine ylide intermediate derived from DA aziridine was very unstable, thereby a 2-fold excess of benzaldehyde 2a was necessary to achieve a satisfying outcome. Furthermore, chromatography purification using basic Al2O3 as the stationary phase instead of silica gel, led to an encouraging outcome with 68% yield and 91% ee (entry 9). Basic Al2O3 might prevent the partial racemization of chiral cis-1,3-oxazolidines that occurs in silica gel. Notably, L-PiPr2 at a loading of 2.5 mol% was able to catalyse the reaction efficiently without any changes to the outcome (entry 10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal salt | Additive | Solvent | Yieldb (%) | eec (%) |
|
a Unless otherwise noted, the reactions were performed with metal salt/L* (10 mol%, 1:1), 4 Å MS (20 mg), aziridine 1a (0.1 mmol) and PhCHO 2a (0.15 mmol) in solvent (1.0 mL) under nitrogen at 35 °C for 12 h.
b Isolated yield by silica gel chromatography.
c The ratio of cis/trans was >19:1 determined by 1H NMR spectroscopy and ee values were determined by chiral HPLC analysis.
d LiNTf2 (10 mol%) was used.
e Nd(OTf)3/L-PiPr2 (5 mol%, 2:1), LiNTf2 (15 mol%), 4 Å MS (100 mg), aziridine 1a (0.1 mmol) and PhCHO 2a (0.2 mmol) in CHCl3 (0.75 mL).
f Isolation by flash chromatography using basic Al2O3.
g Nd(OTf)3/L-PiPr2 (2.5 mol%, 2:1), LiNTf2 (15 mol%).
|
|||||
| 1 | Sc(OTf)3 | — | Toluene | 45 | 0 |
| 2 | Ni(ClO4)2·6H2O | — | Toluene | 20 | −22 |
| 3 | Zn(OTf)2 | — | Toluene | Trace | — |
| 4 | La(OTf)3 | — | Toluene | 14 | 36 |
| 5d | La(OTf)3 | LiNTf2 | Toluene | 24 | 58 |
| 6d | Nd(OTf)3 | LiNTf2 | Toluene | 30 | 71 |
| 7d | Nd(OTf)3 | LiNTf2 | CHCl3 | 35 | 85 |
| 8e | Nd(OTf)3 | LiNTf2 | CHCl3 | 65 | 87 |
| 9e,f | Nd(OTf)3 | LiNTf2 | CHCl3 | 68 | 91 |
| 10e–g | Nd(OTf)3 | LiNTf2 | CHCl3 | 68 | 91 |

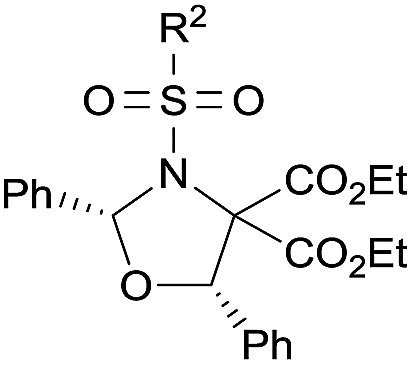
Examples of asymmetric [3 + 2] annulations of DA aziridines 1 with benzaldehyde 2a promoted by the Nd(OTf)3/LiNTf2/L-PiPr2 catalyst system are summarized in Table 2. For the ester moieties, the methyl group had less influence on the outcome, while the isopropyl group was detrimental to both the reactivity and enantioselectivity (entries 1–3). Other benzenesulfonyl motifs were well tolerated except for those with 2-methyl and 2-nitro substituents, which slowed down the reaction and diminished the yield and ee value, perhaps as a result of the steric hindrance at the ortho-position (entries 4–8). It was noteworthy that the methanesulfonyl substituent could make the result rise up to 77% yield and 95% ee (entry 9). Moreover, the 2-trimethylsilylethanesulfonyl group, which proved to be readily cleaved under mild conditions,10 was also a tolerable substituent for the catalyst system (entry 10). Subsequently, a variety of aryl aziridines with electron-withdrawing substituents provided the cycloadducts in 66–98% yields and 87–94% ee (entries 11–17). The position of