
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective assembly of tertiary stereocenters via multicomponent chemoselective cross-coupling of geminal chloro(iodo)alkanes†
X.
Jiang
,
K.
Kulbitski
,
G.
Nisnevich
and
M.
Gandelman
*
Schulich Faculty of Chemistry, Technion-Israel Institute of Technology, Technion City, Haifa-32000, Israel. E-mail: chmark@tx.technion.ac.il
First published on 6th January 2016
Abstract
Tertiary stereocenters represent a ubiquitous and highly important motif in many pharmaceutically active compounds and natural products. Development of efficient and straightforward approaches to their enantioselective construction from readily available simple substrates is an important yet challenging goal for synthetic chemistry. Herein we describe an efficient, versatile and facile method for the highly enantioselective construction of tertiary stereocenters via unprecedented consecutive one-pot Suzuki reactions of non-activated racemic 1-chloro-1-iodoalkanes with alkylboranes. It represents the first cross-coupling approach which employs simple and readily available primary alkyl substrates for the direct multicomponent assembly of enantioenriched tertiary stereocenters. A simple and effective preparation of 1-chloro-1-iodoalkanes from ubiquitous α-chloroalkanoic acids is also described. Collectively, the developed methods open a door to efficient catalytic enantioselective synthesis of alkanes bearing tertiary stereocenters from carboxylic acids just in few steps.
Introduction
Metal catalyzed cross-couplings are extremely useful and efficient reactions for construction of carbon–carbon bonds.1 Contrary to Csp2-based electrophiles, coupling of Csp3-based partners has proven to be a daunting challenge due to the relatively slow oxidative addition of alkyl halides and the undesirable β-hydride elimination that alkyl halides might undergo.2 Development of efficient catalysts for alkyl–alkyl couplings of primary and secondary alkyl electrophiles has emerged as an important frontier with a potential to significantly extend the synthetic power of the cross-coupling transformations.3 In this context, utilization of secondary alkyl electrophiles (or nucleophiles) in cross-couplings is especially attractive as it opens a door to the catalytic asymmetric synthesis of stereogenic tertiary centers.3d–e,4 All-carbon tertiary stereocenters represent an ubiquitous and highly important motif in many pharmaceutically active compounds and natural products, and the development of efficient and straightforward approaches to their construction is an important goal for organic chemistry.5There are two major approaches that utilize cross-coupling reactions of non-activated alkyl partners for the synthesis of enantioenriched products with tertiary stereocenters.4–6 The first stereospecific approach depends on substrate control (electrophile or nucleophile) as the source of stereochemical information.4a This process is mediated by achiral catalysts, and requires use of the enantioenriched secondary electrophiles or nucleophiles in the stoichiometric amount. The second approach employs racemic secondary electrophiles (or nucleophiles) as starting materials, and the stereoselectivity of the product is controlled by substoichiometric amounts of the chiral catalyst (Scheme 1a).4a Recently, remarkable catalyst-controlled enantioselective cross-couplings have been developed for a number of families of non-activated secondary alkyl halides.7 It was demonstrated that this process is stereoconvergent as both stereoisomers of the racemic alkyl halides were converted into a single stereoisomer of product as the reaction proceeds through a common achiral intermediate.7c Notably, all reported cross-coupling processes for the preparation of tertiary stereocenters utilize secondary alkyl partners as starting materials.4a However, synthesis of non-symmetrical secondary alkyl halides (or, alternatively, alkylmetal reagents) might be multistep and tedious, typically involving reaction of Grignard reagents (functional group sensitive) with carbonyls or epoxides. Therefore, alternative approaches which rely on new easily available substrates for stereoselective construction of tertiary centers via cross-coupling reactions could significantly extend the potential of this method in terms of applicability and functional group compatibility.
