
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of the coordination preferences and catalytic pathways of heteroaxial cobalt oximes towards hydrogen generation†
Debashis
Basu‡
a,
Shivnath
Mazumder‡
a,
Jens
Niklas
b,
Habib
Baydoun
a,
Dakshika
Wanniarachchi
a,
Xuetao
Shi
a,
Richard J.
Staples
c,
Oleg
Poluektov
b,
H. Bernhard
Schlegel
*a and
Cláudio N.
Verani
*a
aDepartment of Chemistry, Wayne State University, Detroit, MI 48202, USA. E-mail: cnverani@chem.wayne.edu
bChemical Sciences and Engineering Division, Argonne National Laboratory, Argonne, IL 60439, USA
cDepartment of Chemistry, Michigan State University, Lansing, MI 48824, USA
First published on 2nd February 2016
Abstract
Three new heteroaxial cobalt oxime catalysts, namely [CoIII(prdioxH)(4tBupy)(Cl)]PF6 (1), [CoIII(prdioxH)(4Pyrpy)(Cl)]PF6 (2), and [CoIII(prdioxH)(4Bzpy)(Cl)]PF6 (3) have been studied. These species contain chloro and substituted tert-butyl/pyrrolidine/benzoyl-pyridino ligands axially coordinated to a trivalent cobalt ion bound to the N4-oxime macrocycle (2E,2′E,3E,3′E)-3,3′-(propane-1,3-diylbis(azanylylidene))bis(butan-2-one)dioxime, abbreviated (prdioxH)− in its monoprotonated form. Emphasis was given to the spectroscopic investigation of the coordination preferences and spin configurations among the different 3d6 CoIII, 3d7 CoII, and 3d8 CoI oxidation states of the metal, and to the catalytic proton reduction with an evaluation of the pathways for the generation of H2via CoIII–H− or CoII–H− intermediates by mono and bimetallic routes. The strong field imposed by the (prdioxH)− ligand precludes the existence of high-spin configurations, and 6-coordinate geometry is favored by the LSCoIII species. Species 1 and 3 show a split CoIII/CoII electrochemical wave associated with partial chemical conversion to a [CoIII(prdioxH)Cl2] species, whereas 2 shows a single event. The reduction of these CoIII complexes yields LSCoII and LSCoI species in which the pyridine acts as the dominant axial ligand. In the presence of protons, the catalytically active CoI species generates a CoIII–H− hydride species that reacts heterolytically with another proton to generate dihydrogen. The intermediacy of a trifluoroacetate-bound CoIII/CoII couple in the catalytic mechanism is proposed. These results allow for a generalization of the behavior of heteroaxial cobalt macrocycles and serve as guidelines for the development of new catalysts based on macrocyclic frameworks.
Introduction
Research in molecular catalysts for proton and water reduction has been intensive and in recent years special attention was paid to the use of Earth-abundant metals.1–4 Cobalt ions, with their ability to span redox states from 3+ (3d6) to 1+ (3d8) have been emphasized, and effective catalysts have been designed for both proton and water reduction with oximes.3 Starting with the adventitious observation by Espenson & Connolly5 that biomimetic cobalt oximes are capable of proton reduction in acidic organic media, these catalysts figure among the first observed systems and have been extensively scrutinized. In spite of this interest and of available information for individual cobalt oximes,6 comprehensive spectroscopic studies that interrogate the transformation of oxidation states along the reductive process that leads to the catalytically active CoI species are rare. Furthermore, the vast majority of studied catalytic systems contain equivalent axial ligands, hereafter called homoaxial, and added complexity is observed when non-equivalent axial ligands are present. These heteroaxial cobalt oximes display two CoIII/CoII redox processes associated with the formation of a new CoIII species in solution. These distinct species yield a monovalent catalyst. A previous study to rationalize this behavior was performed by Gerli and Marzilli7 where an exclusively electrochemical pathway was proposed, leading to a square planar CoII species that yields a catalytic 4-coordinate CoI intermediate. However, both experimental8,9 and theoretical6g evidence suggest that this intermediate requires 5-coordination with N-containing donors.Thus, we propose that a unifying investigation based on UV-visible, nuclear magnetic and electron paramagnetic (NMR & EPR) resonance spectroscopies allied with DFT calculations will allow for the understanding of the coordination preferences of heteroaxial cobalt oxime systems and, therefore, of the behavior of the trivalent, divalent, and monovalent species crucial for catalytic H2 generation. This knowledge will enable us to chart the transformations that occur during the catalytic cycle addressing the nature of formed hydrides, the viability of proton-coupled electron transfer, the homo or heterolytic nature of hydrogen formation, and the regeneration of the catalyst.
Continuing a long-standing collaboration on cobalt systems,10 we have recently examined the catalytic activity of cobalt species associated with pendant phenolates10e and pyridines.10f We have also studied in detail a new pentadentate cobalt oxime system10g designed to stabilize the abovementioned preferential 5-coordination favored by CoI in catalysts used for proton/water reduction. We observed that upon metal coordination this new oxime incorporates a molecule of water and enables H+ reduction in presence of weak acids in acetonitrile (CH3CN).
In this paper we present three new heteroaxial catalysts, namely [CoIII(prdioxH)(4tBupy)(Cl)]PF6 (1), [CoIII(prdioxH)(4Pyrpy)(Cl)]PF6 (2), and [CoIII(prdioxH)(4Bzpy)(Cl)]PF6 (3), Fig. 1, in which the axial pyridine displays both electron-donating and electron-withdrawing functionalities. We interrogate by concerted experimental and theoretical methods the transformations in nature and coordination preferences associated with the 3+, 2+, and 1+ oxidation states of the metal, and propose a viable catalytic pathway for hydrogen formation. These results are relevant to the development of strategies for the improvement of new catalysts for proton and water reduction based on heteroaxial cobalt macrocycles.
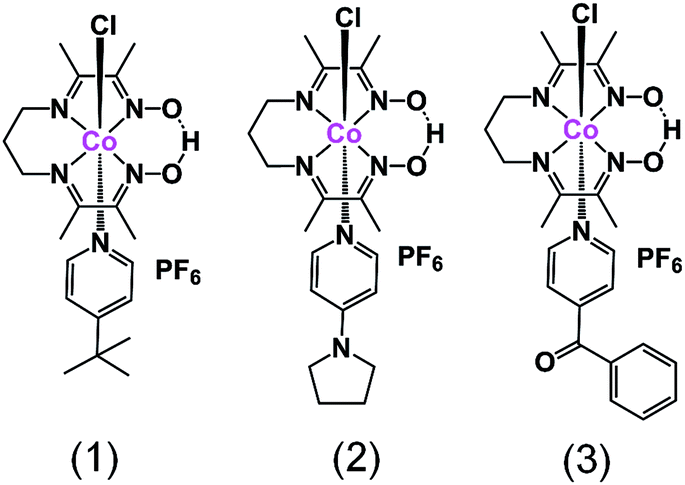 | ||
| Fig. 1 The mononuclear complexes [CoIII(prdioxH)(4tBupy)(Cl)]PF6 (1), [CoIII(prdioxH)(4Pyrpy)(Cl)]PF6 (2), and [CoIII(prdioxH)(4Bzpy)(Cl)]PF6 (3). | ||
Results and discussion
Synthesis and characterization of 1, 2, and 3
The tetradentate ligand prdioxH2 was obtained by literature methods.11,12 Complexes 1, 2, and 3 were generated by stepwise reaction of CoCl2·6H2O with the ligand prdioxH2 to obtain the dichloro complex which was further treated with 4-substituted pyridines and KPF6 to yield 1–3 (Fig. S1†). The FTIR spectra of the complexes reveal a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration mode associated with the oxime ligand at ca. 1600 cm−1 and a strong and broad peak around 850 cm−1 indicative of the presence of PF6− counterions. High-resolution ESI mass spectra (Fig. S2†) display the species [M]+ at 468.1573, 481.1563 and 516.1207 respectively for 1, 2, and 3. Experimental and simulated isotopic distributions are in agreement with the proposed molecular composition. 1H-NMR spectra were taken in CD3CN and the spectrum for 1 is shown in Fig. 2, and the narrow lines confirm the diamagnetic nature of these species, as associated with a 3d6 LSCoIII ion.
N stretching vibration mode associated with the oxime ligand at ca. 1600 cm−1 and a strong and broad peak around 850 cm−1 indicative of the presence of PF6− counterions. High-resolution ESI mass spectra (Fig. S2†) display the species [M]+ at 468.1573, 481.1563 and 516.1207 respectively for 1, 2, and 3. Experimental and simulated isotopic distributions are in agreement with the proposed molecular composition. 1H-NMR spectra were taken in CD3CN and the spectrum for 1 is shown in Fig. 2, and the narrow lines confirm the diamagnetic nature of these species, as associated with a 3d6 LSCoIII ion.
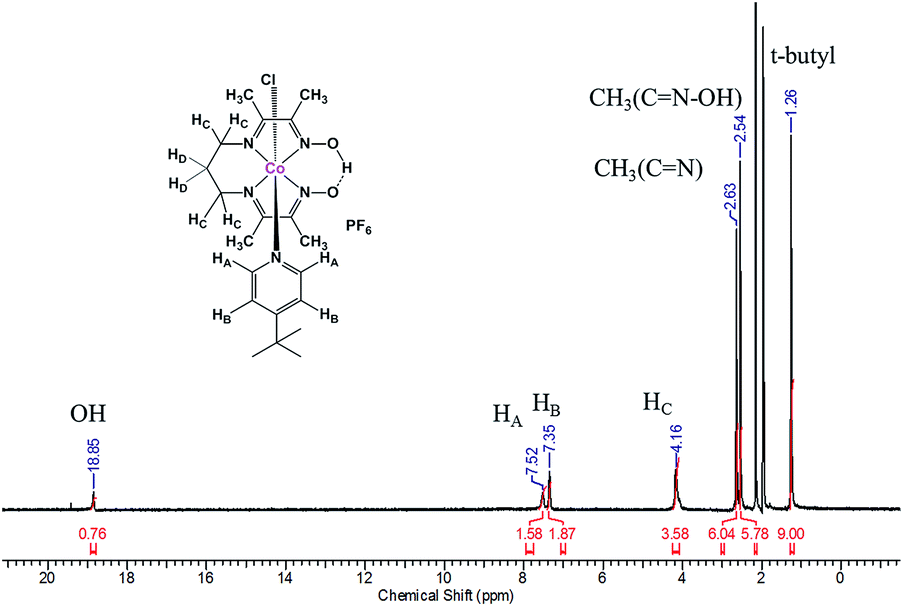 | ||
| Fig. 2 1H-NMR spectra of 1 in CD3CN. | ||
The spectra show appropriate line-splitting patterns for 1 with four aromatic protons between 6 and 8 ppm originating from the pyridine moiety, one OH proton at 18.85 ppm, six methylene protons between 2.0 and 4.5 ppm and 12 methyl protons between 2.4 and 2.6 ppm. The intense peaks found around 1.3 ppm are from the protons (9H) of the t-butyl substituent. Complexes 1 and 3 yielded diffraction-quality crystals whose ORTEP plots are shown in Fig. 3. In both cases a pseudo-octahedral geometry of the cobalt center was found with the oxime ligand occupying the equatorial plane, while chloride and 4tBupyridine/4Bzpyridine ligands occupy the axial positions. One of the oxime-oxygen atoms remains protonated forming an O–H⋯O hydrogen-bonded macrocyclic framework. The Co–Noxime bond lengths (1.88–1.93 Å) are in excellent agreement with other oxime complexes in the literature.3c,7 Comparison of the Co–Cl bond length between 1 and 3 reveals a slightly longer bond in 1 (2.2376 Å) than in 3 (2.2336 Å). On the other hand, the bond length between Co and the NPy atom (Co1–N5) is shorter in 1 (1.975 Å) than in 3 (1.990 Å). These results suggest that incorporation of the electron-withdrawing benzoyl substituent on the pyridine ring of 3 weakens the Co–N5 (pyridine) bond and strengthens the Co–Cl bond.