the substituent had some influence on the enantioselectivity, and the ortho-substituted analogue required an elongated reaction time and gave a reduced ee value (entries 11–13). The aryl substituent could be replaced by biphenyl or naphthalene-2-yl groups and the desired products were obtained in high yields and enantioselectivities, albeit with the need for a 3-fold excess of benzaldehyde 2a (entries 18–19). This large excess of benzaldehyde was beneficial for accomplishing the cycloadditions with more reactive azomethine ylide intermediates of DA aziridines 1r and 1s. Also, it could avoid the background reactions of DA aziridines 1r and 1s with the aldehydes from the decomposition. The absolute configuration of the product 3sa was determined to be (2R, 5S) by X-ray crystal analysis.11 The alkyl substituted aziridine was inert and none of the corresponding 1,3-oxazolidine was generated (entry 20). This result should be caused by the poor stability of its azomethine ylide intermediate compared to the aryl substituted ones. Overall, only cis-1,3-oxazolidines were detected.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 3 | t (h) | Yieldb (%) | eec (%) | |
|
a Unless otherwise noted, the reaction conditions were the same as entry 10, Table 1.
b Isolated yield by flash chromatography using basic Al2O3.
c Determined by chiral HPLC analysis.
d The absolute configuration was assigned as (2R, 5S) by CD analysis and inferred from 3sa whose absolute configuration was determined by X-ray analysis.11
e The yield was determined by 1H NMR.
f Nd(OTf)3/L-PiPr2 (5 mol%, 2:1).
g PhCHO (0.3 mmol, 3 equiv.).
|
|||||
| 1 |
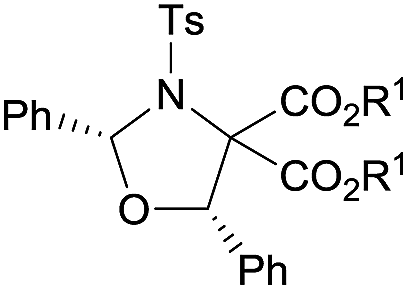
|
Et (3aa) | 12 | 68 | 91 |
| 2d | Me (3ba) | 12 | 62 | 90 | |
| 3d | iPr (3ca) | 14 | 40 | 72 | |
| 4d |
|
4-ClC6H4 (3da) | 12 | 74 | 89 |
| 5 | C6H5 (3ea) | 12 | 70 | 90 | |
| 6 | 4-MeOC6H4 (3fa) | 12 | 60 | 90 | |
| 7 | 2-MeC6H4 (3ga) | 38 | 54 | 76 | |
| 8 | 2-O2NC6H4 (3ha) | 92 | 6e | 62 | |
| 9d | Me (3ia) | 12 | 77 | 95 | |
| 10d |
|
12 | 66 | 93 | |
| 11d |
|
4-ClC6H4 (3ka) | 24 | 80 | 91 |
| 12d | 3-ClC6H4 (3la) | 24 | 78 | 92 | |
| 13d | 2-ClC6H4 (3ma) | 40 | 71 | 88 | |
| 14d | 4-BrC6H4 (3na) | 24 | 70 | 93 | |
| 15d | 4-FC6H4 (3oa) | 24 | 66 | 94 | |
| 16d,f | 4-F3CC6H4 (3pa) | 40 | 98 | 91 | |
| 17 | 4-O2NC6H4 (3qa) | 36 | 84 | 87 | |
| 18g | 4-PhC6H4 (3ra) | 12 | 70 | 93 | |
| 19d,g |
|
12 | 94 | 93 | |
| 20 | Cyclohexyl (3ua) | 36 | 0 | — | |

The reaction also tolerated diverse aromatic aldehydes (see Table 3). In order to reach satisfactory reactivity and enantioselectivity, the amount of Nd(OTf)3, L-PiPr2 and aldehyde needed to be adjusted. A variety of benzaldehydes with electron withdrawing or donating substituents provided cis-1,3-oxazolidines in moderate to high yields (38–73%) with good enantioselectivities (84–94% ee) (entries 1–6). Remarkably, heteroaromatic aldehydes (such as furan-2-carbaldehyde