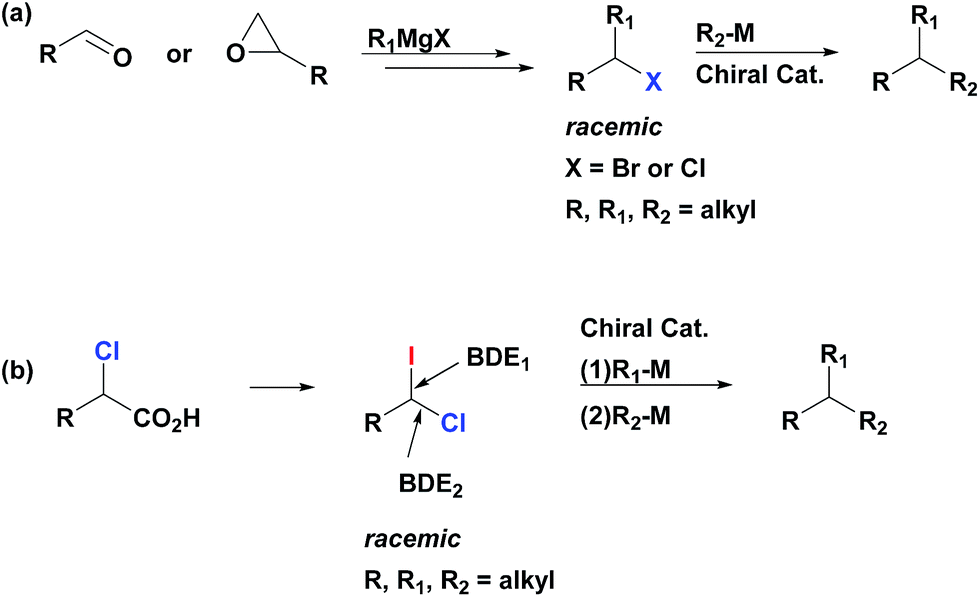 | ||
| Scheme 1 Cross-coupling approaches for stereoconvergent construction of tertiary stereocenters (a) established method employing secondary partners; (b) one-pot sequential cross-couplings of geminal 1-chloro-1-iodoalkanes with primary alkylborons (this work). | ||
Results and discussion
Herein we describe a novel multicomponent method for the facile and highly enantioselective construction of tertiary stereocenters via Suzuki reactions of non-activated racemic geminal dihaloalkanes with primary alkylboranes. This represents the first example of cross-coupling that employs simple primary alkyl electrophiles and nucleophiles for the assembly of tertiary centers (Scheme 1b). We demonstrate the unprecedented use of geminal chloro(iodo)alkanes in cross-coupling reactions, in which the chemoselective functionalization of the C–I bond (bond dissociation energy (BDE1) = 57 kcal mol−1)8 with the first alkylborane, followed by functionalization of the stronger C–Cl bond (BDE2 = 84 kcal mol−1)8 with a second alkylborane in a consecutive step resulting in a one-pot transformation. Importantly, the same catalyst system and reaction conditions are used for both steps of this stereoconvergent transformation. Moreover, we disclose a general and efficient approach to access 1-chloro-1-iodoalkanes. This chemistry relies on the facile iododecarboxylation of α-chloroalkanoic acids, which, in turn, are easily prepared by α-chlorination of the corresponding alkanoic acids.9 Collectively these methods constitute a short, straightforward and efficient approach for the construction of highly enantioenriched alkanes bearing tertiary stereocenters (up to 99% ee) from abundant primary alkanoic acids.1-Chloro-1-iodoalkanes constitute an attractive class of polyfunctional compounds. However, the development of their chemistry has been an uneven process, most likely, due to the lack of selective, practical and high yielding methods of their synthesis.10 Recently, we reported a robust, general and efficient method for synthesis of alkyl iodides from carboxylic acids through their simple treatment with commercially available 1,3-diiodo-5,5-dimethylhydantoin (DIH) under visible light (VIS) irradiation.11 We now report that, contrary to other iododecarboxylation approaches,10d our method can be uniquely extended to α-chloroalkanoic acids. Thus, these compounds react smoothly with DIH under VIS-irradiation to generate the corresponding 1-chloro-1-iodoalkanes (Scheme 2). These products are isolated in an essentially pure form after a simple aqueous work-up of the reaction mixture (see ESI†). All 1-chloro-1-iodoalkanes presented in this paper (Tables 1, 3 and 4) were prepared and isolated by this method, starting from the corresponding α-chloroalkanoic acids, in a selective manner and high yields.