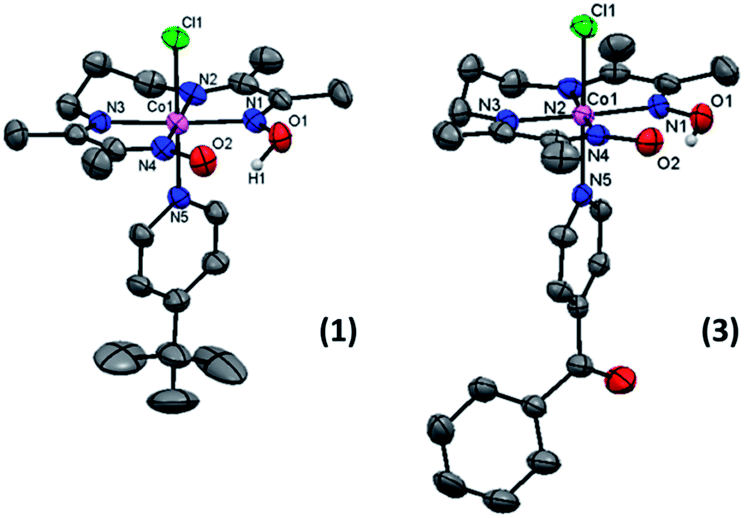 | ||
| Fig. 3 ORTEP representation of the crystal structures for [CoIII(prdioxH)(Xpy)(Cl)]+ cations in 1 (CCDC 1434960) and 3 (CCDC 1434959) at 50% ellipsoid probability. For 1: Co(1)–N(1) = 1.903(3); Co(1)–N(2) = 1.917(2); Co(1)–N(3) = 1.914(2); Co(1)–N(4): 1.898(2) = Co(1)–N(5) = 1.975(3); Co(1)–Cl(1) = 2.237(8) Å. For 3: Co(1)–N(1) = 1.891(3); Co(1)–N(2) = 1.934(3); Co(1)–N(3) = 1.923(3); Co(1)–N(4) = 1.893(3); Co(1)–N(5) = 1.990(3); Co(1)–Cl(1) = 2.2336(11) Å. | ||
Coordination preferences of different oxidation states in the proton reduction pathway
The 1e− and 2e− reduced analogs of 1–3 generated in the electrochemical pathway towards catalysis determine the viability of proton reduction. Therefore, a detailed evaluation of their coordination preferences, spin states, and electronic structures is of the utmost importance in the understanding of catalytic pathways. We probed each of these species by cyclic voltammetry (CV), 1H-NMR, UV-visible, EPR, and DFT methods aiming to characterize comprehensively the nature of these species.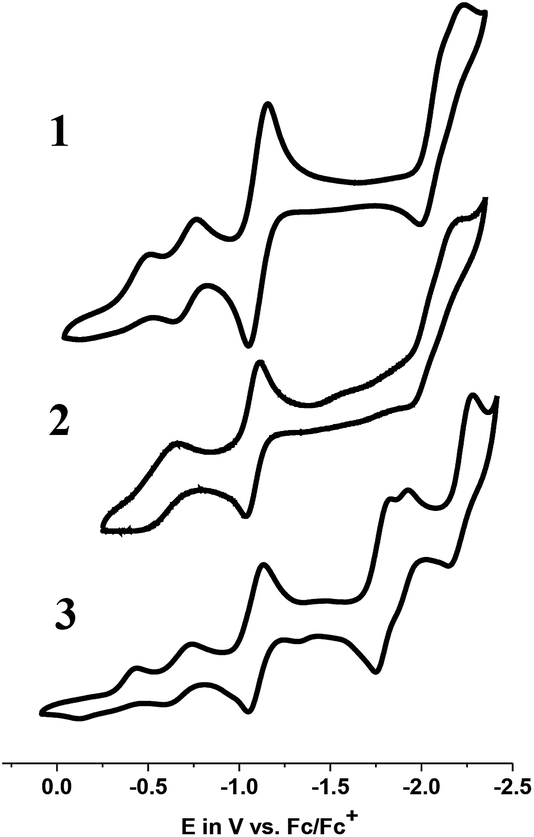 | ||
| Fig. 4 Cyclic voltammograms of 1, 2, and 3 in CH3CN (supporting electrolyte: TBAPF6; scan rate: 100 mV s−1; glassy C: working electrode; Pt-wire: auxiliary electrode; Ag/AgCl: reference electrode). | ||
| CoIII/CoIIE1/2, [Ep,c; Ep,a], V (ΔE, V) |ipc/ipa| |
CoII/CoI
E 1/2, V (ΔE, V) |ipc/ipa| |
CN/C˙–N−E1/2, V (ΔE, V) |ipc/ipa| |
Other L-based eventsE1/2, V (ΔE, V) |ipc/ipa| |
|
|---|---|---|---|---|
| 1 | E p,c: −0.49; Ep,a: −0.19−0.70 (0.10) |0.91| | −1.09 (0.11) |0.99| | −2.11 (0.23) |2.23| | |
| 2 | −0.59 (0.12) |0.45| | −1.07 (0.08) |1.09| | −2.07 (0.23) |N/A| | |
| 3 | E p,c: −0.43; Ep,a: −0.13−0.68 (0.11) |1.41| | −1.09 (0.08) |1.19| | −2.22 (0.12) |5.99| | −1.79 (0.08) |N/A|−1.91 (0.03) |1.03| |
In order to check for potential coordination of the solvent CH3CN, thus generating a new species [CoIII(prdioxH)(CH3CN)2]2+ associated with the second CoIII/CoII redox event observed in 1 and 3, the CV of 1 was repeated in non-coordinating CH2Cl2 (Fig. S3†). Both CoIII/CoII events persisted, thus ruling out the formation of bis-acetonitrile species. The origin of this second CoIII/CoII process will be discussed later.
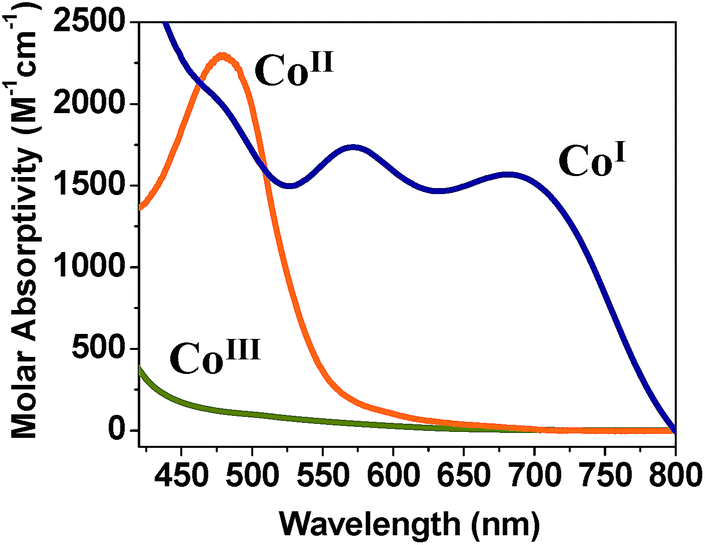 | ||
| Fig. 5 UV-visible absorption spectra of 1 (CoIII) and its reduced CoII analogue with λ = 480 nm (ε = 2288 M−1 cm−1) and CoI analogue with λ = 572 nm (ε = 1738 M−1 cm−1) and λ = 685 nm (ε = 1570 M−1 cm−1)]. | ||
Similar observations were made by Peters et al.9 when studying hydrogen oxidation reactions with CoII homoaxial oxime complexes. The parent yellow solution of CoII species (λmax = 440 nm) turns to CoI upon hydrogen oxidation. The CoI product exhibited a dark blue color and showed two strong absorption bands between 500 and 700 nm.9 Eisenberg et al.3h also detected similar CoI spectra (λmax = 550, 650 nm) for CoII oxime complexes with homoaxial water ligands and with Pt-chromophore and TEOA as the sacrificial donor in the presence of visible light. Thus, the parent compounds 1–3 seem to allow for reduction to CoI at affordable potentials. Knowledge on the nature of this species is relevant for the understanding of competing mechanisms that precede catalytic proton reduction in acidic organic media.
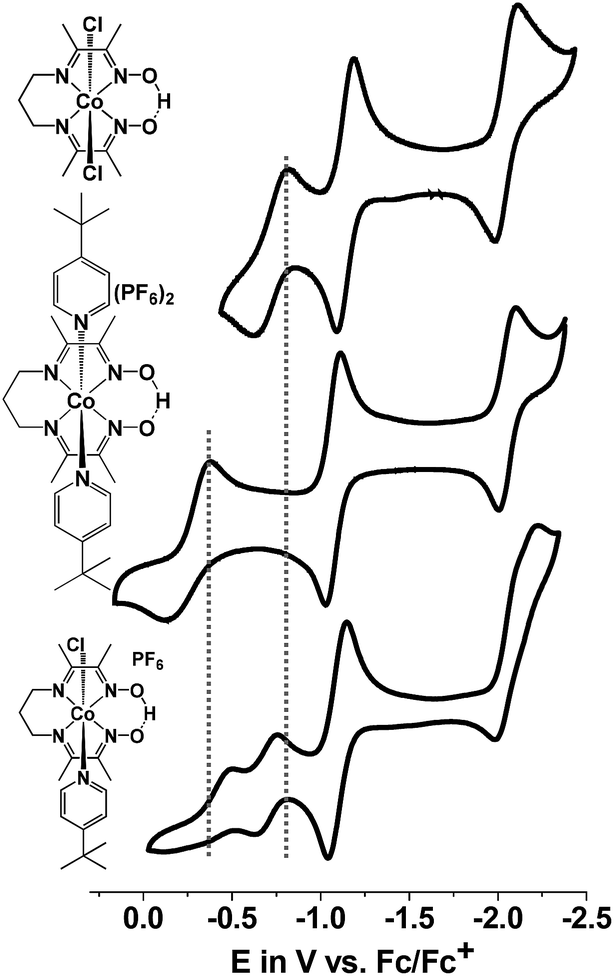 | ||
| Fig. 6 Comparison of CVs for the heteroaxial 1 (bottom), homoaxial [CoIII(prdioxH)(Cl)2] (top), and homoaxial [CoIII(prdioxH)(4tBupy)2]PF6 (middle), in CH3CN. The second CoIII/CoII event appears at similar potentials for 1 and its dichloro analogue, suggesting the formation of the latter species during the reduction of 1. | ||
A potential of −0.70 VFc/Fc+ observed for the second CoIII/CoII process of 1 matched the equivalent process in the dichloro analog, thus suggesting its formation during the CV experiment. A similar result was obtained for 3. On the other hand, there was considerable mismatch between the cathodic potential for 1 at −0.49 VFc/Fc+ and the cathodic CoIII/CoII potential of the bis-pyridine species at −0.37 VFc/Fc+. These combined results are suggestive that the uncoordinated chloride ion is capable of displacing the pyridine coordinated to the Co ion in 1. However formation of a homoaxial bis-pyridine adduct is highly unlikely. In order to determine whether this substitution is driven by electrochemical reduction7 or by chemical lability of the axial ligands in the trivalent state of 1, we performed 1H-NMR experiments in which external sources of chloride or 4tBupyridine were added. In independent experiments we added 1 equiv. 4tBupyridine, tetraethylammonium chloride (Et4NCl) or triethylamine (Et3N) to the CD3CN solution of 1 and compared their 1H-NMR spectra with those of 1 and free 4tBupyridine (Fig. 7a). Note that in these experiments the valence of the CoIII ion remained unchanged. Complete spectra are shown in Fig. S13.†
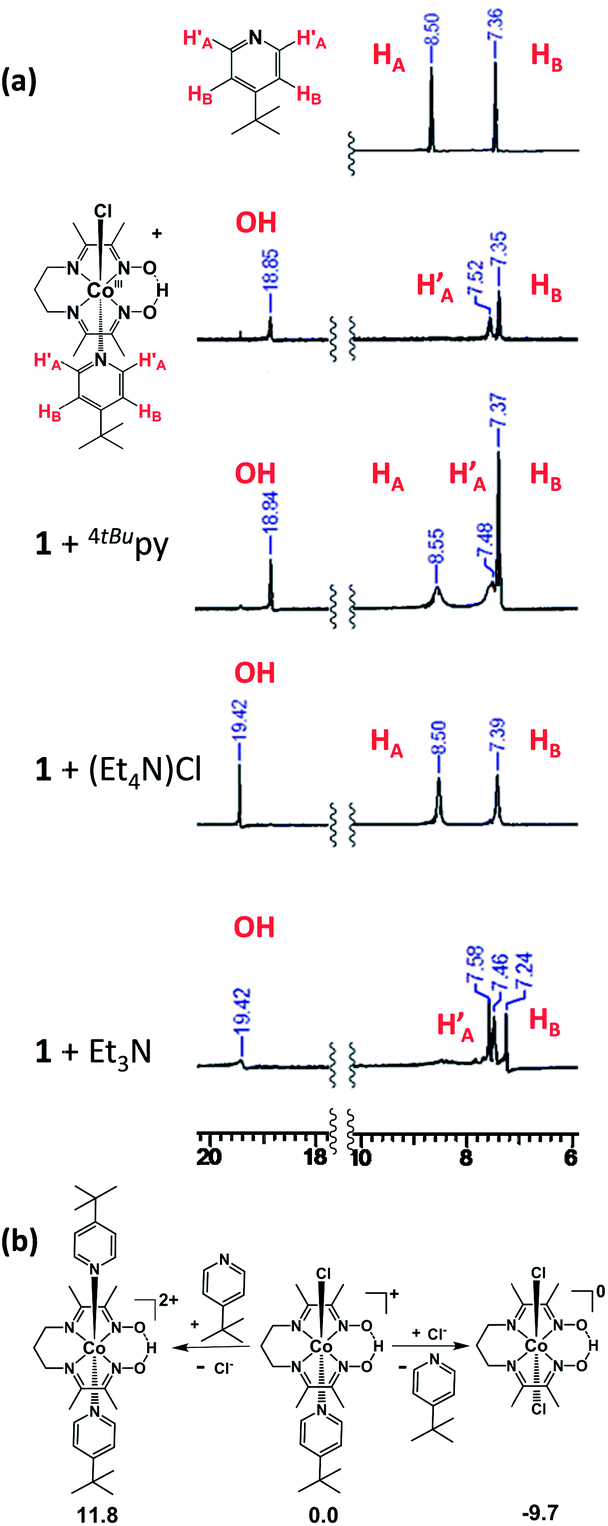 | ||
| Fig. 7 (a) 1H-NMR experiments of 1 in CD3CN upon addition of 1 equiv. of 4tBupyridine, Et4NCl, or Et3N. (b) Energetics of substitution of the axial ligands in 1 as obtained by DFT calculations (energies given in kcal mol−1). As demonstrated by the 1H-NMR spectra and DFT calculations, chloride can replace the 4tBupyridine ligand in 1 but the 4tBupyridine ligand cannot substitute the chloride. | ||