and thiophene-3-carbaldehyde) delivered the desired products in high yields (84–93%) and enantioselectivities (91–94% ee) (entries 7–8). Additionally, cinnamaldehyde displayed high reactivity but gave only a moderate ee value (entry 9). Only trace amounts of the expected adduct were detected when using an aliphatic aldehyde (entry 10). Overall, only cis-1,3-oxazolidines were attained. To evaluate the synthetic utility of this catalyst system, a gram-scale preparation of 3kh was undertaken, resulting in an outcome of 93% yield, >19:1 dr and 93% ee.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 |
1:2 |
t (h) | Yieldb (%) | eec (%) |
|
a Unless otherwise noted, the reactions were performed with Nd(OTf)3 (5.5 mol%), L-PiPr2 (5 mol%), LiNTf2 (15 mol%), 4 Å MS (100 mg), aziridine 1 (0.1 mmol) and aldehyde 2 in CHCl3 (0.75 mL) under nitrogen at 35 °C for the indicated time.
b Isolated yield by flash chromatography using basic Al2O3.
c Determined by chiral HPLC analysis.
d Nd(OTf)3/L-PiPr2 (2.5 mol%, 2:1) was used.
e Nd(OTf)3/L-PiPr2 (5 mol%, 2:1) was used.
f The absolute configuration was assigned as (2R, 5S) by CD analysis referred to 3sa.
g With the absolute configuration of (2R, 5R).
|
||||||
| 1d | H | 4-ClC6H4 | 1:3 |
24 | 70 (3ib) | 90 |
| 2e | 3-Cl | 3-ClC6H4 | 1:2 |
66 | 38 (3lc) | 89 |
| 3f | 4-Cl | 4-MeC6H4 | 1:2 |
24 | 51 (3kd) | 84 |
| 4f | 4-Cl | 3-MeC6H4 | 1:1.5 |
24 | 73 (3ke) | 94 |
| 5f | 3-Me | 3-MeC6H4 | 1:2 |
12 | 51 (3te) | 92 |
| 6f | 4-Cl | 3-MeOC6H4 | 1:2 |
24 | 70 (3kf) | 93 |
| 7f | 4-Cl | 3-Thienyl | 1:1.5 |
27 | 84 (3kg) | 91 |
| 8g | 4-Cl | 2-Furyl | 1:1.5 |
27 | 93 (3kh) | 94 |
| 9f | 4-Cl |

|
1:2 |
30 | 84 (3ki) | 55 |
| 10 | 4-Cl | Cyclohexyl | 1:2 |
40 | Trace | — |
To gain insights into the reaction process, several control experiments were conducted. In the standard reaction conditions, excess aldehyde 2h was used and the desired cis-product 3kh was given in 93% yield and 94% ee (Table 3, entry 8). When the ratio of aziridine 1k to aldehyde 2h was increased, the enantioselectivity of the product 3kh was maintained and the remaining aziridine 1k was racemic (Fig. 1a),12a indicating that the reaction occurs through the formation of an azomethine ylide intermediate.13
 | ||
| Fig. 1 (a) ee traces of the product 3kh and the substrate 1k of the reaction. (b) Reaction time profile of the reactions with or without LiNTf2. | ||
To better understand the catalyst system, HRMS experiments were further explored. ESI-MS species assigned to [L-PiPr2 + Li+]+ and [L-PiPr2 + Nd3+ + OTf−]2+ were detected from separate mixtures of the ligand with each of the metal salts. However, upon mixing the three components together (L-PiPr2/Nd(OTf)3/LiNTf2 = 1:1.1:3), only signals related to the complexes of L-PiPr2/Nd(III) were observed (see the ESI†), suggesting the high stereocontrol might originate from the chiral Nd(III) complex of L-PiPr2.