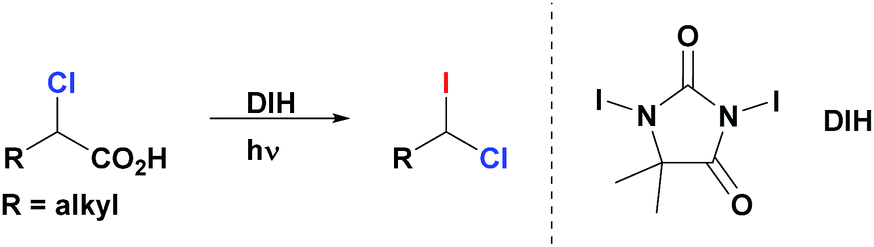 | ||
| Scheme 2 Synthesis of geminal chloro(iodo)alkanes via iododecarboxylation of α-chloroalkanoic acids. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Borane A | Borane B | R1 | R2 | Yieldb, ee (%) |
| a Reactions were conducted with 1a (0.2 mmol), NiCl2·glyme (0.02 mmol), ligand (0.024 mmol), borane A (0.26 mmol), KOtBu (0.2 mmol), i-BuOH (0.26 mmol) in i-Pr2O (2 mL) at RT. After 24 h, borane B (0.4 mmol), KOtBu (0.28 mmol), i-BuOH (0.4 mmol) were added and adjusted the total volume to 4 mL with i-Pr2O. b Isolated yields. | |||||
| 1 | Pr3B | Ph(CH2)3-9-BBN | Pr | Ph(CH2)3 | 73, 86 |
| 2 | Pr-9-BBN | Ph(CH2)3-9-BBN | Pr | Ph(CH2)3 | 84, 96 |
| 3 | Ph(CH2)3-9-BBN | Pr-9-BBN | Ph(CH2)3 | Pr | 70, 82 |

|
|||||
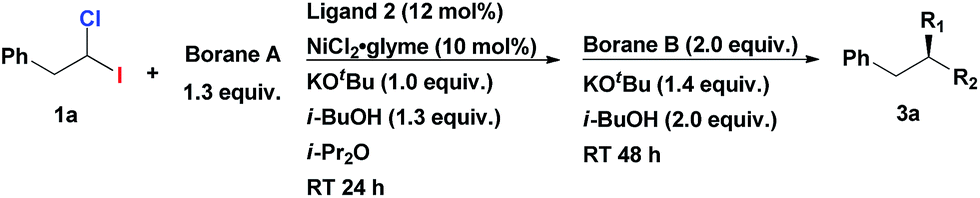
Having an efficient synthetic access to these synthons in hand, we examined their applicability in the construction of tertiary stereocenters through a direct multicomponent reaction. Based on the substantial difference in the BDE's of C–I and C–Cl bonds, we hypothesized that it might be possible to selectively activate the C–I bond in a cross-coupling reaction with alkylmetal R1–M, following by functionalization of the C–Cl bond in a second cross-coupling with a different alkylmetal R2–M (see Scheme 1b). Ideally, one catalyst system might be able to mediate both steps. Since the starting material, 1-chloro-1-iodoalkane, is racemic, a stereoconvergent synthesis of enantiomerically enriched products with tertiary chiral centers may be envisioned if an appropriate chiral catalyst could be identified.
Initiating an active program on studies of this general concept of multicomponent assembly of the tertiary chiral centers, we started from investigating homobenzylic geminal chloroiodides in the asymmetric Suzuki alkyl–alkyl coupling reactions. Recently, Fu et al. reported the first asymmetric Suzuki cross-coupling of secondary homobenzylic bromides with alkyl-9-BBN.7a In this seminal work, it was demonstrated that ArCH2-group in electrophile is necessary for ensuring good enantioselectivity, most likely, due to its weak secondary interaction with the catalyst. We firstly investigated the reaction of homobenzylic dihalide substrate 1a in the reaction with different alkylborane agents in the presence of NiCl2·glyme and diamine ligand 2 (Table 1). Gratifyingly, subjecting 1a to the coupling conditions with tripropylborane, following by addition of Ph(CH2)3-9-BBN to the reaction mixture (after complete conversion of 1a), resulted in formation of the tertiary asymmetric alkane 3a which was isolated in 73% yield and 86% ee (Table 1, entry 1). Remarkably, the same catalyst system is operative in both steps of the transformation. The use of alkyl-9-BBN as the nucleophile source in both stages of the reaction proved to be superior to other tested boron reagents. The replacement of Pr3B with Pr-9-BBN furnished the product in even higher yield and enantioselectivity (84% yield, 96% ee; entry 2). Inversion of the addition sequence, i.e. first adding the bulkier Ph(CH2)3-9-BBN in the first step following by the less bulky Pr-9-BBN, proved to be less efficient (entry 3).