Uncoordinated 4tBupyridine exhibited two sets of 1H-NMR peaks at 7.36 (HB) and 8.50 (H′A) ppm whereas the coordinated 4tBupyridine in 1 displayed peaks at 7.35 (HB) and 7.52 (HA) ppm. The HB protons retain the same chemical shift but the H′A protons shifted from 8.50 to 7.52 ppm on coordination of the 4tBupyridine to the metal center. After 1 equiv. of 4tBupyridine was added to the CD3CN solution of 1, the appearance of signals from the uncoordinated 4tBupyridine protons at 8.55 ppm (2H) was observed and an increase of the intensity of the peak at 7.37 ppm (2H + 2H) was found whereas the proton count at 7.48 ppm of the coordinated 4tBupyridine did not change in intensity suggesting no coordination of the external 4tBupyridine ligand and therefore no replacement of the chloride on cobalt. On the contrary, when 1 equiv. of Et4NCl (the external chloride source) was added to 1, peaks at 7.39 (2H) and 8.50 ppm (2H) were found which are characteristic of the uncoordinated 4tBupyridine ligand. Therefore we conclude that 4tBupyridine in 1 can be replaced by chloride. This substitution is chemical in nature and the Co ion remains trivalent. The opposite, i.e. replacement of chloride by pyridine, is not observed. This substitution pattern was corroborated by DFT calculations13via comparison of the energetics of the different chemical events. The calculations, reported as free energy changes in kcal mol−1, found the low spin states to be energetically favorable (Table S1†). The low spin state was also confirmed by EPR spectroscopy. The substitution of the 4tBupyridine ligand by chloride is favorable by ca. 10 kcal mol−1, while replacement of chloride by external 4tBupyridine is unfavorable by ca. 12 kcal mol−1 (Fig. 7b). The energetics of pyridine substitution are shown in Table 2 for 1–3 and the proposed mechanism involves an unsaturated five-coordinate intermediate. This event requires ca. 14 kcal mol−1, is energetically unfavorable and the limiting step for 1 and 3. Chloride addition to the five-coordinate intermediate is favored by 23 kcal mol−1 and drives the overall (A + B) substitution process forward by 9–10 kcal mol−1.
On the other hand, complex 2 with an electron-donating pyrrolidine ligand has the most energy-demanding first step (20 kcal mol−1) and shows little preference for pyridine substitution with a total energy of the substitution process at −3.2 kcal mol−1. This is in agreement with absence of a second CoIII/CoII process during the CV experiment. We also probed the acidic nature of the oxime-OH hydrogen as it can contribute to the CV response and found out that no deprotonation, i.e. no change of the proton count or shift of the oxime-OH hydrogen at 18.84 ppm was observed upon addition of 4tBupyridine. On the contrary, when 1 equiv. of Et3N was added to 1 in CD3CN, we observed near complete disappearance of the oxime-OH peak intensity and slight shift of the position of the signal from the OH suggesting that Et3N can deprotonate the ligand. In summary, these results allow us to conclude that the second CoIII/CoII redox event observed for 1 and 3 is associated with the chemical formation of [CoIII(prdioxH)(Cl)2]. The chemical, rather than electrochemical basis for this process is supported by the fact that external 4tBupyridine is unable to replace the chloride in the trivalent Co ion nor to deprotonate the oxime –O–H⋯O– moiety. On the other hand, facile replacement of 4tBupyridine by chloride on Co(III) was observed. This observation of a chemical, rather than electrochemical process diverges from the mechanism proposed by Gerli and Marzilli.7 Furthermore, the presence of an electron-donating moiety attached to the pyridine strengthens the Py–Co bond and prevents substitution in 2.
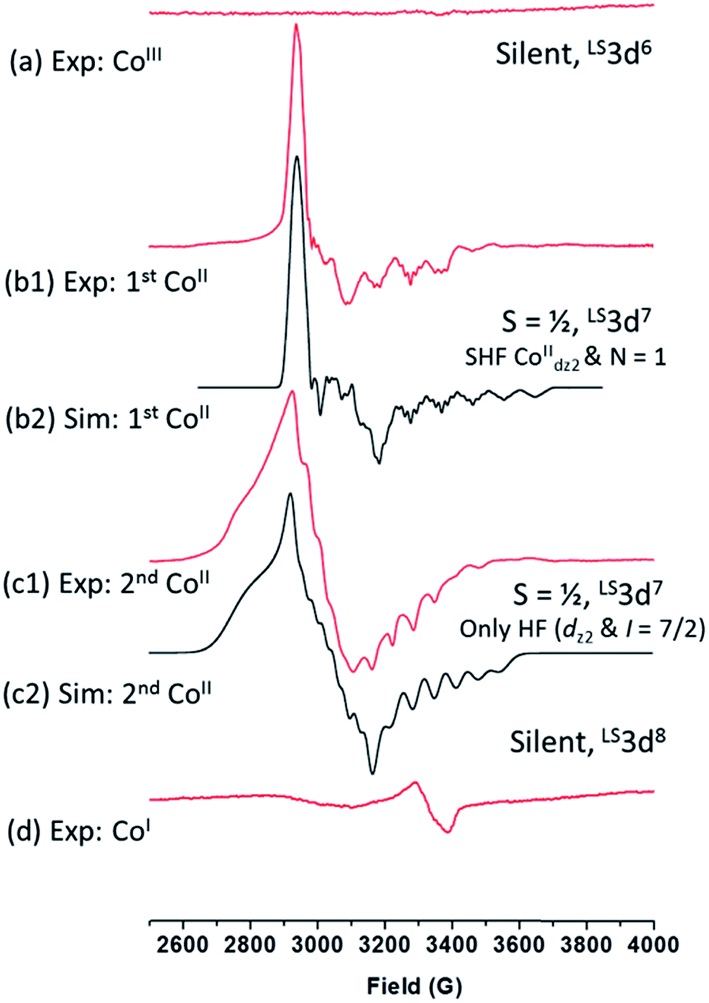 | ||
| Fig. 8 CW X-band EPR experiments to probe the spin-states and coordination environments of the complexes (CoIII, CoII, and CoI) generated during the electrochemistry of 1 in CH3CN. Spectra (b1) and (c1) were obtained after bulk electrolysis of 1 at −0.20 VAg/AgCl and −0.46 VAg/AgCl, respectively. | ||
Based on the EPR data alone it is not possible to pinpoint the nature of these axial ligands, however we rule out the presence of two 4tBupyridines based on the mismatch of observed redox potentials between 1 and [CoIII(prdioxH)(4tBupy)2]2+ in Fig. 6. Furthermore, DFT calculations suggest that formation of a [CoII(prdioxH)(CH3CN)2]+ from [CoII(prdioxH)(CH3CN)]+ is unfavorable by about 9 kcal mol−1 in good agreement with results reported by Artero et al.6m Therefore, we propose that once the chloride is lost, a five-coordinate species [CoII(prdioxH)(4tBupy)]+ is formed as an intermediate. Loss of chloride following the reduction of 1 leads to minor conformational changes, where the CoII ion is displaced by 0.15 Å out of the plane of the macrocyclic ligand and towards the remaining 4tBupyridine (Fig. 9).
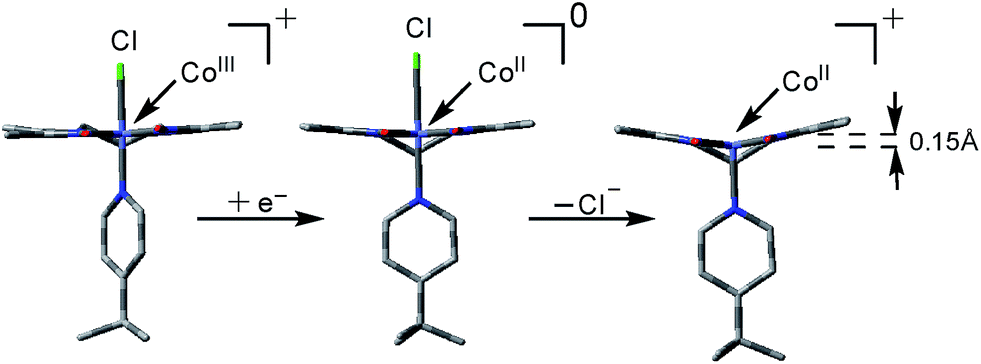 | ||
| Fig. 9 DFT-optimized geometries of [CoIII(prdioxH)(4tBupy)(Cl)]+, [CoII(prdioxH)(4tBupy)(Cl)], and [CoII(prdioxH)(4tBupy)]+. The metal center is displaced by 0.15 Å off of the plane of the macrocyclic oxime ligand in [CoII(prdioxH)(4tBupy)]+. | ||
This change likely sterically precludes coordination of a second 4tBupyridine, but may allow for coordination of a linear CH3CN molecule. This coordination to [CoII(prdioxH)(4tBupy)]+ species is, in principle, thermodynamically unfavorable in good agreement with the literature.6m However it may take place considering the large excess of CH3CN as solvent. The CoII-species resulting from the second redox event at −0.46 VAg/AgCl equally supports a LS3d7 center with S = 1/2, but with a different g-tensor, hyperfine structure, and no superhyperfine splitting (Fig. 8(c1)). The simulation of this spectrum (Fig. 8(c2)) yields the following magnetic resonance parameters: g = 2.320, 2.200, 2.017; A(59Co) = 120, 95, 180 MHz. These results suggest that axial coordination by nitrogen atoms is unlikely.14a Similarly, the magnetic resonance parameters, especially the hyperfine splitting in the high field region due to 59Co, do not support coordination by oxygen, e.g., from adventitious H2O. We tentatively assign this spectrum to a Co-species with one or two axial chloride ions. Titration experiments were carried out with 1 to evaluate the effects of adding chloride and 4tBupyridine ligands during the electrochemical experiment (Fig. 10a).