Next, the primary actions of the catalyst components were studied. When mixtures of NdCl3/AgNTf2/L-PiPr2 or LiNTf2/L-PiPr2 were used, the desired [3 + 2] cycloadducts were not observed but byproducts were detected (Scheme 2a and b). In addition, the utilization of NaNTf2 in place of LiNTf2 deteriorated the results (41% yield/73% ee vs. 77% yield/95% ee, Scheme 2c), which implies that the lithium salt does not merely provide a counter-anion and participate in the cycloaddition step. A reaction time profile of the transformation between aziridine 1i and benzaldehyde 2a shows that the cis-1,3-oxazolidine 3ia formed gradually accompanied by the consumption of aziridine 1i. It is also evident from Fig. 1b that the reaction rate was faster in the presence of, rather than the absence of, LiNTf2. A 1H NMR spectroscopy study of aziridine 1r revealed that LiNTf2 could obviously accelerate the generation of 4-PhC6H4CHO and MsNHCH(CO2Et)2, which forms when the azomethine ylide is trapped by water. Therefore, it is reasonable to conclude that LiNTf2 could promote the cleavage of the aziridine to generate the azomethine ylide intermediate (see the ESI†).14 Moreover, competition reactions show that the rate of cycloaddition was more sensitively influenced by the electronic nature of the aldehydes (Scheme 2d).12b,15 This implies that the aldehydes function as electron rich dipolarphiles and the cycloaddition step is more likely to be the rate-determining step in comparison with the carbon–carbon cleavage in this case.
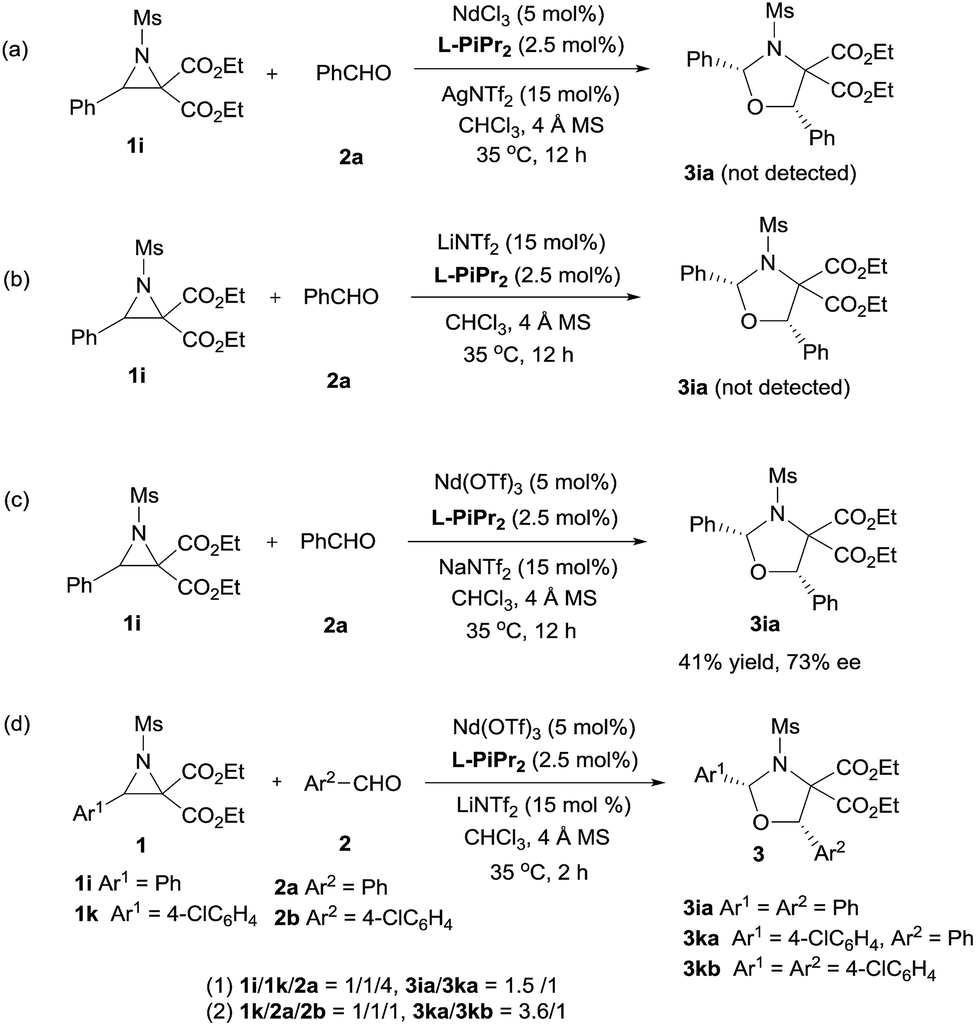 | ||
| Scheme 2 Control experiments. | ||