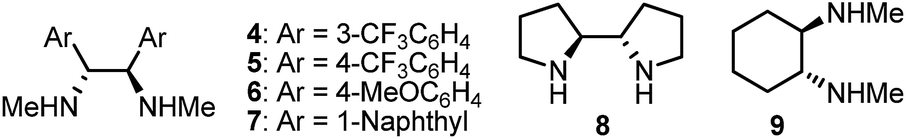
Any deviation from these reaction conditions resulted in inferior results (Table 2). We tested the chiral secondary diamine ligands 4–9 in the multicomponent cross-coupling of 1a with Pr-9-BBN and Ph(CH2)3-9-BBN; however, none of these outperformed the original ligand 2 (compare entry 1 with entries 2–7). Reaction using ligand 6, bearing electron rich aryl substituents on the ethylenediamine backbone, provided 3a with enantioselectivity comparable to ligand 2 (94% ee, entry 4). However analogs of 6 with electron poor aryls or bulky naphthyl substituents gave the product in significantly lower ee (entries 2, 3 and 5).
|
|
||
|---|---|---|
| Entry | Variation from the “standard” conditions | Yieldb, ee (%) |
| a Reactions were conducted with 1a (0.2 mmol), Ni salt (0.02 mmol), ligand (0.024 mmol), propyl-9-BBN (0.26 mmol), KOtBu (0.2 mmol), i-BuOH or hexanol (0.26 mmol) in i-Pr2O or dioxane (2 mL) RT. After 24 h, Ph(CH2)3-9-BBN (0.4 mmol), KOtBu (0.28 mmol), i-BuOH or hexanol (0.4 mmol) were added and adjusted the total volume to 4 mL with the corresponding solvent. b Isolated yield. | ||
| 1 | 84, 96 | |
| 2 | 4 as ligand instead of 2 | 79, 86 |
| 3 | 5 as ligand instead of 2 | 71, 73 |
| 4 | 6 as ligand instead of 2 | 80, 94 |
| 5 | 7 as ligand instead of 2 | 82, 68 |
| 6 | 8 as ligand instead of 2 | 65, 90 |
| 7 | 9 as ligand instead of 2 | 67, 78 |
| 8 | NiBr2·diglyme instead of NiCl2·glyme | 77, 89 |
| 9 | Ni(cod)2 instead of NiCl2·glyme | 68, 87 |
| 10 | Hexanol instead of i-BuOH | 80, 92 |
| 11 | Et2O instead of i-Pr2O | 81, 96 |
| 12 | Dioxane instead of i-Pr2O | 75, 80 |
| 13 | 6% cat. and 8% ligand instead of 10%/12% | 61, 84 |
|
||

Interestingly, bis-pyrrolidine ligand 8, which was recently used by us for the efficient synthesis of chiral secondary fluoroalkanes via Suzuki cross-coupling of 1-bromo-1-fluoroalkanes,12 also afforded a product with high enantioselectivity, albeit with considerably lower yield (entry 6). Utilizing other sources of Ni(II) or Ni(0) instead of NiCl2·glyme, as well as lower loadings of Ni salt and ligand, eroded the reaction efficiency (entries 8, 9 and 13). While dioxane is unsuitable as a solvent (entry 12), diethyl ether proved to be as efficient as diisopropyl ether (entry 11). We preferred to use the higher boiling diisopropyl ether in order to ensure a constant concentration of the reaction partners in the course of the process.
Having arrived at the optimal reaction conditions, we explored the substrates scope of this multicomponent coupling (Table 3 and 4). The transformation is very versatile as the R, R1 and R2 fragments can be easily varied upon assembly of tertiary center; therefore, diverse combinations can be envisioned (see Scheme 1b). We found that the introduction of a relatively compact alkyl group in the first step ensures overall high enantioselectivity (vide supra; see Table 1).