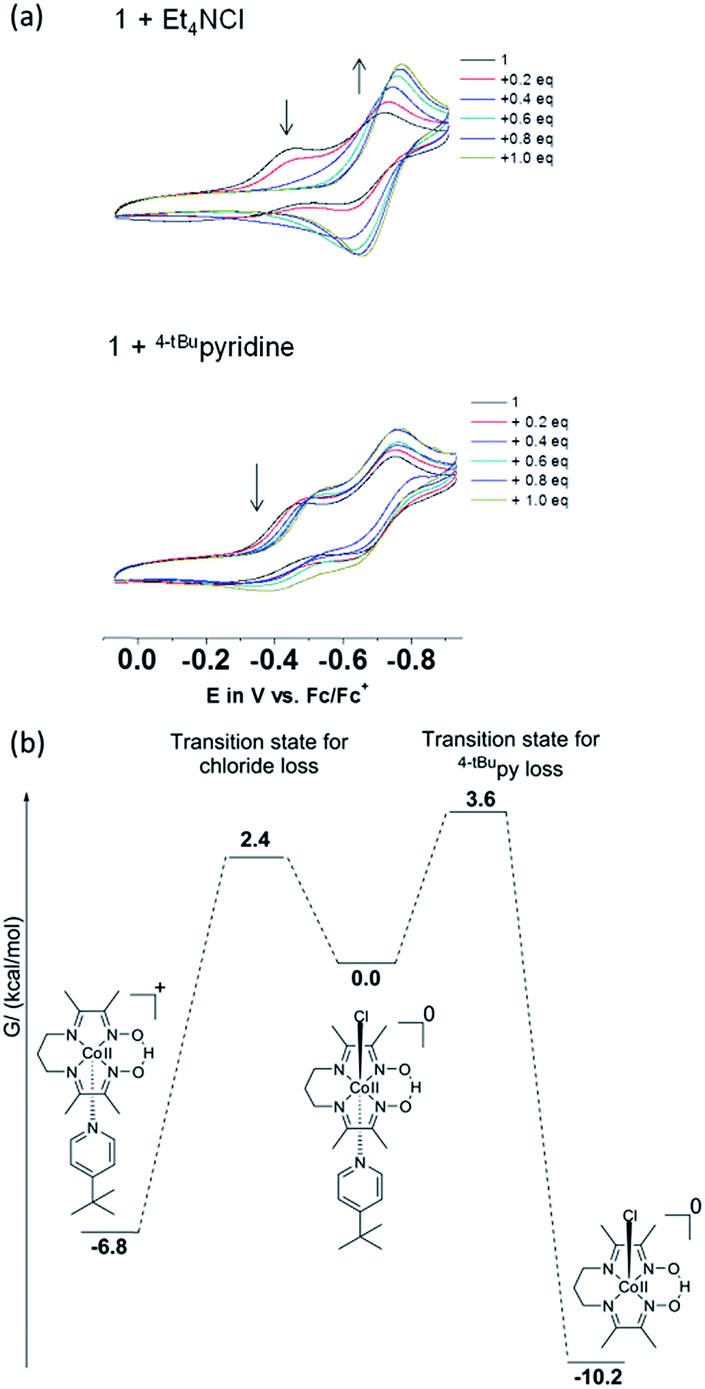 | ||
| Fig. 10 (a) Changes of the CVs of 1 upon addition of Et4NCl or 4tBupyridine in CH3CN. The first CoIII/CoII event gradually disappears with increasing Cl−, whereas 4tBuPy has no effect. (b) Calculated free energies and activation barriers for 1. Dissociation of Cl− is more favorable than that of 4tBuPy yielding [CoIII(prdioxH)(4tBupy)]+. | ||
We observed that in presence of increasing amounts of Et4NCl, the first CoIII/CoII process decreases while the second process increases in intensity. While the first process disappeared after ca. 0.6 equivalents of Et4NCl, addition of 4tBupyridine showed only minor cathodic shifts for both the CoIII/CoII processes. The CV (Fig. S5†) of a fresh solution prepared with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Et4NCl and 1 confirmed the disappearance of the first CoIII/CoII process and an increase in intensity of the second process. As expected, both the CoIII/CoII processes retained their intensities in a similarly prepared solution of 1:1 4tBupyridine and 1 (Fig. S5†). These results confirm the CV experiments in Fig. 10a and suggest that the chloride ion is involved in both the CoIII/CoII reduction events of complex 1. On the other hand, 4tBupyridine has little or no effect on the CoIII/CoII reduction. DFT calculations support that loss of chloride after the first CoIII/CoII reduction of 1 is thermodynamically favorable by 6.8 kcal mol−1 and yields the five-coordinate [CoII(prdioxH)(4tBupy)]+. Alternatively, dissociation of the 4tBupyridine ligand is calculated to be lower in energy by 10.2 kcal mol−1. These results demonstrate that there is a greater thermodynamic preference of [CoII(prdioxH)(4tBupy)(Cl)] to release 4tBupyridine rather than chloride giving rise to [CoII(prdioxH)(Cl)]. However, calculations of the transition states of the axial ligand dissociation find that loss of chloride is associated with a smaller kinetic barrier than that of the dissociation of 4tBupyridine (Fig. 10b). The difference between the activation energies is 1.2 kcal mol−1. The Arrhenius model, k = Ae(−E/RT), estimates that the rate constant k of a chemical reaction increases by 10 times with a decrease of the activation energy E by 1.36 kcal mol−1 at 298 K. Therefore, we propose that the dissociation of the axial ligands from [CoII(prdioxH)(4tBupy)(Cl)] is kinetically controlled favoring the formation of [CoII(prdioxH)(4tBupy)(CH3CN)]+ which is confirmed by EPR as well (Fig. 8). A comparison of X-ray crystal and DFT-calculated metal–ligand bond distances for 1 is shown in Table 3 along with the DFT bond distances for the 1e− reduced analog of 1. The CoIII reduction is associated with filling of the dz2 orbital which is antibonding in nature. This process increases the bond distance along the z axis of the CoII complex facilitating dissociation of the axial ligands. The Co–Cl bond elongates by 0.441 Å, thus considerably more than the 0.300 Å observed for the Co–N5 (4tBupy) bond and becomes more labile so that loss of chloride is invoked upon reduction of 1. The dichloro analog [CoII(prdioxH)(Cl)2]−, obtained from the reduction of the in situ generated [CoIII(prdioxH)(Cl)2], is negatively charged and chloride loss is expected to be even more favorable. This ligand loss is calculated at 9.7 kcal mol−1 and yields the five-coordinate neutral [CoII(prdioxH)(Cl)] species. Uptake of a CH3CN molecule by the latter species followed by substitution of the remaining chloride by 4tBupyridine yields [CoII(prdioxH)(4tBupy)(CH3CN)]+. These events are calculated at 10.0 and 3.6 kcal mol−1, respectively. These substitution processes are thermodynamically less favorable and may be slow, as suggested by the absence of hyperfine structure in the EPR spectrum of the CoII-species obtained after the second CoIII reduction of 1. We conclude that a six-coordinate geometry with 4tBupyridine as one axial ligand and a CH3CN ligand as second is favored by the CoII species formed after the first reduction.
:1 Et4NCl and 1 confirmed the disappearance of the first CoIII/CoII process and an increase in intensity of the second process. As expected, both the CoIII/CoII processes retained their intensities in a similarly prepared solution of 1:1 4tBupyridine and 1 (Fig. S5†). These results confirm the CV experiments in Fig. 10a and suggest that the chloride ion is involved in both the CoIII/CoII reduction events of complex 1. On the other hand, 4tBupyridine has little or no effect on the CoIII/CoII reduction. DFT calculations support that loss of chloride after the first CoIII/CoII reduction of 1 is thermodynamically favorable by 6.8 kcal mol−1 and yields the five-coordinate [CoII(prdioxH)(4tBupy)]+. Alternatively, dissociation of the 4tBupyridine ligand is calculated to be lower in energy by 10.2 kcal mol−1. These results demonstrate that there is a greater thermodynamic preference of [CoII(prdioxH)(4tBupy)(Cl)] to release 4tBupyridine rather than chloride giving rise to [CoII(prdioxH)(Cl)]. However, calculations of the transition states of the axial ligand dissociation find that loss of chloride is associated with a smaller kinetic barrier than that of the dissociation of 4tBupyridine (Fig. 10b). The difference between the activation energies is 1.2 kcal mol−1. The Arrhenius model, k = Ae(−E/RT), estimates that the rate constant k of a chemical reaction increases by 10 times with a decrease of the activation energy E by 1.36 kcal mol−1 at 298 K. Therefore, we propose that the dissociation of the axial ligands from [CoII(prdioxH)(4tBupy)(Cl)] is kinetically controlled favoring the formation of [CoII(prdioxH)(4tBupy)(CH3CN)]+ which is confirmed by EPR as well (Fig. 8). A comparison of X-ray crystal and DFT-calculated metal–ligand bond distances for 1 is shown in Table 3 along with the DFT bond distances for the 1e− reduced analog of 1. The CoIII reduction is associated with filling of the dz2 orbital which is antibonding in nature. This process increases the bond distance along the z axis of the CoII complex facilitating dissociation of the axial ligands. The Co–Cl bond elongates by 0.441 Å, thus considerably more than the 0.300 Å observed for the Co–N5 (4tBupy) bond and becomes more labile so that loss of chloride is invoked upon reduction of 1. The dichloro analog [CoII(prdioxH)(Cl)2]−, obtained from the reduction of the in situ generated [CoIII(prdioxH)(Cl)2], is negatively charged and chloride loss is expected to be even more favorable. This ligand loss is calculated at 9.7 kcal mol−1 and yields the five-coordinate neutral [CoII(prdioxH)(Cl)] species. Uptake of a CH3CN molecule by the latter species followed by substitution of the remaining chloride by 4tBupyridine yields [CoII(prdioxH)(4tBupy)(CH3CN)]+. These events are calculated at 10.0 and 3.6 kcal mol−1, respectively. These substitution processes are thermodynamically less favorable and may be slow, as suggested by the absence of hyperfine structure in the EPR spectrum of the CoII-species obtained after the second CoIII reduction of 1. We conclude that a six-coordinate geometry with 4tBupyridine as one axial ligand and a CH3CN ligand as second is favored by the CoII species formed after the first reduction.
| Co–N1 | Co–N2 | Co–N3 | Co–N4 | Co–N5 | Co–Cl | ||
|---|---|---|---|---|---|---|---|
| a Values are in Å. | |||||||
| Experiment | [CoIII(prdioxH)(4tBupy)(Cl)]+ | 1.903 | 1.917 | 1.914 | 1.898 | 1.975 | 2.237 |
| Theory | [CoIII(prdioxH)(4tBupy)(Cl)]+ | 1.907 | 1.925 | 1.928 | 1.905 | 1.988 | 2.304 |
| Theory | [CoII(prdioxH)(4tBupy)(Cl)] | 1.904 | 1.927 | 1.928 | 1.904 | 2.288 | 2.745 |
A five-coordinate geometry with 4tBupyridine as the axial ligand maybe present in a certain admixture. Even though the binding of CH3CN is – according to DFT calculations – energetically not favorable, the large excess of CH3CN as solvent shifts the equilibrium towards the six-coordinate species. The 3d7 CoII species are low-spin. The second reduction involves conversion of [CoIII(prdioxH)(Cl)2] to [CoII(prdioxH)(Cl)2]− that yields [CoII(prdioxH)(Cl)] and possibly [CoII(prdioxH)(CH3CN)(Cl)]. In the divalent state chloride is amenable to substitution by 4tBupyridine ligand thus forming [CoII(prdioxH)(4tBupy)]+ and possibly [CoII(prdioxH)(4tBupy)(CH3CN)]+. Therefore, prior to the reduction from CoII → CoI a single species [CoII(prdioxH)(4tBupy)(CH3CN)]+ is present in solution, as evidenced by the presence of a single wave in Fig. 4. Like for the first reduction, the 3d7 CoII species are low-spin.