Based on above mentioned results and our previous study on N,N′-dioxide–metal complex catalysis,8 we propose a dual Lewis acids relay catalysis process (Scheme 3).16 Firstly, with the assistance of LiNTf2, the carbon–carbon bond of the DA aziridine is cleaved to form a dipolar intermediate. It is then caught by the chiral Nd(III)/L-PiPr2 complex due to the strong bidentate coordination of the two ester groups to the metal center. A concerted [3 + 2] cycloaddition occurs enantioselectively to give cis-(2R,5S)-1,3-oxazolidine 3sa, liberating the catalysts.
 | ||
| Scheme 3 A plausible catalytic cycle. | ||
Conclusions
In summary, we have disclosed an enantioselective [3 + 2] annulation of donor–acceptor aziridines with aldehydes through C–C bond cleavage. LiNTf2 and a chiral N,N′-dioxide/Nd(OTf)3 complex worked as relay catalysts to promote the reaction under mild reaction conditions. The protocol allowed the efficient production of a variety of enantiomerically enriched cis-1,3-oxazolidines. Additional research on expanding the asymmetric transformations of DA aziridines to other dipolarphiles is under way.Acknowledgements
The study was funded by the National Natural Science Foundation of China (No. 21432006, 21321061, and 21172151).Notes and references
- For selected reviews: (a) E. A. Ilardi and J. T. Njardarson, J. Org. Chem., 2013, 78, 9533 CrossRef CAS PubMed; (b) H. Ohno, Chem. Rev., 2014, 114, 7784 CrossRef CAS PubMed; (c) G. Callebaut, T. Meiresonne, N. D. Kimpe and S. Mangelinckx, Chem. Rev., 2014, 114, 7954 CrossRef CAS PubMed . For recent examples; (d) H. Guo, Q. Xu and O. Kwon, J. Am. Chem. Soc., 2009, 131, 6318 CrossRef CAS PubMed; (e) P. A. Wender and D. Strand, J. Am. Chem. Soc., 2009, 131, 7528 CrossRef CAS PubMed; (f) B. M. Trost, M. Osipov and G. Dong, J. Am. Chem. Soc., 2010, 132, 15800 CrossRef CAS PubMed; (g) J.-J. Feng, T.-Y. Lin, H.-H. Wu and J. Zhang, J. Am. Chem. Soc., 2015, 137, 3787 CrossRef CAS PubMed; (h) L. Wang, D. Yang, F. Han, D. Li, D. Zhao and R. Wang, Org. Lett., 2015, 17, 176 CrossRef CAS PubMed; (i) A. Rai and L. D. S. Yadav, Tetrahedron Lett., 2013, 54, 3127 CrossRef CAS.
- (a) B. M. Trost and D. R. Fandrick, J. Am. Chem. Soc., 2003, 125, 11836 CrossRef CAS PubMed; (b) C.-F. Xu, B.-H. Zheng, J.-J. Suo, C.-H. Ding and X.-L. Hou, Angew. Chem., Int. Ed., 2015, 54, 1604 CrossRef CAS PubMed.
- Z. Chai, Y.-M. Zhu, P.-J. Yang, S. Wang, S. Wang, Z. Liu and G. Yang, J. Am. Chem. Soc., 2015, 137, 10088 CrossRef CAS PubMed.
- For relative reports: (a) R. Huisgen, W. Scheer and H. Huber, J. Am. Chem. Soc., 1967, 89, 1753 CrossRef CAS; (b) T. DoMinh and A. M. Trozzolo, J. Am. Chem. Soc., 1972, 94, 4046 CrossRef; (c) B. R. Henke, A. J. Kouklis and C. H. Heathcock, J. Org. Chem., 1992, 57, 7056 CrossRef CAS; (d) A. S. Pankova and M. A. Kuznetsov, Tetrahedron Lett., 2014, 55, 2499 CrossRef CAS.
- For relevant examples: (a) P. D. Pohlhaus, R. K. Bowman and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 2294 CrossRef CAS PubMed; (b) X. Wu, L. Li and J. Zhang, Chem. Commun., 2011, 47, 7824 RSC; (c) Z. Jiang, J. Wang, P. Lu and Y. Wang, Tetrahedron, 2011, 67, 9609 CrossRef CAS; (d) X. Wu and J. Zhang, Synthesis, 2012, 44, 2147 CrossRef CAS; (e) L. Li, X. Wu and J. Zhang, Chem. Commun., 2011, 47, 5049 RSC; (f) L. Li and J. Zhang, Org. Lett., 2011, 13, 5940 CrossRef CAS PubMed; (g) H. Liu, C. Zheng and S.-L. You, J. Org. Chem., 2014, 79, 1047 CrossRef CAS PubMed; (h) H. Liu, C. Zheng and S.-L. You, Chin. J. Chem., 2014, 32, 709 CrossRef CAS; (i) A. Ghosh, A. K. Pandey and P. Banerjee, J. Org. Chem., 2015, 80, 7235 CrossRef CAS PubMed; (j) T. Soeta, Y. Miyamoto, S. Fujinami and Y. Ukaji, Tetrahedron, 2014, 70, 6623 CrossRef CAS; (k) R. A. Craig II, N. R. O'Connor, A. F. G. Goldberg and B. M. Stoltz, Chem.–Eur. J., 2014, 20, 4806 CrossRef PubMed.
- For selected reviews: (a) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef CAS PubMed; (b) F. de Nanteuil, F. De Simone, R. Frei, F. Benfatti, E. Serrano and J. Waser, Chem. Commun., 2014, 50, 10912 RSC . For selected examples; (c) A. T. Parsons and J. S. Johnson, J. Am. Chem. Soc., 2009, 131, 3122 CrossRef CAS PubMed; (d) H. Xu, J.-P. Qu, S. Liao, H. Xiong and Y. Tang, Angew. Chem., Int. Ed., 2013, 52, 4004 CrossRef CAS PubMed; (e) H. Xiong, H. Xu, S. Liao, Z. Xie and Y. Tang, J. Am. Chem. Soc., 2013, 135, 7851 CrossRef CAS PubMed; (f) F. de Nanteuil, E. Serrano, D. Perrotta and J. Waser, J. Am. Chem. Soc., 2014, 136, 6239 CrossRef CAS PubMed; (g) H. Xu, J.-L. Hu, L. Wang, S. Liao and Y. Tang, J. Am. Chem. Soc., 2015, 137, 8006 CrossRef CAS PubMed.