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Product | Yieldb, ee (%) |
| a Reactions were conducted with 1a (0.5 mmol), Ni salt (0.05 mmol), ligand 2 (0.06 mmol), R1-9-BBN (0.65 mmol), KOtBu (0.5 mmol), i-BuOH (0.65 mmol) in i-Pr2O 5 mL at RT. After 24 h, R2-9-BBN (1.0 mmol), KOtBu (0.7 mmol), i-BuOH (1.0 mmol) were added and adjusted the total volume to 10 mL with i-Pr2O. b Isolated yields. | ||||
| 1 | Et | Ph(CH2)3 | 3b | 79, 92 |
| 2 | Pr | Ph(CH2)3 | 3a | 85, 96 |
| 3 | Bu | Ph(CH2)3 | 3c | 80, 98 |
| 4 | Pentyl | Ph(CH2)3 | 3d | 81, 80 |
| 5 | Hexyl | Ph(CH2)3 | 3e | 83, 89 |
| 6 | Ph(CH2)3 | 4-MeOC6H4(CH2)3 | 3f | 68, 78 |

|
|
|||||
|---|---|---|---|---|---|
| Entry | Ar | Substrate | R | Product | Yieldb, ee (%) |
| a Reactions were conducted with 1 (0.5 mmol), Ni salt (0.05 mmol), ligand 2 (0.06 mmol), n-propyl-9-BBN (0.65 mmol), KOtBu (0.5 mmol), i-BuOH (0.65 mmol) in i-Pr2O (5 mL) at RT. After 24 h, R-9-BBN (1.0 mmol), KOtBu (0.7 mmol), i-BuOH (1.0 mmol) were added and adjusted the total volume to 10 mL with i-Pr2O. b Isolated yields. | |||||
| 1 | Ph | 1a | Ph(CH2)3 | 3a | 85, 96 |
| 2 | Ph | 1a | 4-FC6H4(CH2)3 | 10a | 81, 92 |
| 3 | Ph | 1a | 4-MeC6H4(CH2)3 | 10b | 85, 87 |
| 4 | Ph | 1a | 4-MeOC6H4(CH2)3 | 10c | 88, 82 |
| 5 | Ph | 1a | Ph(CH2)4 | 10d | 81, 78 |
| 6 | 4-MeOC6H4 | 1b | Ph(CH2)3 | 10e | 82, >99.5 |
| 7 | 4-MeOC6H4 | 1b | 2-MeOC6H4(CH2)3 | 10f | 88, 99.5 |
| 8 | 4-MeOC6H4 | 1b | 4-MeOC6H4(CH2)3 | 10g | 87, 94 |
| 9 | 4-MeOC6H4 | 1b | 4-CF3C6H4(CH2)3 | 10h | 82, 95 |
| 10 | 4-MeOC6H4 | 1b | 4-FC6H4(CH2)3 | 10i | 85, 94 |
| 11 | 4-MeOC6H4 | 1b | 4-MeC6H4(CH2)3 | 10j | 86, 92 |
| 12 | 4-MeOC6H4 | 1b | TBSO(CH2)3 | 10k | 80, 81 |
| 13 | 4-MeOC6H4 | 1b | Cyclohexyl(CH2)3 | 10l | 84, 78 |
| 14 | 2-MeC6H4 | 1c | Ph(CH2)3 | 10m | 78, 87 |
| 15 | 3-MeC6H4 | 1d | TBSO(CH2)3 | 10n | 84, 80 |
| 16 | 4-MeC6H4 | 1e | Ph(CH2)3 | 10o | 86, 78 |
| 17 | 2-FC6H4 | 1f | Ph(CH2)3 | 10p | 83, 72 |
| 18 | 3-FC6H4 | 1g | Ph(CH2)3 | 10q | 80, 82 |
| 19 | 4-FC6H4 | 1h | TBSO(CH2)3 | 10r | 79, 38 |
| 20 | 4-CF3C6H4 | 1i | Ph(CH2)3 | 10s | 81, 89 |
| 21 | 2-Napthyl | 1j | Ph(CH2)3 | 10t | 58, 71 |
| 22 | 3-MeOC6H4 | 1k | Ph(CH2)3 | 10u | 78, 88 |

As such, several unbulky alkyl-9-BBN were initially examined as trans-metallation partners in the first step (Table 3). Although a very good to high enantioselectivity is obtained in each case, the variation of the chain length in R1 group has a distinguishable influence. While the shorter ethyl, propyl and butyl motifs ensure highly enantioenriched products (entries 1–3), the longer pentyl, hexyl and 3-phenylpropyl units lead to slightly diminished ee (entries 4–6).