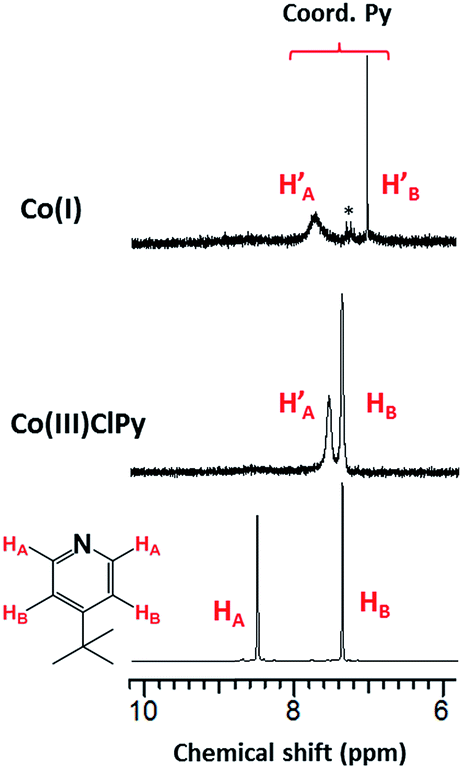 | ||
| Fig. 11 1H-NMR spectra of [CoI(prdioxH)(4tBupy)], in CD3CN. The asterisk denotes the presence of adventitious toluene. See ESI† for the complete spectra. | ||
Well-defined peaks between 1 and 9 ppm confirmed the low spin nature of the species. Three peaks associated with the aromatic protons of the 4tBupyridine ligand appear between 7 and 9 ppm. The peaks observed at 7.70 and 7.00 ppm are associated with the HA and HB protons of the coordinated 4tBupyridine, as discussed in Fig. 7. This species is described as [CoI(prdioxH)(4tBupy)] (4). Five-coordinate CoI complexes similar to 4 have been reported.8,9,16
While running the previous NMR experiments we were able to grow crystals from the deuterated solvent and solve the structure of a dimeric product in Fig. 12. The structure confirms the presence of an apical pyridine on the catalytically active CoI species thus validating the proposed mechanism beyond reasonable doubt. In the time scale of the cyclic voltammetric and electrocatalytic experiment, this species is clearly monomeric, suggesting that the observed dimer is the result of a kinetically sluggish reaction. Dimer formation resulting from reduction with cobaltocene derivatives has been observed in related oxime species17 and might point out to possible deactivation pathways for the catalyst.
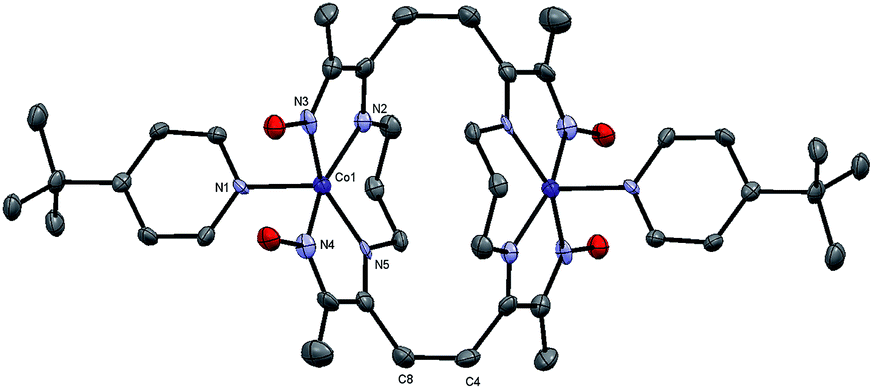 | ||
| Fig. 12 ORTEP representation of the crystal structure for the neutral dimer of 4 (CCDC 1448834) at 50% ellipsoid probability. Hydrogen atoms omitted for clarity. Co(1)–N(1) = 1.997(4); Co(1)–N(2) = 1.902(4); Co(1)–N(3) = 1.866(4); Co(1)–N(4): 1.850(4); Co(1)–N(5) = 1.890(4); C(4)–C(8) = 1.592(6). | ||
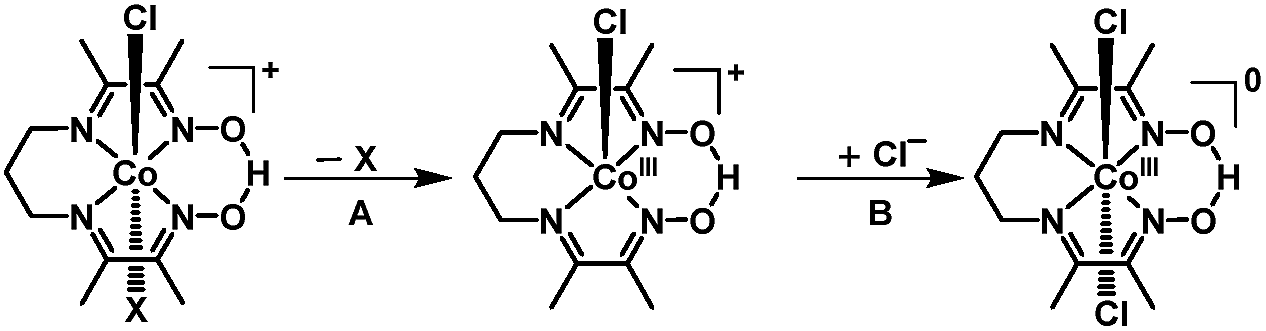
In light of the comprehensive UV-visible, EPR, NMR, and DFT data accumulated in our study, we propose a viable chemical and electrochemical pathway starting from the parent 1 and reducing to the CoII counterpart and to the catalytically active monovalent species (Scheme 1). The first CoIII/CoII reduction shown in step 1 is followed by loss of a chloride to yield the five-coordinate CoII species [CoII(prdioxH)(4tBupy)]+. The latter species incorporates a CH3CN molecule to form the six-coordinate CoII complex [CoII(prdioxH)(4tBupy)(CH3CN)]+ described in step 2. The released chloride in step 1 replaces the 4tBupyridine in the remaining [CoIII(prdioxH)(4tBupy)(Cl)]+ (1) as observed generating a second six-coordinate CoIII species with two chlorides occupying the axial positions, step 3, along with uncoordinated 4tBupyridine. This is a chemical, rather than electrochemical step. The second CoIII/CoII reduction, shown in step 4, yields the six-coordinate [CoII(prdioxH)(Cl)2]− that is subsequently converted into the five-coordinate [CoII(prdioxH)Cl]. Uptake of a solvent CH3CN molecule by the latter species (step 5) may take place and is followed by replacement of the remaining chloride with one 4tBupyridine present in solution to give rise to the species [CoII(prdioxH)(4tBupy)(CH3CN)]+ in step 6. This species is then reduced and transforms into the five-coordinate and catalytically active [CoI(prdioxH)(4tBupy)] with release of the CH3CN molecule in step 7. It is important to mention that Marzilli7 proposed generation of a CoII–Cl species already in step 1. It was further proposed that an outer sphere electron transfer mechanism involving the newly generated CoII–Cl species and the parent CoIII complex, and chloride transfer from one metal center to another could yield a CoIII dichloro species. The chloride transfer event is calculated to be favorable by 6.7 kcal mol−1. The present study offers an alternative pathway where the CoIII dichloro species is generated via substitution of 4tBupy already on the parent CoIII complex by an external chloride, this process being favorable by 9.7 kcal mol−1.
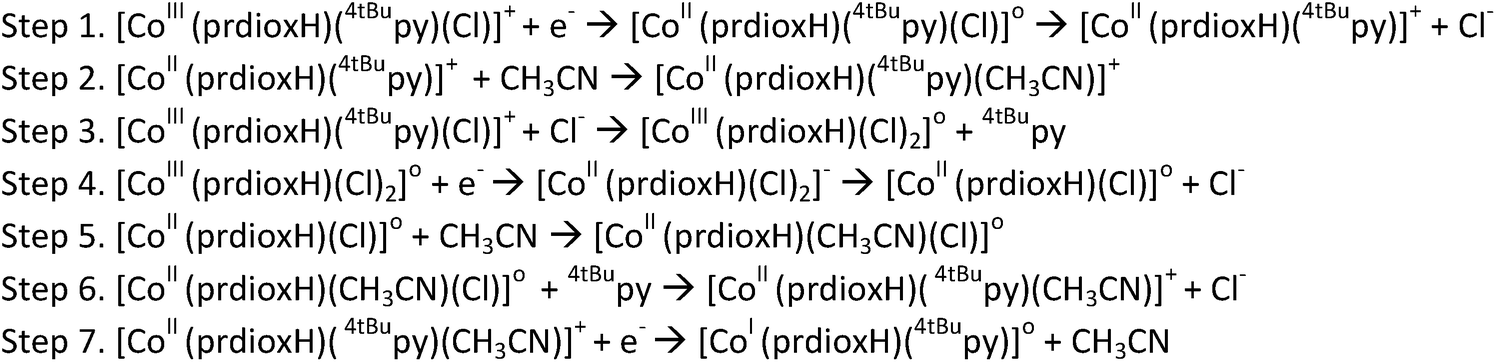 | ||
| Scheme 1 Proposed chemical and electrochemical pathway from 1 the catalytically active CoI species. | ||
Catalytic activity and proposed mechanisms
The optimization of proton-reducing catalysts requires a detailed study of catalytic parameters such as overpotentials (η), turnover numbers (TON), and faradaic efficiencies (% FE) allied to an understanding of the plausible catalytic mechanisms. In the next paragraphs we discuss the experimental and theoretical results observed for complexes 1–3.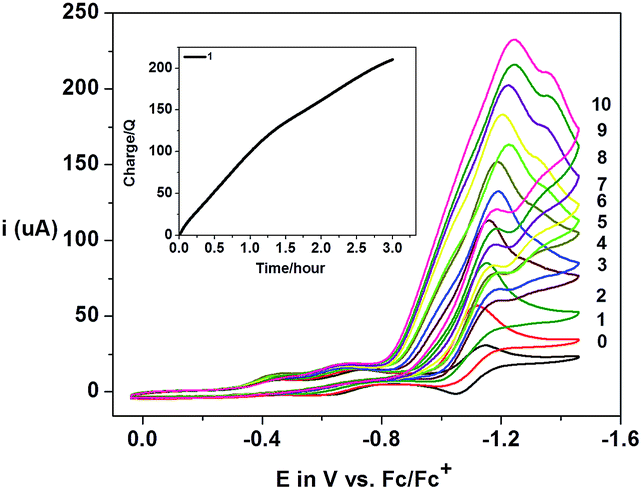 | ||
| Fig. 13 CV experiments showing the electrocatalytic behavior of 1 towards H2 generation in the presence of HTFA in CH3CN solution. The numbers 0–10 indicate the number of equiv. of HTFA. (Inset) Plot of charge consumed against time during the bulk-electrolysis experiment for dihydrogen generation by 1 after 3 h. | ||
:100
| Catalytic parameters | 1 | 2 | 3 |
|---|---|---|---|
| E(H+/H2) (V) vs. Fc/Fc+ | −1.03 | −1.04 | −1.03 |
| Overpotential (V) (10 eq. HTFA) | 0.35 | 0.36 | 0.35 |
| TON/3 h (4.0 × 10−5 mol) | 18.7 | 13.8 | 13.8 |
| Faradaic efficiency (%) | 77 | 84 | 75 |
In presence of p-cyanoanilinium (pKa 7.6 in CH3CN), the electrocatalytic wave of a cobalt oxime complex was observed at a potential of 320 mV more positive than that required for the CoII/CoI reduction6m and a pathway involving the protonation of the oxime bridge was proposed to explain the observed potential shift. Although the thermodynamics of these pathways have been studied in reasonable detail, the free energy barriers for the proton transfer and H2 production steps are considerably less understood.
Recent studies consider the protonation of the glyoxime bridge by a CoIII–H− moiety19 or even the lack of formation of CoIII–H−.17 Although potentially relevant—especially to evaluate catalyst decomposition mechanisms—these pathways will require further validation and for the purposes of this study we will assume the need for formation of the CoIII–H− hydride intermediate. The protonation of the oxime bridge in [CoII(prdioxH)(4tBupy)]+ species seems energetically unfavorable by 19 kcal mol−1. This is consistent with a previous study where it was proposed that the weak acid HTFA cannot protonate the oxime moiety.6f
Complexes 1–3 displayed electrocatalytic waves in the presence of 10 equiv. HTFA at potentials close to the reduction of CoII/CoI. The respective E1/2(H+/H2) are −1.03, −1.04 and −1.03 VFc/Fc+ and the CoII/CoI reduction potentials are −1.09, −1.07 and −1.09 VFc/Fc+ for 1, 2, and 3, respectively. The catalytic pathway calculated for 1 is shown in Fig. 14. The calculated potential for the CoII/CoI reduction of 1 is −0.93 VFc/Fc+ and the species obtained after reduction, [CoI(prdioxH)(4tBupy)], is neutral and can easily take up a proton upon reaction with HTFA. This cobalt protonation is favorable by 8.4 kcal mol−1 and yields the cobalt-hydride species described as [CoIII(H−)(prdioxH)(4tBupy)]+. This is expected because CoI is a closed-shell 3d8 center and can behave as a good Lewis base.