- (a) W. L. Chen, L. L. Lin, Y. F. Cai, Y. Xia, W. D. Cao, X. H. Liu and X. M. Feng, Chem. Commun., 2014, 50, 2161 RSC; (b) W. L. Chen, X. Fu, L. L. Lin, X. Yuan, W. W. Luo, J. H. Feng, X. H. Liu and X. M. Feng, Chem. Commun., 2014, 50, 11480 RSC.
- For reviews on chiral N,N′-dioxides: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS PubMed; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298 RSC . Selected examples; (c) K. Shen, X. H. Liu, W. T. Wang, G. Wang, W. D. Cao, W. Li, X. L. Hu, L. L. Lin and X. M. Feng, Chem. Sci., 2010, 1, 590 RSC; (d) Y. Y. Chu, X. H. Liu, W. Li, X. L. Hu, L. L. Lin and X. M. Feng, Chem. Sci., 2012, 3, 1996 RSC; (e) Y. B. Liu, H. P. Hu, H. F. Zheng, Y. Xia, X. H. Liu, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2014, 53, 11579 CrossRef CAS PubMed; (f) Y. Xia, X. H. Liu, H. F. Zheng, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2015, 54, 227 CrossRef CAS PubMed; (g) W. Li, F. Tan, X. Y. Hao, G. Wang, Y. Tang, X. H. Liu, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2015, 54, 1608 CrossRef CAS PubMed.
- For selected examples: (a) S. Hirayama, N. Wada, T. Nemoto, T. Iwai, H. Fujii and H. Nagase, ACS Med. Chem. Lett., 2014, 5, 868 CrossRef CAS PubMed; (b) S.-L. Shi, L.-W. Xu, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 6638 CrossRef CAS PubMed.
- The SES group have been attempted to cleave by use of TBAF but with the complicated side products obtained. For selected reports: (a) J. J. Caldwell and D. Craig, Angew. Chem., Int. Ed., 2007, 46, 2631 CrossRef CAS PubMed; (b) Y. Fukunaga, T. Uchiba, Y. Ito, K. Matsumoto and T. Katsuki, Org. Lett., 2012, 14, 4658 CrossRef CAS PubMed.
- CCDC 1057118 ((2R, 5S)-3sa).
- For a similar mechanistic consideration: (a) M. Jin, L. Adak and M. Nakamura, J. Am. Chem. Soc., 2015, 137, 7128 CrossRef CAS PubMed; (b) J.-L. Hu, L. Wang, H. Xu, Z. Xie and Y. Tang, Org. Lett., 2015, 17, 2680 CrossRef CAS PubMed.
- The computation revealed that the DA aziridine went through the azomethine ylide intermediate during the [3 + 2] cycloaddition, see: A. Paasche, M. Arnone, R. F. Fink, T. Schirmeister and B. Engels, J. Org. Chem., 2009, 74, 5244 CrossRef CAS PubMed.
- The azomethine ylide intermediate has been detected by NMR through mixing LiClO4 with donor–acceptor N-arylaziridines, see: M. Vaultier and R. Carrie, Tetrahedron Lett., 1978, 19, 1195 CrossRef.
- The desired yield sharply decreased in 100% aziridine consumption when the electron density of aryl groups on aldehydes was reduced (Table 2, entries 9 and 12 vs.Table 3, entry 2).
- For selected reviews: (a) D.-F. Chen, Z.-Y. Han, X.-L. Zhou and L.-Z. Gong, Acc. Chem. Res., 2014, 47, 2365 CrossRef CAS PubMed . For selected examples; (b) X. Wang, S. Dong, Z. Yao, L. Feng, P. Daka, H. Wang and Z. Xu, Org. Lett., 2014, 16, 22 CrossRef CAS PubMed; (c) J. B. Gianino, C. A. Campos, A. J. Lepore, D. M. Pinkerton and B. L. Ashfeld, J. Org. Chem., 2014, 79, 12083 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1057118. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04151a |
| This journal is © The Royal Society of Chemistry 2016 |