Selecting propyl-9-BBN as a representative nucleophile for the first cross-coupling event, we demonstrated the flexibility of the reaction with respect to the alkylborane, utilized in the second step (R2-9-BBN), and 2-aryl-1-chloro-1-iodoethanes (ArCH2CHClI; 1a–k). These results are summarized in Table 4. A wide array of non-activated homoaryl geminal chloroiodides was tolerated in the reaction. Both phenyl as well as ortho-, meta- and para-substituted phenyls with both electron donating and withdrawing groups participated in the reaction affording the products in high yields and enantioselectivities with variety of organoboron partners. Interestingly, having an electron donating methoxy-group in the para-position of the aromatic ring in homoaryl dihalide (i.e. compound 1b) was beneficial providing 10e–10l in enantioselectivities up to 99% ee (entries 6–13).
An increase of electron density in the aromatic ring possibly strengthens its secondary interaction with the catalyst in the enantiodiscriminative step. Indeed, 1-chloro-1-fluoro-2-(p-fluorophenyl)ethane (1h), bearing a strongly withdrawing fluoride substituent in para-position, reacts with considerably lower enantioselectivity (compare entries 6 vs. 20; 12 vs. 19). 1-Chloro-1-iodo-2-(meta-methoxyphenyl)ethane (1k) leads to the tertiary alkane 10u with lower optical purity (88% ee) than the analogous para-substituted substrate 1b (compare entries 6 and 22). It is conceivable that the secondary interactions between the aromatic system and the catalyst are relatively weak in case of the substrate 1k due to steric congestion imposed by the meta-methoxy substituent. Noteworthy, the high yields and enantioselectivities are obtained in these reactions under the mild conditions and at room temperature.
When gem-dihaloalkane 1a was cross-coupled with hexyl-9-BBN under the standard reaction conditions, the corresponding unsymmetrical secondary alkyl chloride 11 was isolated in 90% yield (Scheme 3a). However, the product was a racemic mixture of enantiomers. A complete absence of ee in 11 was surprising, since when such cross-coupling was performed with the corresponding fluoro(halo)alkanes as substrates the resulting secondary alkyl fluorides were isolated in highly enantioenriched form.12 This difference can be explained by the assumption that the C–Cl bond in secondary alkyl chlorides (e.g., 11) undergoes fast reversible oxidative addition/reductive elimination process under our cross-coupling conditions which leads to epimerization. This process is apparently impossible in case of stronger C–F bond. Since the first coupling step leads to racemic alkyl chlorides, we can conclude that the second cross-coupling event represents an enantiodiscriminative step under the given developed reaction conditions.13
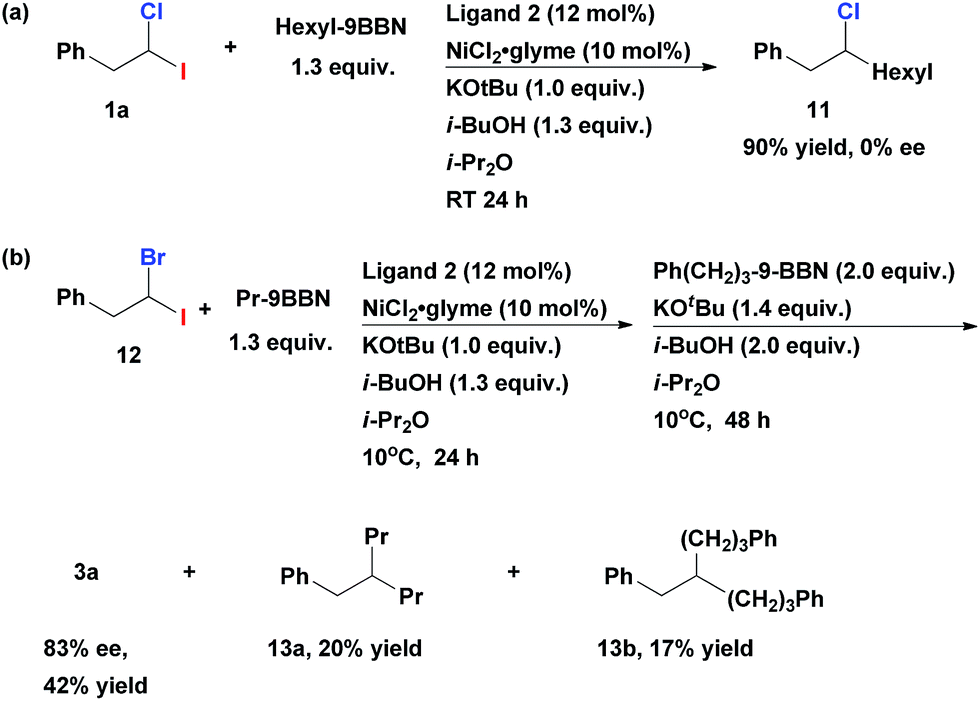 | ||
| Scheme 3 (a) Generation of the racemic intermediate 11 (b) enantioselective alkyl–alkyl Suzuki cross-coupling of geminal bromo-iodoalkane 12. | ||