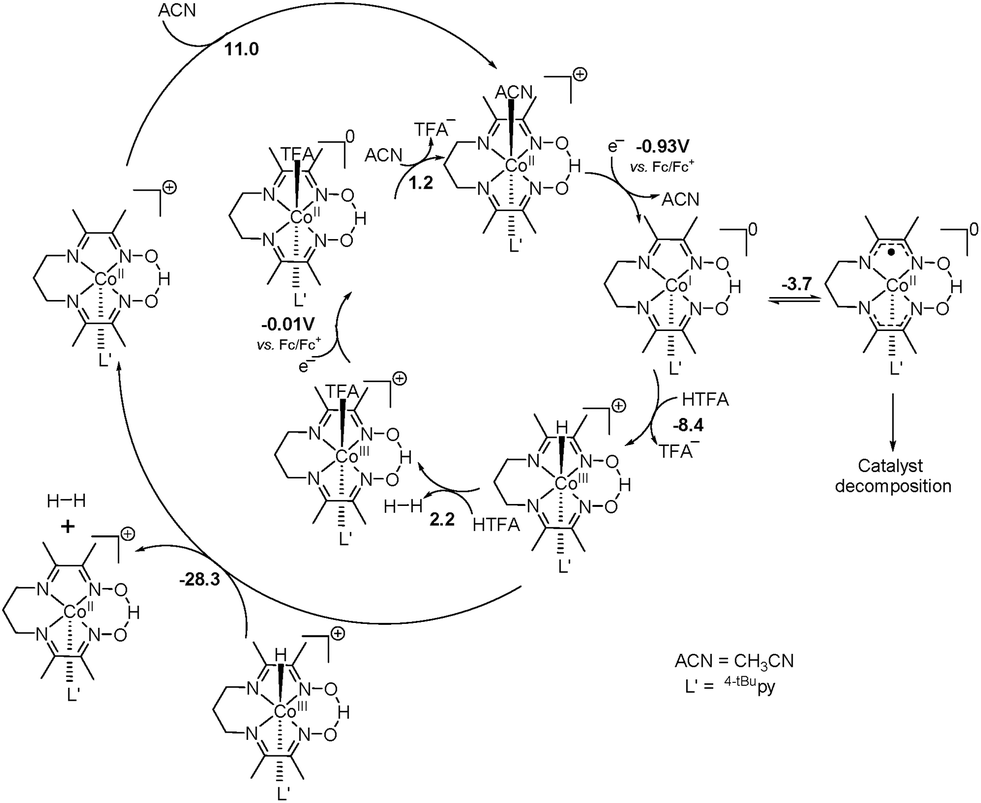 | ||
| Fig. 14 The catalytic pathways calculated for H2 evolution by 1 in the presence of HTFA in CH3CN (ACN). The formation of a CoIII–H− species is invoked. This complex can react either heterolytically with a proton or homolytically with another CoIII–H− complex to yield H2. The heterolytic pathway involves an acetate-bound Co complex. The homolytic mechanism is more likely at low concentrations of acid. Energies given in kcal mol−1 as calculated at the B3PW91//TZVP/6-311++G(d,p) level of theory. | ||
The reduction potential for the generation of a CoII–H− from CoIII–H− is −1.43 VFc/Fc+ and it is in agreement with the appearance of a shoulder at about −1.40 VFc/Fc+ at higher concentrations of acid in Fig. 13. This potential is more negative than that of the CoII/CoI couple of −0.93 VFc/Fc+. Similarly, a PCET event for conversion of the CoI complex into the CoII–H− species is calculated to be −1.39 VFc/Fc+. Because electrocatalysis occurs near the measured value of E(CoII/CoI) (Fig. 13), the involvement of a CoII–H− species is not invoked in the catalytic mechanism. The electrocatalytic wave did not show an anodic shift with the decrease of pH, when compared to the reduction potential of the CoII/CoI couple (Fig. 13) and therefore, a PCET mechanism from the CoII complex, [CoII(prdioxH)(4tBupy)(CH3CN)]+, to the CoIII–H− species is not considered. The protonation of the latter CoIII–H− species by HTFA leads to the generation of H2. This event is calculated to be slightly uphill by 2.2 kcal mol−1 and is facilitated by the excess of acid present. The CoIII complex obtained after the evolution of H2 contains a trifluoroacetate ligand bound to the metal center.6l The reduction of this species is calculated at −0.01 VFc/Fc+ and yields the corresponding CoII complex, [CoII(prdioxH)(4tBupy)(TFA)], which can exchange the trifluoroacetate ligand by a CH3CN molecule, reentering the catalytic cycle. This ligand exchange event is nearly isoenergetic at about 1 kcal mol−1 and is expected to be favored by the large concentration of the solvent.
The bimolecular mechanism involving two [CoIII(H−)(prdioxH)(4tBupy)]+ complexes can generate H2 favorably at −28.3 kcal mol−1. However, this pathway may have a moderate kinetic barrier arising from the electrostatic repulsion between two cationic species and is likely plausible at lower concentrations of acid. It is also noteworthy that the complex [CoI(prdioxH)(4tBupy)] obtained after the reduction of [CoII(prdioxH)(4tBupy)(CH3CN)]+ can undergo an intramolecular electron transfer from metal to the oxime ligand framework giving rise to a radical-containing [CoII(prdioxH˙)(4tBupy)]. Similar to other pathways17,19 where the ligand is involved, the latter species may contribute to catalyst decomposition. A detailed study about the role of this species is currently underway.
Conclusions
A series of heteroaxial complexes in a trivalent cobalt/oxime macrocyclic framework was synthesized and characterized by multiple physico-chemical techniques. The electrochemistry of these species exhibited complex behavior with two CoIII/CoII and one CoII/CoI processes; the origin of which has been identified with multiple redox and spectroscopic methods like CV, 1H-NMR, UV-vis, EPR, and DFT calculations. A detailed understanding of the chemical and electrochemical pathways with distinct coordination preferences was revealed: (i) the first CoIII/CoII reduction involves chloride loss followed by incorporation of CH3CN to form a six-coordinate CoII complex; (ii) the free chloride replaces 4tBupyridine in the remaining unreduced 1, and forms a second CoIII species with chlorides occupying the axial positions; (iii) this complex is then reduced and yields a five-coordinate CoII species that accommodates a solvent molecule at its sixth axial position; (iv) available 4tBupyridine replaces the remaining chloride; (v) the resulting species is then reduced, transforming into the five-coordinate and catalytically active [CoI(prdioxH)(4tBupy)]. The complexes exhibited catalytic properties towards proton reduction in HTFA/CH3CN with low overpotentials and good turnover numbers. Variation of the substituent groups in the pyridine ligand affected the final TONs; however overpotentials remained relatively unchanged. A detailed catalytic mechanism favors a heterolytic pathway for hydrogen generation, and involves: (i) a favorable CoI protonation that yields the required CoIII–H− species; (ii) this complex reacting heterolytically with an available proton from HTFA, generating H2, and therefore regenerating CoIII; (iii) this CoIII species first incorporating TFA and undergoing reduction followed by CH3CN exchange prior to reentering the catalytic cycle. Bimolecular mechanisms seem favorable only at lower acid concentrations, and both the CoII/CoIII–H− and CoI/CoII–H− PCET events seem unlikely. These results allow for some generalizations on the behavior of heteroaxial cobalt macrocycles and serve as guidelines for the development of new catalysts based on macrocyclic frameworks.Materials, methods and synthetic procedures
General
Reagents and solvents were used as received from commercial sources. Infrared spectra were recorded from 4000 to 650 cm−1 as KBr pellets on a Bruker Tensor 27 FTIR spectrophotometer. 1H-NMR spectroscopy was carried out with a Mercury FT-NMR 400 MHz setup using CD3CN as solvent at 25 °C. Chemical shifts are plotted vs. TMS. ESI-(+) mass spectrometry was measured in a triple quadrupole Micromass Quattro LC equipment where experimental mass patterns were fitted with the theoretical isotopic distribution. Elemental analysis (C, H, and N) was performed using an Exeter analytical CHN analyzer by Midwest Microlab, an independent lab located in Indianapolis, Indiana, USA. UV-visible spectra were obtained using quartz cells at room temperature in a Varian Cary 50 spectrophotometer operating in the range of 200 to 1000 nm. Values of ε are given in M−1 cm−1. HPLC-grade CH3CN solutions were used for cyclic voltammetry and UV-absorption spectroscopy studies.Electrochemistry and bulk electrolysis
The electrochemical behavior of complexes 1–3 was investigated with a BAS 50W potentiostat/galvanostat. Cyclic voltammograms were obtained at room temperature in CH3CN solutions containing 0.1 M of n-Bu4NPF6 as the supporting electrolyte under an argon atmosphere. The electrochemical cell was comprised of three electrodes: glassy-carbon (working), platinum wire (auxiliary) and Ag/AgCl (reference). The ferrocene/ferrocenium redox couple Fc/Fc+ (Eo = 400 mV vs. NHE)20 was used as the internal standard. Peak to peak potential separation (ΔEp = |Ep,c − Ep,a|) and |ipa/ipc| values were measured to evaluate the reversibility of the redox processes. Bulk electrolysis was performed in a custom-made air-tight H-type cell with vitreous carbon as the working electrode, Ag/AgCl as the reference electrode placed in the same compartment and a Pt-coil used as the auxiliary electrode placed in another compartment separated by a frit. Controlled potential electrolysis of the complex has been done in 20 mL CH3CN with tetrabutyl ammonium hexafluorophosphate (TBAPF6) as supporting electrolyte until the calculated final charge is attained. Potentials were applied to the cell by a BAS 50W potentiostat/galvanostat and measured against the Ag/AgCl reference electrode.X-ray structural determination
For complex 1 an orange needle-shaped crystal with dimensions 0.40 × 0.16 × 0.11 mm was mounted on a Nylon loop using a small amount of paratone oil. Data were collected in a Bruker CCD (charge coupled device) based diffractometer equipped with an Oxford Cryostream low-temperature apparatus operating at 173 K. Data were measured using omega and phi scans of 0.5° per frame for 30 s. The total number of images was based on results from the program COSMO V1.58 (ref. 21a) where redundancy was expected to be 4.0 and completeness to 0.83 Å to 100%. Cell parameters were retrieved using APEX II software21b and refined using the SAINT software21c for all observed reflections. Data reduction was performed using SAINT which corrects for Lp. Scaling and absorption corrections were applied using the SADABS21d multi-scan technique, supplied by George Sheldrick. The structure was solved by the direct method using the SHELXS-97 program and refined by the least squares method on F2, SHELXL-97, which is incorporated in SHELXTL-PC V 6.10.21e The structure was solved in the space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) (# 82). All the non-hydrogen atoms are refined anisotropically. Hydrogens were calculated by geometrical methods and refined as a riding model. Solvent molecules were found to reside in the lattice void, which is presumably octane resulting from crystallization. Attempts to model these solvents and/or to redefine as CH2Cl2 failed to generate a chemically sensible model. The SQUEEZE (P. V. D. Sluis & Spek, 1990)21f function of PLATON (Spek, 2003)21g was used to eliminate the contribution of the electron density in the void from the intensity data. The total solvent area volume was found to be 625 Å3, with electron count of about 80 electrons. This corresponds to approximately two molecules of CH2Cl2 residing in the cell. The calculated F(000) and density were obtained for the cell containing two molecules of CH2Cl2 per cell or one per molecule of interest. The refinement was carried out on the new reflection file generated by PLATON. The crystal used for the diffraction study showed no decomposition during data collection. All the drawings are done at 50% ellipsoids. In the case of complex 3, a yellow plate-shaped crystal with dimensions 0.16 × 0.10 × 0.04 mm was mounted on a Nylon loop using a small amount of paratone oil. Data were collected using a Bruker CCD (charge coupled device) based diffractometer equipped with an Oxford Cryostream low-temperature apparatus operating at 173 K. Data were measured using omega and phi scans of 0.5° per frame for 30 s. The total number of images was based on results from the program COSMO V1.61 (ref. 21h) where redundancy was expected to be 4.0 and completeness to 0.83 Å to 100%. Cell parameters were retrieved using APEX II software21i and refined using SAINT for all observed reflections. Data reduction was performed using the SAINT software.21j Scaling and absorption corrections were applied using the SADABS21d multi-scan technique. The structure was solved by the direct method using the SHELXS-97 program and refined by the least squares method on F2, SHELXL-97,21e which is incorporated in OLEX2.21k The structure was solved in the space group P21/c (# 14). All the non-hydrogen atoms are refined anisotropically. Hydrogens were calculated by geometrical methods and refined as a riding model. All the drawings are done at 50% ellipsoids.
