
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Successive C–C bond cleavage, fluorination, trifluoromethylthio- and pentafluorophenylthiolation under metal-free conditions to provide compounds with dual fluoro-functionalization†
Ibrayim
Saidalimu
a,
Shugo
Suzuki
b,
Etsuko
Tokunaga
a and
Norio
Shibata
*ab
aDepartment of Nanopharmaceutical Sciences, Nagoya Institute of Technology, Gokiso, Showa-ku, Nagoya 466-8555, Japan
bDepartment of Frontier Materials, Nagoya Institute of Technology, Gokiso, Showa-ku, Nagoya 466-8555, Japan. E-mail: nozshiba@nitech.ac.jp
First published on 9th December 2015
Abstract
The selective C–C bond cleavage and simultaneous formation of two C–F bonds and one C–S bond in β-keto esters with nucleophilic fluorination reagents such as DAST under metal-free/catalyst-free conditions is disclosed. Double fluorination at two remote carbons with additional dialkylamino-sulfenylation provided unique fluorinated compounds in good to high yields. This method can be applied for the successive C–C bond cleavage/fluorination/trifluoromethylthiolation of β-keto esters using trifluoromethyl-DAST (CF3-DAST) providing different type of fluorinated and trifluoromethylthiolated compounds via a shunt pathway. Doubly fluoro-functionalized compounds obtained in these reactions are unique and difficult to synthesize by other methods.
The selective cleavage/activation of carbon–carbon (C–C) bonds during chemical transformations poses a significant synthetic challenge in traditional organic synthesis.1 Due to the inherent solidity and stability or unreactivity of the C–C bond, this transformation requires harsh conditions. Moreover, following simultaneous chemical transformations, including the formation of new C–X bond(s), the process can be applied to more complex tasks. In recent years, significant achievements and progress have been reported in the area of transition metal catalysis.2 However, metal-free conditions to accomplish this, including C–C bond cleavage followed by C–X bond(s) formation, have clear advantages from a green chemistry viewpoint.3,4 Here we disclose the selective C–C bond cleavage and simultaneous formation of two C–F bonds and one C–S bond in β-keto esters with nucleophilic fluorination reagents such as diethylaminosulfur trifluoride (DAST) under metal- or catalyst-free conditions (Scheme 1). Double fluorination at two remote carbons with additional dialkylamino-sulfenylation provided unique acid fluorides with a tetra-substituted fluorinated/sulfenylated carbon center at a remote position in good to high yields (Scheme 1a). This method can be applied for the successive C–C bond cleavage, fluorination and trifluoromethylthiolation of β-keto esters using trifluoromethyl-DAST (CF3-DAST) to provide different types of fluorinated and trifluoromethylthiolated compounds with a tri-substituted carbon center (Scheme 1b). Doubly fluoro-functionalized compounds obtained in these reactions are unique and are difficult to synthesize by other methods. A pentafluorophenyl-thiolated analogue was also synthesized using pentafluorophenyl-DAST (C6F5-DAST). Our results suggest that unique sequential transformation that provides attractive fluorinated compounds is possible without any state-of-the-art catalyst, energy of the ring-strain or heating. Instead, it simply involves a suitable choice of substrates and reagents.