It should be mentioned, that the developed multicomponent transformation is efficient and chemoselective only for geminal alkanes possessing a combination of chloro- and iodo-substituents. Geminal homo-dihaloalkanes are not suitable substrates for this method. It was previously reported, that CH2Cl2 undergoes dialkylation with BuMgBr via Kumada cross-couplings to result in nonane even if a large excess of dichloromethane is used and reaction is carried out at −20 °C.14 Similarly, reaction of 1,1-diiodo-2-phenylethane under our coupling conditions mainly resulted in double alkylation already in the first step.
When 1-bromo-1-iodo-2-phenylethane 12 was tested in the representative multicomponent cross-coupling, the asymmetric alkane 3a was obtained in 42% yield and 83% ee (Scheme 3b). However, the main side products were doubly alkylated compounds 13a and 13b even if this reaction is performed at 10 °C. As such, the obtained exclusive chemoselectivity (in the first step) in our method is quite surprising, given that both coupling steps are mediated with the same catalyst under the same reaction conditions.
Conclusions
In conclusion, we have developed an efficient, versatile and facile method for the highly enantioselective construction of tertiary stereocenters through an unprecedented Suzuki reactions of non-activated racemic geminal chloro(iodo)-alkanes with alkylboranes. We have established the first cross-coupling approach which employs simple and readily available primary alkyl substrates for the direct multicomponent assembly of enantioenriched tertiary stereocenters. The ability to selectively functionalize an alkyl C–I bond in the presence of geminal C–Cl bond is an important factor for the success of this one-pot two-step transformation. Importantly, the same catalyst system is used for both steps of this stereoconvergent transformation, and the selectivity is achieved under mild conditions and at room temperature. In addition, we have disclosed a general and efficient preparation of 1-chloro-1-iodoalkanes, the electrophiles used in the described cross-coupling transformation. These highly functional compounds have become readily available due to developed direct iododecarboxylation of the corresponding ubiquitous α-chloroalkanoic acids. Collectively, the developed methods open a door to efficient catalytic enantioselective synthesis of alkanes bearing tertiary stereocenters from carboxylic acids just in few steps. Further studies on the reactivity of unactivated geminal dihaloalkanes are underway in our labs.Acknowledgements
We acknowledge the financial support from German-Israeli Project Cooperation (DIP) and Israel Science Foundation personal grant (1890/15).Notes and references
- (a) Metal-Catalyzed cross-coupling reactions, ed. A. De Meijere and F. Diederich, Wiley-VCH, 2004 Search PubMed; (b) Transition metals for organic synthesis, ed. M. Beller and C. Bolm, Wiley-VCH, 2004 Search PubMed.