(# 82). All the non-hydrogen atoms are refined anisotropically. Hydrogens were calculated by geometrical methods and refined as a riding model. Solvent molecules were found to reside in the lattice void, which is presumably octane resulting from crystallization. Attempts to model these solvents and/or to redefine as CH2Cl2 failed to generate a chemically sensible model. The SQUEEZE (P. V. D. Sluis & Spek, 1990)21f function of PLATON (Spek, 2003)21g was used to eliminate the contribution of the electron density in the void from the intensity data. The total solvent area volume was found to be 625 Å3, with electron count of about 80 electrons. This corresponds to approximately two molecules of CH2Cl2 residing in the cell. The calculated F(000) and density were obtained for the cell containing two molecules of CH2Cl2 per cell or one per molecule of interest. The refinement was carried out on the new reflection file generated by PLATON. The crystal used for the diffraction study showed no decomposition during data collection. All the drawings are done at 50% ellipsoids. In the case of complex 3, a yellow plate-shaped crystal with dimensions 0.16 × 0.10 × 0.04 mm was mounted on a Nylon loop using a small amount of paratone oil. Data were collected using a Bruker CCD (charge coupled device) based diffractometer equipped with an Oxford Cryostream low-temperature apparatus operating at 173 K. Data were measured using omega and phi scans of 0.5° per frame for 30 s. The total number of images was based on results from the program COSMO V1.61 (ref. 21h) where redundancy was expected to be 4.0 and completeness to 0.83 Å to 100%. Cell parameters were retrieved using APEX II software21i and refined using SAINT for all observed reflections. Data reduction was performed using the SAINT software.21j Scaling and absorption corrections were applied using the SADABS21d multi-scan technique. The structure was solved by the direct method using the SHELXS-97 program and refined by the least squares method on F2, SHELXL-97,21e which is incorporated in OLEX2.21k The structure was solved in the space group P21/c (# 14). All the non-hydrogen atoms are refined anisotropically. Hydrogens were calculated by geometrical methods and refined as a riding model. All the drawings are done at 50% ellipsoids.
In the case of complex 4, blue needle-shaped crystals were grown via slow evaporation from deuterated acetonitrile. A single weakly diffracting crystal with dimensions 0.05 × 0.08 × 0.13 mm was mounted on a Nylon loop using a small amount of paratone oil. Data were collected using a Bruker CCD (charge coupled device) based diffractometer equipped with an Oxford Cryostream low-temperature apparatus operating at 100 K. Data were measured using omega and phi scans of 0.5° per frame for 90 s. Cell parameters were retrieved using APEX II software21i and refined using SAINT for all observed reflections. Data reduction was performed using the SAINT software.21j Scaling and absorption corrections were applied using SADABS21d multi-scan technique. The structure was solved by the direct method using the SHELXS-97 program and refined by the least squares method on F2, SHELXL-97,21e which is incorporated in OLEX2.21k The structure was solved in the space group C2/c. All the non-hydrogen atoms are refined anisotropically. Hydrogens were calculated by geometrical methods and refined as a riding model. All the drawings are done at 50% ellipsoids. We present this structure as a dimer molecule with a C8–C4 bond distance of 1.592 through symmetry of the unit cell. Although we are confident that our interpretation is correct, analysis of the raw data suggests that the crystal chosen is of a multiple nature with more than 3 domains, which may indeed mean that we have not correctly identified the correct unit cell. Use of different indexing programs, and thresholding levels along with viewing the data with a reciprocal viewer program provides us with what we present here as the most reasonable unit cell.
EPR spectroscopy
Continuous wave (CW) X-band (9–10 GHz) EPR experiments were carried out with a Bruker ELEXSYS E580 EPR spectrometer (Bruker Biospin, Rheinstetten, Germany), equipped with a TE102 rectangular EPR resonator (Bruker ER 4102st) and a helium gas-flow cryostat (ICE Oxford, UK). An intelligent temperature controller (ITC503) from Oxford Instruments, UK, was used. Data processing was done using Xepr (Bruker BioSpin, Rheinstetten) and Matlab 7.11.1 (the MathWorks, Inc., Natick) environment. Simulations were performed using the EasySpin software package (version 4.5.5) (Stoll & Schweiger, J. Magn. Reson., 2006).Computational methods
Calculations were performed with a development version of the Gaussian suite of programs,22a using B3PW91 (ref. 22b–d) functional with double-zeta SDD basis set on cobalt and D95 (ref. 22e and f) basis on the other atoms. All optimized structures were confirmed as minima by analyzing the harmonic vibrational frequencies. Solvation effects in CH3CN were estimated using the IEF polarizable continuum model (PCM)22g–j and were included during structure optimization. Single-point energies were reevaluated with triple-zeta TZVP basis22k on the metal atom and 6-311++G(d,p) basis22l on the other atoms in presence of the continuum solvation model. The free energies were calculated using the triple-zeta SCF energy while the zero-point energy and thermal corrections were included from the double-zeta calculations. The standard states of 1 M concentration were considered for all the reactants and products for calculating the free energies of reactions. Low-spin configurations yielded lower energies for all the species. The wave functions of the optimized structures were tested for SCF stability.22m–o Isosurface plots of orbitals and spin densities were visualized using GaussView.22p The calculation of the reduction potentials of the complexes included zero-point energy and thermal corrections and standard thermodynamic equation ΔG = −nFE was used. The calculated potentials were referenced to a value of E1/2 = 4.678 V for the ferrocene/ferrocenium couple calculated under our level of theory.Catalytic activity
Proton reduction electrocatalysis was evaluated for 1–3 by cyclic voltammetry in the presence of trifluoroacetic acid (HTFA) using a glassy carbon as working electrode, platinum wire as auxiliary and Ag/AgCl as reference electrodes with tetra-butyl ammonium tetrafluoroborate (TBABF4) as supporting electrolyte. Overpotential (η), ic/ip, and kinetic rate constant, kobs, have been calculated from the observed changes in the voltammogram. To determine the amount of dihydrogen release, bulk electrolysis has been performed in a custom-made air-tight H-type cell in the presence of a mercury-pool as working electrode, Ag/AgCl as reference electrode placed in the same compartment whereas Pt-coil used as the auxiliary electrode placed in the other compartment separated by a frit. TBABF4 was used as the supporting electrolyte. The main chamber was filled with an electrolyte solution and proton source (TBABF4: 1.317 g; TFA: 0.456 g [4 mmol], 20 mL CH3CN) and the glass-fitted chamber was filled with another electrolyte solution (TBABF4: 0.329 g; 5 mL CH3CN). In a standard experiment, the cell was purged with N2 gas for 20 minutes followed by sample head space gas (100 μL) to ensure O2-free environment in the gas chromatograph (GC). The solution was electrolyzed in the absence of the catalyst for 180 minutes at −1.0 V vs. Ag/AgCl. The head space gas was again injected into the GC and the amount of dihydrogen was recorded. Then, the cell was purged with N2 gas for another 20 minutes followed by injection of the catalyst (0.04 mmol) dissolved in CH3CN. Bulk electrolysis was conducted for another 180 minutes and the head space gas (100 μL) was injected into the GC to record the amount of dihydrogen produced. After the background subtraction, the turnover number was calculated as the ratio of the moles of dihydrogen produced over the moles of catalyst used. Faradaic efficiency was calculated from the gas chromatography measurements using the equation % FE = [(nH2)/(Q/2)] × 100, where Q is in Faraday unit. The GC is a Gow-Mac 400 instrument equipped with a thermal conductivity detector. An 8′ × 1/8′′ long 5 Å molecular sieve column operating at 60 °C was used with the carrier gas N2. The calibration was carried out with dihydrogen gas (dihydrogen GC grade 99.99%, Scotty analyzed gases, Sigma Aldrich).Synthetic procedures
The synthesis of the oxime ligand, prdioxH2,11 and the precursor [CoIII(prdioxH)(Cl)2],12 followed the literature procedure. The complex [CoIII(prdioxH)(4tBupy)2](PF6)2 used for electrochemical comparisons was obtained by modifications to the literature procedure7 and described in Scheme S1.† The synthetic procedure and characterization of [CoIII(prdioxH)(Cl)2] are described in Scheme S2.†:1) mixture. Yield: 75%. IR (KBr, cm−1) 3645 (w) (OH); 3234 (w) (aromatic-CH); 2969 (s), 2907 (m), 2871 (m) (aliphatic CH); 1620 (s) (CN); 1432 (m) (CC); 847 (s) (PF6−). 1H-NMR [400 MHz, CD3CN, 300 K] δ/ppm = 1.249 [s, 9H (t-butyl)]; 2.534 [s, 6H (CH3)]; 2.631 [s, 6H (CH3)]; 4.173 [m, 4H (CH2)]; 7.358 [d, 2H (aryl)]; 7.526 [d, 2H (aryl)]. ESI pos. in MeOH: m/z = 468.1573 for [CoIII(prdioxH)(4tBupy)(Cl)]+. Anal. calcd for C20H32ClCoF6N5O2P: C: 39.13; H: 5.25; N: 11.41; found: C: 39.05; H: 5.33; N: 11.37.
N); 1461 (m) (CC); 835 (s) (PF6−). ESI pos. in MeOH: m/z = 481.1563 for [CoIII(prdioxH)(4Pyrpy)(Cl)]+. Anal. calcd for C20H31ClCoF6N6O2P: C: 38.32; H: 4.98; N: 13.41; found: C: 38.25; H: 4.91; N: 13.36.
:1) mixture. Yield: 80%. IR (KBr, cm−1) 3509 (w) (OH); 3141 (w), 3090 (w) (aromatic-CH); 2934 (w), 2850 (w) (aliphatic CH); 1674 (m) (CO); 1613 (m), 1598 (m) (CN); 1448 (m) (CC); 840 (s) (PF6−). ESI pos. in MeOH: m/z = 516.1207 for [CoIII(prdioxH)(4Bzpy)(Cl)]+. Anal. calcd for C23H28ClCoF6N5O3P: C: 41.74; H: 4.26; N: 10.58; found: C: 41.60; H: 4.30; N: 10.16.
Acknowledgements
This research was made possible by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy through the Single-Investigator and Small-Group Research (SISGR) – Solar Energy program grants DE-SC0001907 and DE-FG02-09ER16120 to C. N. V. and H. B. S., including financial support to D. B., S. M., H. B., D. W., and X. S. Work at the Argonne National Laboratory (J. N. and O. G. P.) was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, under contract number DE-AC02-06CH11357. D. B. and S. M. contributed equally to this work. We thank Prof. J. Endicott, WSU, for insightful discussions and suggestions. We also acknowledge the WSU-Computing Grid for high-level DFT calculations.References
- (a) J. A. Turner, Science, 2004, 305, 972 CrossRef CAS PubMed; (b) N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed; (c) A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022 CrossRef CAS PubMed; (d) T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474 CrossRef CAS PubMed; (e) M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed; (f) P. D. Tran, V. Artero and M. Fontecave, Energy Environ. Sci., 2010, 3, 727 RSC; (g) J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865 RSC.
- (a) Y. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 9212 CrossRef CAS PubMed; (b) J. P. Bigi, T. E. Hanna, W. H. Harman, A. Chang and C. J. Chang, Chem. Commun., 2010, 46, 958 RSC; (c) M. J. Rose, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 2012, 134, 8310 CrossRef CAS PubMed; (d) E. Deponti, A. Luisa, M. Natali, E. Iengo and F. Scandola, Dalton Trans., 2014, 43, 16345 RSC; (e) P. Zhang, M. Wang, F. Gloaguen, L. Chen, F. Quentelb and L. Sun, Chem. Commun., 2013, 49, 9455 RSC; (f) M. Natali, A. Luisa, E. Iengo and F. Scandola, Chem. Commun., 2014, 50, 1842 RSC; (g) C. Bachmann, B. Probst, M. Guttentag and R. Alberto, Chem. Commun., 2014, 50, 6737 RSC; (h) K. Kawano, K. Yamauchi and K. Sakai, Chem. Commun., 2014, 50, 9872 RSC; (i) W. M. Singh, M. Mirmohades, R. T. Jane, T. A. White, L. Hammarström, A. Thapper, R. Lomoth and S. Ott, Chem. Commun., 2013, 49, 8638 RSC; (j) J. Xie, Q. Zhou, C. Li, W. Wang, Y. Hou, B. Zhanga and X. Wang, Chem. Commun., 2014, 50, 6520 RSC; (k) Y. Sun, J. Sun, J. R. Long, P. Yang and C. J. Chang, Chem. Sci., 2013, 4, 118 RSC.