 | ||
| Scheme 1 Sequential carbon–carbon bond cleavage, fluorination and fluorination, trifluoromethylthiolation or pentafluorophenylthiolation under a metal-free system. | ||
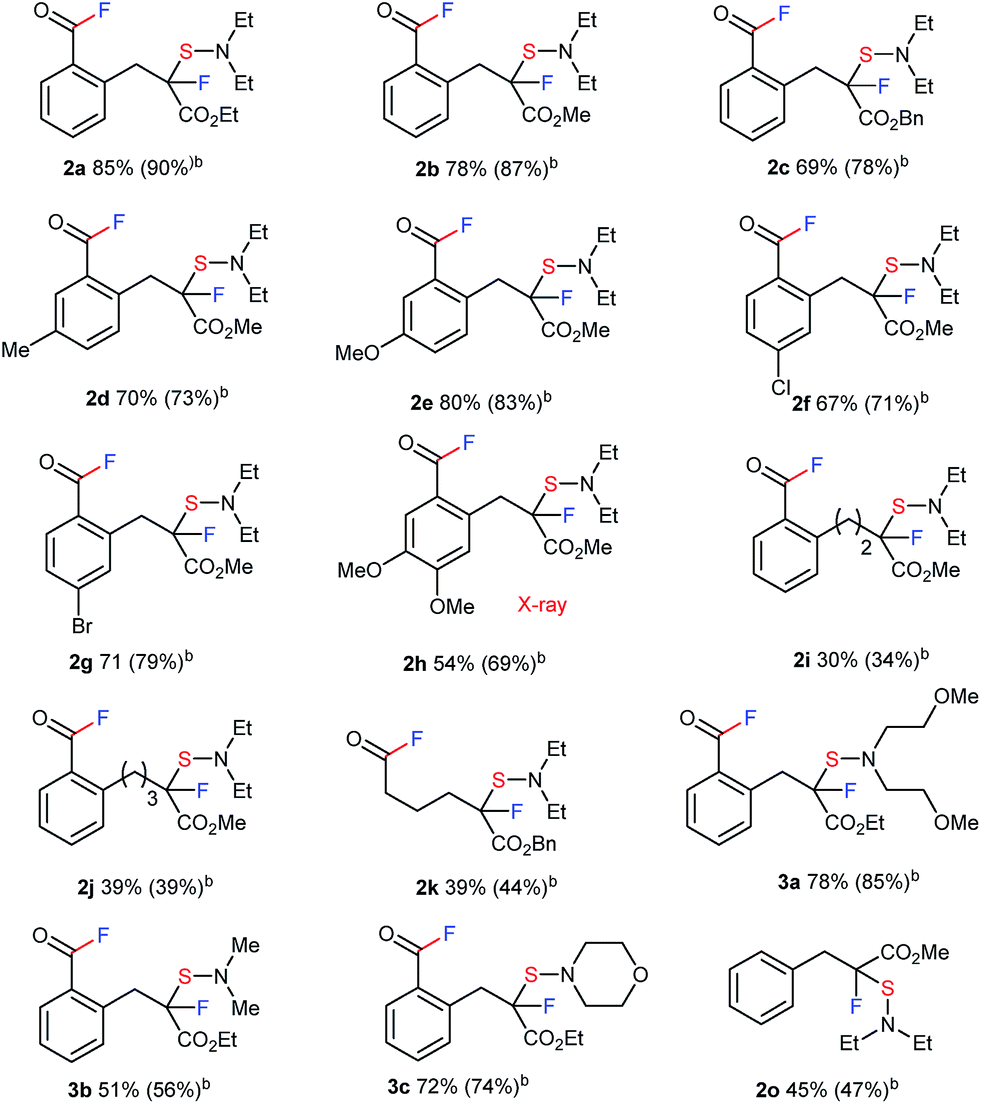
A large number of commercial applications for fluorinated organic compounds have induced much interest in developing novel synthetic methods to incorporate fluorine or fluorinated groups into organic compounds.5 Fluorination (F),6 trifluoromethylation (CF3),7 and trifluoromethylthiolation (SCF3)8,9 reactions are among the three most important chemical transformations investigated in recent years due to the impressive electron-withdrawing effects and lipophilicity of the groups being introduced. While developing novel methodologies for fluoro-functionalization reactions, we unexpectedly transformed ethyl indanone carboxylate (1a) with DAST10 in CH2Cl2 to acyclic acid fluoride 2a with a tetra-substituted carbon center with C–F and C–S bonds. Although the chemical yield was low, only 31%, the reaction was unique enough for further investigation since it effected four important chemical transformations without any catalysis: C–C bond cleavage, the formation of two C–F bonds at remote positions, and a C–S bond.11 We thus envisioned that this strategy might be viable for the synthesis of new types of fluoro-functionalized acid fluorides from ubiquitous carboxylic esters. With this idea in mind, we set out to investigate the use of β-keto ester 1a and DAST.12,13 After thoroughly surveying reaction conditions, including temperature, solvent, concentration, etc. (see ESI, Table S1†), we found that the use of 2.0 equivalents of DAST in THF at room temperature gave the best result (2a, 85% yield).
We proceeded to evaluate the scope of these four metal-free, sequential transformations by DAST with a wide variety of β-keto esters 1 (Table 1). The sequential transformation of indanone substrates with DAST was in general independent of the size of the ester moiety (Me, Et, Bn), and a substitution on the benzene ring (MeO, Me, Br, Cl) provided the corresponding products 2a–2g in good to high yields. Substrate 1h, which is very rich in electrons, also underwent the same four sequential transformations to give the corresponding product in good yield (2h, 54%). Tetralone carboxylate 1i and benzosuberanone carboxylate 1j were also good substrates for transformation to furnish the desired products 2i and 2j in 30% and 39% yield, respectively. Non-aromatic benzyl 2-oxocyclopentanecarboxylate (1k) was also converted to fluorinated-sulfenylated acid fluoride 2k in 39% yield. Other nucleophilic fluorination reagents such as (MeOCH2CH2)2NSF3 (DeoxoFluor®),10c,14 4-morpholinylsulfur trifluoride (Morph-DAST),10a,15 and N,N-dimethylaminosulfur trifluoride (Me-DAST)10a were equally effective for these transformations, yielding the corresponding fluorinated dialkylaminosulfenylated acid fluoride products 3a–3c in moderate to high yields. Finally, this strategy was also effective for an acyclic substrate, methyl 2-benzyl-3-oxobutanoate (1o) in DMF at 50 °C to provide the C–C bond cleavage/fluorination/sulfenylation product 2o in moderate yield, while the acetyl fluoride moiety produced was separated due to its acyclic system. Information gleaned from 1H NMR, 13C NMR, 19F NMR, IR, and mass spectra led to the formulation of a unique fluorinated acid fluoride product, 2. Finally, the structure of 2 was confirmed unambiguously by single crystal X-ray structure analysis of 2h (CCDC 1415530).†
|
|
|---|
| a The reaction of 1 with DAST or its derivatives (2.0 equiv.) was carried out overnight in THF (0.1 M) at room temperature. Isolated yields are indicated. For detailed reaction conditions, see ESI. b 19F NMR yields. c The reaction of 1o with DAST (2.0 equiv.) was carried out overnight in DMF (0.1 M) at 50 °C. |
|

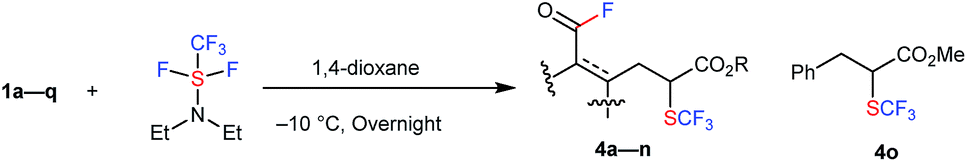
More unexpected supersizing results were obtained when a similar reaction of 1 with trifluoromethyl-DAST16 (CF3-DAST reagent) was attempted. The CF3-DAST reagent was readily prepared by mixing Ruppert–Prakash reagent (CF3SiMe3) with DAST under basic conditions, but it was not stable enough to be isolated. Thus, we directly used in situ generated CF3-DAST in CH2Cl2 instead of DAST for our reaction system with 1a in THF at room temperature overnight. Acid fluoride 4a with a trifluoromethylthiolated tri-substituted carbon center was detected in 28% yield. With this result in hand, the reaction conditions, including solvent, temperature, reagent equivalents, etc. (see ESI, Tables S2 and S3†), were further optimized. A set of optimal reaction conditions was screened: 2.0 equivalents of CF3-DAST (0.5 M mixed in DCM) and overnight reaction at −10 °C in 1,4-dioxane as solvent (up to 61% yield of 4a). The substrate scope of the reaction is shown in Table 2. A variety of alkyl indanone carboxylates 1 (R = Me, Et, Bn) with different substitutions on the benzene ring (MeO, Me, Br, Cl, di-MeO), tetralone carboxylate 1q, benzosuberanone carboxylate 1j and benzyl 2-oxocyclopentanecarboxylate 1k were employed under the same conditions to provide the corresponding sequential C–C bond cleavage, fluorination or trifluoromethylthiolation products 4a–4k in moderate to high yields. A sterically demanding secondary ester, tertiary ester, and electron-withdrawing p-nitrobenzyl ester (R = iPr, 1-admantanyl, CH2C6H4p-NO2) were also successfully converted into the desired products 4l–4n under the same conditions. Acyclic methyl 2-benzyl-3-oxobutanoate 1o was converted into the desired C–C bond cleavage trifluoromethylthiolation product 4o in acceptable yield (19%) after the release of the acid fluoride part. The isolated yields are somewhat lower than the NMR yields due to instability during purification by silica-gel chromatography. The structures of the trifluoromethylthiolated acid fluorides were assigned by spectroscopy and clearly determined by X-ray crystallographic analysis of 4h (CCDC 1415531).†
|
|
|---|
| a The reaction of 1 with 2.0 equivalents of CF3-DAST (0.5 M mixture in DCM) was carried out overnight in 1,4-dioxane at −10 °C. Isolated yields are indicated. For detailed reaction conditions, see ESI. b 19F NMR yields. c 4k is too unstable to be isolated after purification. d The reaction with 2.0 equivalents of CF3-DAST (0.5 M mixture in DCM) was carried out overnight in DMF at 50 °C. |

|
Acid fluorides are versatile building blocks.17 In particular, they are popular for peptide coupling reactions without epimerization, and thus a range of more complex fluorinated compounds can be synthesized. As shown in Scheme 2, 2b and 4b easily underwent alkylation, amination, and esterification to form the corresponding fluorinated and sulfenylated products 5a,b and trifluoromethylthiolated products 6a–6c in good to high yields (Scheme 2).
 | ||
| Scheme 2 Transformation of acid fluorides 2b and 4b to 5a,b and 6a–6c. | ||
It is interesting to note that this methodology was effectively extended to the reaction of 1b with in situ generated, previously unknown pentafluorophenyl-DAST (C6F6-DAST) to provide SC6F5-analogue 7b in 53% isolated yield (Scheme 3).
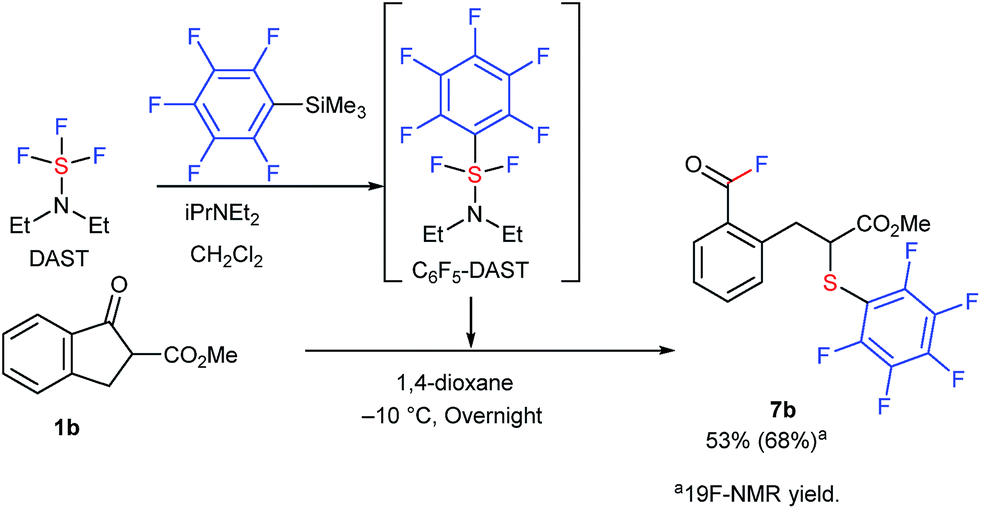 | ||
| Scheme 3 Reaction of 1b with C6F6-DAST. Reaction details are shown in ESI.† | ||
Moreover, 1,3-diketone 8 also reacted with DAST or CF3-DAST to provide the corresponding unexpected fluorinated or sulfenylated product 9 or trifluoromethylthiolated product 10 in 63% and 54% yield, respectively. Although it was possible to isolate both compounds, 9 was not very stable during silica-gel column chromatography. Deacetylation was observed in this case, similar to the reaction of acyclic substrates 1o to 2o or 1o to 4o (Scheme 4).
 | ||
| Scheme 4 Reaction of 1,3-diketone 8 with DAST or CF3-DAST. Reaction details are shown in ESI.† | ||
A possible reaction mechanism (Fig. 1) is based on the unexpected formation of two different types of products 2 and 4. Initially, the fluorine anion generated from DAST or CF3-DAST selectively attacks the ketone moiety of 1a to give the acid fluoride enolate A via a ring-opening reaction through a retro-Dieckmann18,19 type reaction (for acyclic substrates 2o and 1,3-diketone 8, a “retro-Claisen”19c type reaction might be suitable due to the de-acetylation). The enolate rapidly attacks the sulfur atom of the DAST or CF3-DAST residue providing unstable intermediate B. In the case of the reaction with DAST (X = F), intermediate B promptly releases HF initiated by the attack from the internal nitrogen moiety. This is followed by intramolecular fluoro-Pummerer-type rearrangement20 to furnish final product 2a as an HF salt via thionium intermediate C (route a). On the other hand, the reaction with CF3-DAST (X = CF3) enters route b instead of route a due to the presence of diisopropylethylamine (iPr2NEt). CF3-DAST should be prepared in situ from an equivalent mixture of DAST, CF3SiMe3, and iPr2NEt. A molar equivalent of iPr2NEt is crucial for complete transformation to CF3-DAST, and iPr2NEt is presumably required to initiate the reaction and stabilize the generated CF3-DAST.16 The acidic proton in intermediate B needs to be removed by iPr2NEt to furnish D rather than the elimination of HF before heading into route a. Unstable intermediate D promptly releases HF as an N-ethylidene ethanamine salt,16 resulting in trifluoromethylthiolation product 4avia enolate E.
 | ||
| Fig. 1 A plausible reaction mechanism. | ||
Conclusions
In summary, we have efficiently synthesized acid fluorides with a tetra-substituted fluorinated and sulfenylated carbon center at a remote position via a metal- or catalyst-free ring opening reaction of β-keto esters with DAST. The chemical transformation undergoes a sequence of C–C bond cleavages, two C–F bonds form at the remote positions of C1 to C5–C6 and C–S bond formation affords a wide range of unique fluorinated acid fluorides in good to high yields under mild reaction conditions. This sequential transformation was extended to the reaction of β-keto esters with CF3-DAST. More interestingly, trifluoromethylthiolated acid fluorides with a tri-substituted carbon center were produced under the same reaction conditions. 1,3-Diketones are also acceptable substrates in these transformations with DAST and CF3-DAST. All these reactions are triggered by an attack by fluoride on the carbonyl through a retro-Dieckmann or retro-Claisen type reaction. Both fluoro-functionalized compounds unexpectedly obtained here are otherwise difficult to prepare. Although a large number of reactions have been reported using DAST and related reagents with a variety of substrates10,17,18 including β-keto esters,12,13 the present sequential reaction has never been reported. The reaction mechanism, the utility of this strategy for the development of new chemical transformations and the synthesis of biologically attractive molecules using these fluorinated products are under investigation.Acknowledgements
This research is partially supported by the Platform Project for Supporting in Drug Discovery and Life Science Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), the Japan Agency for Medical Research and Development (AMED), the Advanced Catalytic Transformation (ACT-C) from the Japan Science and Technology (JST) Agency, and the Kobayashi International Foundation.Notes and references
- G. Dong, C–C Activation (Topics in Current Chemistry), Springer-Verlag, Berlin, Heidelberg, 2014 Search PubMed.
- (a) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410 CrossRef CAS PubMed; (b) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613 CrossRef CAS PubMed; (c) I. Marek, A. Masarwa, P.-O. Delaye and M. Leibeling, Angew. Chem., Int. Ed., 2015, 54, 414 CAS.
- X. Sun, M. Wang, P. Li, X. Zhang and L. Wang, Green Chem., 2013, 15, 3289 RSC.
- B. Tiwari, J. Zhang and Y. R. Chi, Angew. Chem., Int. Ed., 2012, 51, 1911 CrossRef CAS PubMed.
- P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, Germany, 2013 Search PubMed.
- (a) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612 CrossRef CAS PubMed; (b) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed; (c) J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1 CrossRef CAS PubMed; (d) A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073 CrossRef PubMed.
- (a) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (b) X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683 CrossRef CAS PubMed; (c) P. Gao, X.-R. Song, X.-Y. Liu and Y.-M. Liang, Chem.–Eur. J., 2015, 21, 7648 CrossRef CAS PubMed; (d) Y.-Y. Huang, X. Yang, Z. Chen, F. Verpoort and N. Shibata, Chem.–Eur. J., 2015, 21, 8664 CrossRef CAS PubMed; (e) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (f) C. Alonso, de. M. E. Martinez, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed; (g) N. Shibata, A. Matsnev and D. Cahard, Beilstein J. Org. Chem., 2010, 6(65) CAS; (h) G. K. S. Prakash and A. K. Yudin, Chem. Rev., 1997, 97, 757 CrossRef CAS PubMed.
- X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef CAS PubMed.
- C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed.
- (a) L. N. Markovski, V. E. Pashinnik and A. V. Kirsanov, Synthesis, 1973, 787 CrossRef; (b) W. J. Middleton, J. Org. Chem., 1975, 40, 574 CrossRef CAS; (c) R. P. Singh and J. M. Shreeve, Synthesis, 2002, 2561 CAS.
- (a) H. Zhao, X. Fan, J. Yu and C. Zhu, J. Am. Chem. Soc., 2015, 137, 3490 CrossRef CAS PubMed; (b) I. Saidalimu, X. Fang, X. He, J. Liang, X. Yang and F. Wu, Angew. Chem., Int. Ed., 2013, 52, 5566 CrossRef CAS PubMed; (c) I. Saidalimu, X. Fang, W. Lv, X. Yang, X. He, J. Zhang and F. Wu, Adv. Synth. Catal., 2013, 355, 857 CrossRef CAS; (d) Y. Chen, Y. Wang, X. Sun and D. Ma, Org. Lett., 2008, 10, 625 CrossRef CAS PubMed; (e) S. Ventre, F. R. Petronijevic and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 5654 CrossRef CAS PubMed.
- A. E. Asato and R. S. H. Liu, Tetrahedron Lett., 1986, 27, 3337 CrossRef CAS.
- S. Bildstein, J.-B. Ducep, D. Jacobi and P. Zimmermann, Tetrahedron Lett., 1995, 36, 5007 CrossRef CAS.
- G. S. Lal, G. P. Pez, R. J. Pesaresi, F. M. Prozonic and H. Cheng, J. Org. Chem., 1999, 64, 7048 CrossRef CAS.
- P. K. Mykhailiuk, S. V. Shishkina, O. V. Shishkin, O. A. Zaporozhets and I. V. Komarov, Tetrahedron, 2011, 67, 3091 CrossRef CAS.
- A. Ferry, T. Billard, B. R. Langlois and E. Bacque, J. Org. Chem., 2008, 73, 9362 CrossRef CAS PubMed.
- (a) Y. Zhang and T. Rovis, J. Am. Chem. Soc., 2004, 126, 15964 CrossRef CAS PubMed; (b) C. Kaduk, H. Wenschuh, M. Beyermann, K. Forner, L. A. Carpino and M. Bienert, Lett. Pept. Sci., 1996, 2, 285 CrossRef CAS; (c) S. S. Corinna, M. F. Patrik and M. C. Erick, Org. Lett., 2010, 12, 4102 CrossRef PubMed.
- The retro-Dieckmann and related ring-opening reactions require ring-strain, strong bases, and/or a high reaction temperature for initiation. Our case corresponds to a “fluoride-induced retro-Dieckmann-type reaction” and the first fluorination triggers the following successive reactions including ring-opening.
- (a) E. M. Clavijo, A. J. M. Vargas, A. T. Carmona and I. Robina, Org. Biomol. Chem., 2013, 11, 7016 RSC; (b) S. Sano, H. Shimizu and Y. Nagao, Tetrahedron Lett., 2005, 46, 2883 CrossRef CAS; (c) A. Kawata, K. Takata, Y. Kuninobu and K. Takai, Angew. Chem., Int. Ed., 2007, 46, 7793 CrossRef CAS PubMed.
- (a) J. R. McCarthy, N. P. Peet, M. E. Letourneau and M. Inbasekaran, J. Am. Chem. Soc., 1985, 107, 735 CrossRef CAS; (b) S. F. Wnuk and M. J. Robins, J. Org. Chem., 1990, 55, 4757 CrossRef CAS; (c) H. Chen, Z. Hua, J. Zhang, G. Liang and B. Xu, Tetrahedron, 2015, 71, 2089 CrossRef CAS; (d) V. Kumar and M. R. Bell, Heterocycles, 1992, 34, 1289 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1415530 and 1415531. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04208a |
| This journal is © The Royal Society of Chemistry 2016 |