- For pioneering examples, see: (a) A. Devasagayaraj, T. Stüdemann and P. Knochel, Angew. Chem., 1995, 107, 2952 ( Angew. Chem., Int. Ed. , 1995 , 34 , 2723 ) CrossRef; (b) T. Ishiyama, A. Shigeru, N. Miyaura and A. Suzuki, Chem. Lett., 1992, 21, 691 CrossRef; (c) J. K. Kochi and M. Tamura, J. Am. Chem. Soc., 1971, 93, 1483 CrossRef CAS.
- (a) A. C. Frisch and M. Beller, Angew. Chem., 2005, 117, 680 ( Angew. Chem., Int. Ed. , 2005 , 44 , 674 ) CrossRef; (b) A. Fürstner and R. Martin, Chem. Lett., 2005, 34, 624 CrossRef; (c) M. R. Netherton and G. C. Fu, Adv. Synth. Catal., 2004, 346, 1525 CrossRef CAS; (d) A. Rudolph and M. Lautens, Angew. Chem., 2009, 121, 2694 ( Angew. Chem., Int. Ed. , 2009 , 48 , 2656 ) CrossRef; (e) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1471 CrossRef PubMed.
- (a) E. C. Swift and E. R. Jarvo, Tetrahedron, 2013, 69, 5799 CrossRef CAS PubMed; (b) F. Glorius, Angew. Chem., 2008, 120, 8474 ( Angew. Chem., Int. Ed. , 2008 , 49 , 8347 ) CrossRef.
- (a) Comprehensive asymmetric catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer-Verlag, 2004 Search PubMed; (b) Asymmetric catalysis on industrial scale: challenges, approaches and solutions, ed. H.-U. Blaser and E. Schmidt, Wiley-VCH, 2004 Search PubMed.
- For construction of tertiary carbon stereocenters by methods alternative to cross-coupling reactions of non-activated alkyl partners, see: (a) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (b) D. McCartney and P. J. Guiry, Chem. Soc. Rev., 2011, 40, 5122 RSC; (c) R. S. Harutyunyan, T. D. Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824 CrossRef PubMed.
- (a) B. Saito and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 6994 CrossRef PubMed; (b) N. A. Owston and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 11908 CrossRef CAS PubMed; (c) Z. Lu, A. Wilsily and G. C. Fu, J. Am. Chem. Soc., 2011, 133, 8154 CrossRef CAS PubMed; (d) S. L. Zultanski and G. C. Fu, J. Am. Chem. Soc., 2011, 133, 15362 CrossRef CAS PubMed; (e) A. Wilsily, F. Tramutola, N. A. Owston and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 5794 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255 CrossRef CAS PubMed.
- R. J. Crawford, J. Org. Chem., 1983, 48, 1364 CrossRef CAS.
- (a) R. W. Hoffmann, G. P. Nell, R. Leo and K. Harms, Chem.–Eur. J., 2000, 6, 3359 CrossRef CAS PubMed; (b) C. D. Lima, M. Julia and J.-N. Verpeaux, Synlett, 1992, 2, 133 CrossRef; (c) A. J. Fry and J. N. Cawse, J. Org. Chem., 1967, 32, 1677 CrossRef CAS; (d) R. C. Neuman Jr and M. L. Rahm, J. Org. Chem., 1966, 31, 1857 CrossRef.
- K. Kulbitski, G. Nisnevich and M. Gandelman, Adv. Synth. Catal., 2011, 353, 1438 CrossRef CAS.
- (a) X. Jiang, S. Sakthivel, K. Kulbitski, G. Nisnevich and M. Gandelman, J. Am. Chem. Soc., 2014, 136, 9548 CrossRef CAS PubMed; (b) X. Jiang and M. Gandelman, J. Am. Chem. Soc., 2015, 137, 2542 CrossRef CAS PubMed.
- A stereoconvergent mechanism of Ni-catalysed cross-couplings of secondary alkyl halides was previously suggested by Fu et al. For discussion of the origin of enantioselectivity in products, see ref. 4a, 7d and e.
- Z. Csok, O. Vechorkin, S. B. Harkins, R. Scopelliti and X. Hu, J. Am. Chem. Soc., 2008, 130, 8156 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04378f |
| This journal is © The Royal Society of Chemistry 2016 |