- (a) L. A. Berben and J. C. Peters, Chem. Commun., 2010, 398 RSC; (b) C. C. McCrory, L. C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164 CrossRef CAS PubMed; (c) P. A. Jacques, V. Artero, J. Pecaut and M. Fontecave, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20627 CrossRef CAS PubMed; (d) M. Razavet, V. Artero and M. Fontecave, Inorg. Chem., 2005, 44, 4786 CrossRef CAS PubMed; (e) C. Baffert, V. Artero and M. Fontecave, Inorg. Chem., 2007, 46, 1817 CrossRef CAS PubMed; (f) A. Fihri, V. Artero, M. Razavet, C. Baffert, W. Leibl and M. Fontecave, Angew. Chem., Int. Ed., 2008, 47, 564 CrossRef CAS PubMed; (g) A. Fihri, V. Artero, A. Pereira and M. Fontecave, Dalton Trans., 2008, 5567 RSC; (h) P. Du, J. Schneider, G. Luo, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2009, 48, 4952 CrossRef CAS PubMed; (i) T. Lazarides, T. McCormick, P. Du, G. Luo, B. Lindley and R. Eisenberg, J. Am. Chem. Soc., 2009, 131, 9192 CrossRef CAS PubMed; (j) T. M. McCormick, B. D. Calitree, A. Orchad, N. D. Kraut, F. V. Bright, M. R. Detty and R. Eisenberg, J. Am. Chem. Soc., 2010, 132, 15480 CrossRef CAS PubMed; (k) T. M. McCormick, Z. Han, D. J. Weinberg, W. W. Brennessel, P. L. Holland and R. Eisenberg, Inorg. Chem., 2011, 50, 10660 CrossRef CAS PubMed; (l) B. Probost, A. Rodenberg, M. Guttentag, P. Hamm and R. Alberto, Inorg. Chem., 2010, 49, 6453 CrossRef PubMed; (m) L. M. Utschig, S. C. Silver, K. L. Mulfort and D. M. Tiede, J. Am. Chem. Soc., 2011, 133, 16334 CrossRef CAS PubMed; (n) K. L. Mulfort and D. M. Tiede, J. Phys. Chem. B, 2010, 114, 14572 CrossRef CAS PubMed; (o) C. Li, M. Wang, J. Pan, P. Zhang, R. Zhang and L. Sun, J. Organomet. Chem., 2009, 694, 2814 CrossRef CAS; (p) F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381 CrossRef CAS PubMed; (q) F. Lakadamyali and E. Reisner, Chem. Commun., 2011, 47, 1695 RSC; (r) J. Willkomm, N. M. Muresan and E. Reisner, Chem. Sci., 2015, 6, 2727 RSC.
- (a) C. N. Valdez, J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15589 CrossRef CAS PubMed; (b) S. C. Marinescu, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15127 CrossRef CAS PubMed; (c) E. A. Mallart, C. Costentin, M. Fournier, S. Nowak, M. Robert and J. M. Saveant, J. Am. Chem. Soc., 2012, 134, 6104 CrossRef PubMed; (d) W. M. Singh, T. Baine, S. Kudo, S. Tian, X. A. N. Ma, H. Zhou, N. J. DeYonker, T. C. Pham, J. C. Bollinger, D. L. Baker, B. Yan, C. E. Webster and X. Zhao, Angew. Chem., Int. Ed., 2012, 51, 5941 CrossRef CAS PubMed.
- P. Connolly and J. H. Espenson, Inorg. Chem., 1986, 25, 2684 CrossRef CAS.
- (a) V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238 CrossRef CAS PubMed; (b) P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012 RSC; (c) M. Wang, L. Chen and L. Sun, Energy Environ. Sci., 2012, 5, 6763 RSC; (d) B. H. Solis and S. Hammes-Schiffer, J. Am. Chem. Soc., 2011, 133, 19036 CrossRef CAS PubMed; (e) C. F. Leung, Y. Z. Chen, H. Q. Yu, S. M. Yiu, C. C. Ko and T. C. Lau, Int. J. Hydrogen Energy, 2011, 36, 11640 CrossRef CAS; (f) B. Solis, Y. Yu and S. Hammes-Schiffer, Inorg. Chem., 2013, 52, 6994 CrossRef CAS PubMed; (g) J. T. Muckermann and E. Fujita, Chem. Commun., 2011, 47, 12456 RSC; (h) M. T. D. Nguyen, M. F. Charlot and A. Aukauloo, J. Phys. Chem. A, 2011, 115, 911 CrossRef CAS PubMed; (i) B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2011, 50, 11252 CrossRef CAS PubMed; (j) J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995 CrossRef CAS PubMed; (k) J. L. Dempsey, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2010, 132, 16774 CrossRef CAS PubMed; (l) A. E. King, Y. Surendranath, N. A. Piro, J. P. Bigi, J. R. Long and C. J. Chang, Chem. Sci., 2013, 4, 1578 RSC; (m) A. Bhattacharjee, E. S. Andreiadis, M. C. Kerlidou, M. Fontecave, M. J. Field and V. Artero, Chem.–Eur. J., 2013, 19, 15166 CrossRef CAS PubMed; (n) M. Nippe, R. S. Khnayzer, J. A. Panetier, D. Z. Zee, B. S. Olaiya, M. Head-Gordon, C. J. Chang, F. N. Castellano and J. R. Long, Chem. Sci., 2013, 4, 3934 RSC.
- A. Gerli and L. G. Marzilli, Inorg. Chem., 1992, 31, 1152 CrossRef CAS.
- S. Shi, L. M. Daniels and J. H. Espenson, Inorg. Chem., 1991, 30, 3407 CrossRef CAS.
- X. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988 CrossRef CAS PubMed.
- (a) R. Shakya, C. Imbert, H. P. Hratchian, M. Lanzaster, M. J. Heeg, B. R. McGarvey, M. Allard, H. B. Schlegel and C. N. Verani, Dalton Trans., 2006, 21, 2517 RSC; (b) R. Shakya, S. S. Hindo, L. Wu, M. Allard, M. J. Heeg, H. P. Hratchian, B. R. McGarvey, S. R. P. Da Rocha and C. N. Verani, Inorg. Chem., 2007, 46, 9808 CrossRef CAS PubMed; (c) F. D. Lesh, R. L. Lord, M. J. Heeg, H. B. Schlegel and C. N. Verani, Eur. J. Inorg. Chem., 2012, 03, 463 CrossRef; (d) M. M. Allard, F. R. Xavier, M. J. Heeg, H. B. Schlegel and C. N. Verani, Eur. J. Inorg. Chem., 2012, 4622 CrossRef CAS; (e) D. Basu, M. M. Allard, F. R. Xavier, M. J. Heeg, H. B. Schlegel and C. N. Verani, Dalton Trans., 2015, 44, 3454 RSC; (f) D. Basu, S. Mazumder, X. Shi, H. Baydoun, J. Niklas, O. Poluektov, H. B. Schlegel and C. N. Verani, Angew. Chem., Int. Ed., 2015, 54, 2105 CrossRef CAS PubMed; (g) D. Basu, S. Mazumder, X. Shi, R. Staples, H. B. Schlegel and C. N. Verani, Angew. Chem., Int. Ed., 2015, 54, 7139 CrossRef CAS PubMed.
- E. Uhlig and M. Friedrich, Z. Anorg. Allg. Chem., 1966, 343, 299 CrossRef.
- G. Costa, G. Mestroni and E. D. Savorgnani, Inorg. Chim. Acta, 1969, 323 CrossRef CAS.
- A. Bhattacharjee, M. Chavarot-Kerlidou, E. S. Andreiadis, M. Fontecave, M. J. Field and V. Artero, Inorg. Chem., 2012, 51, 7087 CrossRef CAS PubMed.
- (a) M. Rangel, A. Leite, J. Gomes and B. Castro, Organometallics, 2005, 24, 3500 CrossRef CAS; (b) J. Niklas, K. L. Mardis, R. R. Rakhimov, K. L. Mulfort, D. M. Tiede and O. G. Poluektov, J. Phys. Chem. B, 2012, 116, 2943 CrossRef CAS PubMed.
- (a) M. D. Wirt, C. J. Bender and J. Peisach, Inorg. Chem., 1995, 34, 1663 CrossRef CAS; (b) J. Hermer, S. V. Doorslaer, I. Gromov and A. Schweiger, Chem. Phys. Lett., 2002, 358, 8 CrossRef.
- T. J. R. Weakley, J. Marks and R. G. Finke, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 1690 CrossRef.
- D. C. Lacy, G. M. Roberts and J. C. Peters, J. Am. Chem. Soc., 2015, 137, 4860 CrossRef CAS PubMed.
- V. Fourmond, P. A. Jacques, M. Fontecave and V. Artero, Inorg. Chem., 2010, 49, 10338 CrossRef CAS PubMed.
- D. P. Estes, D. C. Grills and J. R. Norton, J. Am. Chem. Soc., 2014, 136, 17362 CrossRef CAS PubMed.
- R. Gagne, C. Koval and G. Licenski, Inorg. Chem., 1980, 19, 2854 CrossRef CAS.
- (a) COSMO V1.58, Software for the CCD Detector Systems for Determining Data Collection Parameters. Bruker Analytical X-ray Systems, Madison, WI, 2008 Search PubMed; (b) APEX2 V2008.5-0 Software for the CCD Detector System; Bruker Analytical X-ray Systems, Madison, WI, 2008 Search PubMed; (c) SAINT V 7.34 Software for the Integration of CCD Detector System Bruker Analytical X-ray Systems, Madison, WI, 2008 Search PubMed; (d) SADABS V2.008/2 Program for absorption corrections using Bruker-AXS CCD based on the method of Robert Blessing; R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, A51, 33 CrossRef CAS; (e) G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed; (f) P. V. D. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef; (g) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS , aObtained with graphite monochromated Mo Kα (l = 0.71073 Å) radiation. bR1 = ∑||Fo|−|Fc||/∑|Fo|. cwR2 = {∑[w(Fo2 − Fc2)2/{∑[w(Fo2)2]}1/2; (h) COSMO V1.61, Software for the CCD Detector Systems for Determining Data Collection Parameters. Bruker Analytical X-ray Systems, Madison, WI, 2009 Search PubMed; (i) APEX2 V2010.11-3. Software for the CCD Detector System; Bruker Analytical X-ray Systems, Madison, WI, 2010 Search PubMed; (j) SAINT V 7.68A Software for the Integration of CCD Detector System Bruker Analytical X-ray Systems, Madison, WI, 2010 Search PubMed; (k) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- (a) M. J. Frisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian Development Version; Revision H.35 ed., Gaussian, Inc., Wallingford, CT 2010 Search PubMed; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (c) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef; (d) J. P. Perdew, K. Burke and Y. Wang, Phys. Rev., 1996, 54, 16533 CrossRef CAS; (e) T. H. Dunning Jr and P. J. Hay, in Modern Theoretical Chemistry, ed. H. F. Schaefer III, Plenum, New York, 1977, vol. 3, pp. 1–28 Search PubMed; (f) M. Dolg, H. Wedig and H. Preuss, J. Chem. Phys., 1987, 86, 866 CrossRef CAS; (g) S. Miertus, E. Scrocco and J. Tomasi, J. Chem. Phys., 1981, 55, 117 CAS; (h) G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed; (i) G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, J. Chem. Phys., 2006, 124, 9410 CrossRef PubMed; (j) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed; (k) A. Schaefer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef CAS; (l) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS; (m) R. Seeger and J. A. Pople, J. Chem. Phys., 1977, 66, 3045 CrossRef CAS; (n) H. B. Schlegel and J. J. McDouall, Computational Advances in Organic Chemistry, ed. C. Ögretir and I. G. Csizmadia, Kluwer Academic, Amsterdam, The Netherlands, 1991 Search PubMed; (o) R. Bauernschmitt and R. Ahlrichs, J. Chem. Phys., 1996, 104, 9047 CrossRef CAS; (p) R. Pennington, T. Keith and J. M. Millam, GaussView V. 5.0, Semichem, Inc., Shawnee Mission, KS, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1434959, 1434960, 1448834 and 1449138. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04214c |
| ‡ D. B. and S. M. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |