
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulating the cobalt redox potential through imidazole hydrogen bonding interactions in a supramolecular biomimetic protein-cofactor model†
Marjorie
Sonnay
,
Thomas
Fox
,
Olivier
Blacque
and
Felix
Zelder
 *
*
Department of Chemistry, University of Zurich, Winterthurerstr. 190, CH-8057, Zurich, Switzerland. E-mail: felix.zelder@chem.uzh.ch
First published on 23rd February 2016
Abstract
A realistic model for the active site of histidine-on cobalamin@protein complexes is reported and studied under homogeneous and immobilized conditions. Analysis of lower ligand modulation and its influence on the properties of the biomimetic compound are presented. The cofactor attachment by a protein's histidine residue was imitated by covalently linking an artificial imidazole-containing linker to cobyric acid. The resulting intramolecular coordination complex is an excellent structural model of its natural archetype, according to 2D 1H-NMR studies and molecular modeling. The effect of deprotonation of the axially coordinating imidazole ligand – as proposed for natural cofactor complexes – tunes significantly the position of the cathodic peak (ΔV = −203 mV) and stabilizes thereby the CoIII form. Partial deprotonation of the imidazole moiety through hydrogen bonding interactions was then achieved by immobilizing the biomimetic model on hydrophobic C18 silica, which yielded an unprecedented insight on how this class of Cbl-dependent proteins may fine-tune their properties in biological systems.
The vitamin B12 (“B12”) cofactors methylcobalamin (MeCbl) and adenosylcobalamin (AdoCbl) represent organometallic CoIII–corrinoids (Fig. 1 left) that catalyze biologically important methyl transfer and difficult [1, 2] rearrangement reactions.1 In MeCbl-dependent enzymatic reactions, the cobalt–carbon bond of MeCbl is cleaved heterolytically, which generates a square planar Cob(I)alamin species. Alternatively, in the case of AdoCbl-dependent enzymes, homolytic bond scission leads to a protein bound cofactor in the CoII state and an organic 5′-deoxyadenosyl radical (Ado˙). This difference in organometallic reactivity is remarkable, considering that MeCbl and AdoCbl cofactors share identical features, as (i) the redox-active cobalt ion, (ii) the equatorially chelating corrin ligand, (iii) the lower coordinating dimethylbenzimidazole (Dmbz) base and (iv) the upper cobalt–carbon bond (Fig. 1, left).2,3 Obviously, a prominent role of the surrounding protein has been anticipated for triggering cobalt–carbon bond activation and enzymatic catalysis.4–9
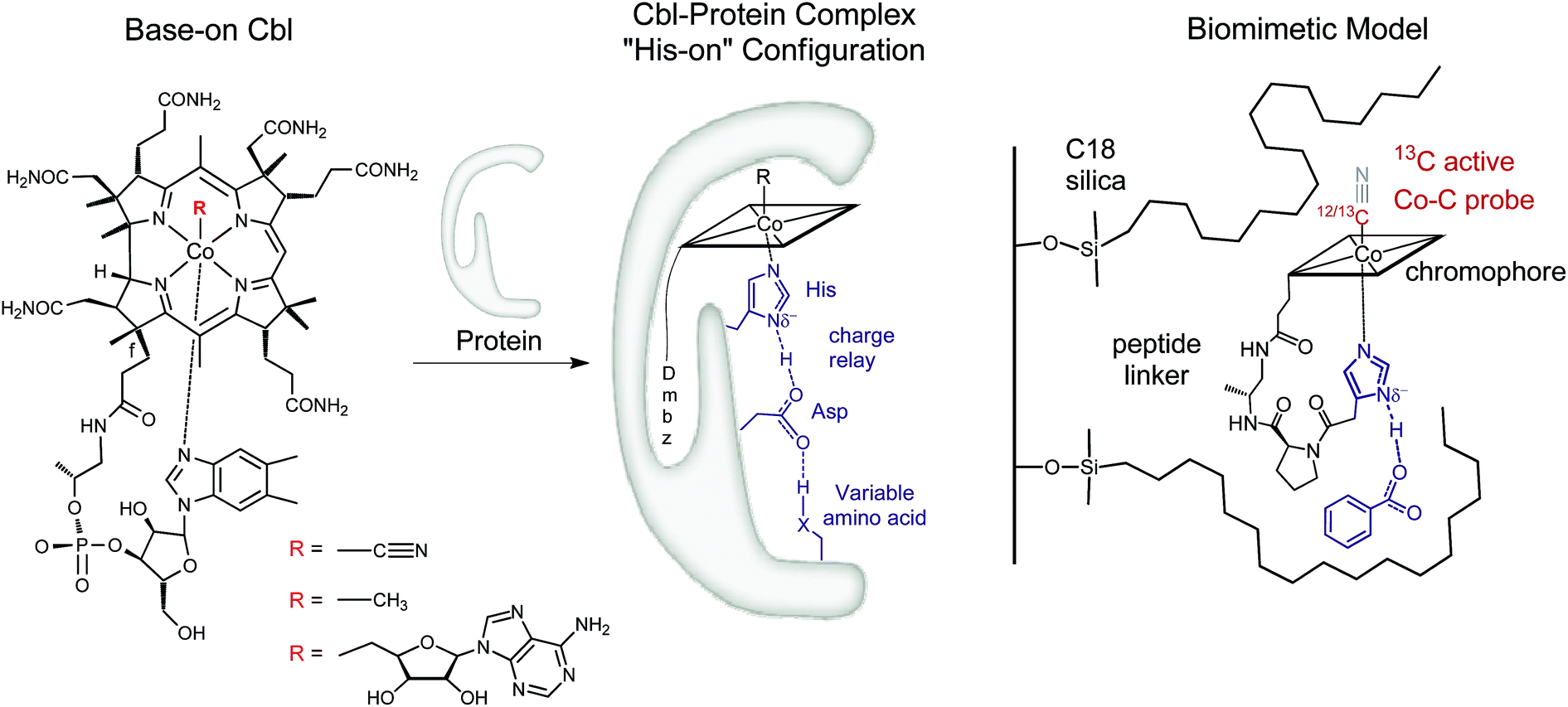 | ||
| Fig. 1 Left: Structure of vitamin B12 and Cbl cofactors having an intramolecularly bound dimethylbenzimidazole (“Dmbz”) (“base-on”) and distinct upper ligands (R = CN, B12; R = Me, MeCbl; R = 5′-deoxyadenosyl, AdoCbl). Middle: Cbl bound to the regulatory triad in the “histidine (“His”)-on” form, as found in several B12-dependent enzymes (X = donor atom, Table 1). Right: The corresponding biomimetic supramolecular model 1 with imidazole hydrogen-bonding interaction (charges are omitted). | ||
In this context, most studies have focused so far on the impact of protein interactions with the upper axial ligand of the cofactor, whereas the interactions of the protein from the opposite site are less considered.10–14 Indeed, B12 cofactors are anchored to B12-dependent enzymes in two different ways: (i) the cofactor retains its intramolecularly coordinating Dmbz base (“base-on” form) during cofactor incorporation into the protein (e.g. class II ribonucleotide reductase or diol dehydratase) or (ii) the intramolecularly coordinating Dmbz base of the cofactor is replaced by a protein histidine (“His”) anchoring group (“His-on” configuration). This mode of protein-cofactor binding is shown in Fig. 1 (ref. 10) and encountered, amongst others, in MeCbl-dependent methionine synthase (MetH) as well as AdoCbl dependent methylmalonyl CoA mutase (MCM) and glutamate mutase (GluM) (Table 1).15–17
| Enzyme complex | Protein designation | Organism | Cofactor | Cofactor attachment and lower part hydrogen bonding networka | Reference |
|---|---|---|---|---|---|
| a The cobalt anchoring group is underlined. b The Asp residue in GluM forms H-bonds with different residues (Leu and Tyr proposed here). | |||||
| Methionine synthase | MetH | Escherichia coli, Homo sapiens | MeCbl | Asp-X-![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) ![[s with combining low line]](https://www.rsc.org/images/entities/char_0073_0332.gif) -XX-Ser -XX-Ser |
18 |
| Metyhmalonyl CoA mutase | MCM | Propionibacterium shermanii | AdoCbl | Asp-X--XX-Lys |
19 and 20 |
| Homo sapiens, Ascaris lumbricoides | |||||
| Glutamate mutase | GluM | Clostridium tetanomorphum | AdoCbl | Asp-X--XX-Leu-X-Tyrb |
21 and 22 |
| Clostridium cochlearium | |||||
Furthermore, the Co-bound His is part of a hydrogen-bonded amino acid chain, together with an aspartic acid (Asp) and a variable third residue (lysine for MCM or serine for MetH, Table 1).11,23 The importance of the cofactor anchoring and the hydrogen bonding network for catalysis has been controversially discussed.13,23 For example, mutation studies with MetH indicated a dramatic loss of activity while replacing either His or Asp by other amino acids in the hydrogen bonded network.23 A different study focusing on “His-on” AdoCbl dependent isomerases indicate a structural reorganization of the hydrogen bonding network after substrate binding.13,24 This behavior is suggested to stabilize the catalytically active cob(II)alamin intermediates by reducing the basicity of the anchoring His ligand.13 Such behaviour seems very appealing and could represent a general mode of remote control in reactions depending on “His-on” configured cofactor B12. Interestingly, a large number of heme-proteins such as globins, cytochromes c and horse radish peroxidase exhibit also a “His-on” type of cofactor attachment.25–28 Partial deprotonation of the proximal His ligand within a lower part hydrogen bonding network enables the heme-dependent enzymes to fine-tune their properties and reactivity (“push effect”).29–33 This type of redox control has been thoroughly investigated for heme proteins, as the FeIII–13CN protoporphyrin IX complexes,34–36 but was so far not studied for related mimics of “His on” Cbl@protein complexes.12,13,24,37–44 In this context, only one example reported so far the heterogeneous model of a Cob(III)alamin–protein complex,14 whereas some fascinating immobilized models of heme–protein complexes have recently attracted considerable attention.45–48 Insights into the roles of the “His-on” cofactor attachment and the lower part hydrogen bonding network in B12-dependent enzymes are therefore still required. In this context, the development of molecular mimics,13,40,42 enzyme models44,49 and new supramolecular biomimetic architectures14 represent attractive assets.
We envisaged developing a new type of immobilized biomimetic model of the hydrogen bonding network, as found in “His-on” B12 cofactor complexes. This supramolecular assembly (Fig. 1 right) consists of three independent subunits: (i) a structurally modified metal cofactor complex, (ii) a carboxylate ion and (iii) a hydrophobic silica C18 solid support. In particular, we envisaged to induce intermolecular hydrogen bonding between the immobilized CoIII-coordinated imidazole ligand and the carboxylate anion through hydrophobic interactions on the solid support.30
With this supramolecular immobilization strategy in mind, we began by developing the most important component of the supramolecular assembly: the molecular B12 derivative, mimicking the “His-on” CoIII-complex. This intramolecular coordination compound consists of three subunits: (i) the tetradentate CoIII–corrin complex, (ii) a lower (α-) coordinating imidazole unit and (iii) a trans-located cyanide ligand (Fig. 1, right). Such a molecular model allows a thorough study of the imidazole-deprotonation influence on the redox properties (cyclic voltammetry), on the chromophore (UV-vis) and on the trans-influence exercised on the upper (β-) axial CN group (13C-NMR). A meticulous structural modeling using QM/MM calculations (ESI, Fig. S1†) suggested the imidazole backbone derivative of B121-H+ as a close biomimetic of the “His-on” configuration in B12-cofactor protein complexes (Fig. 2).
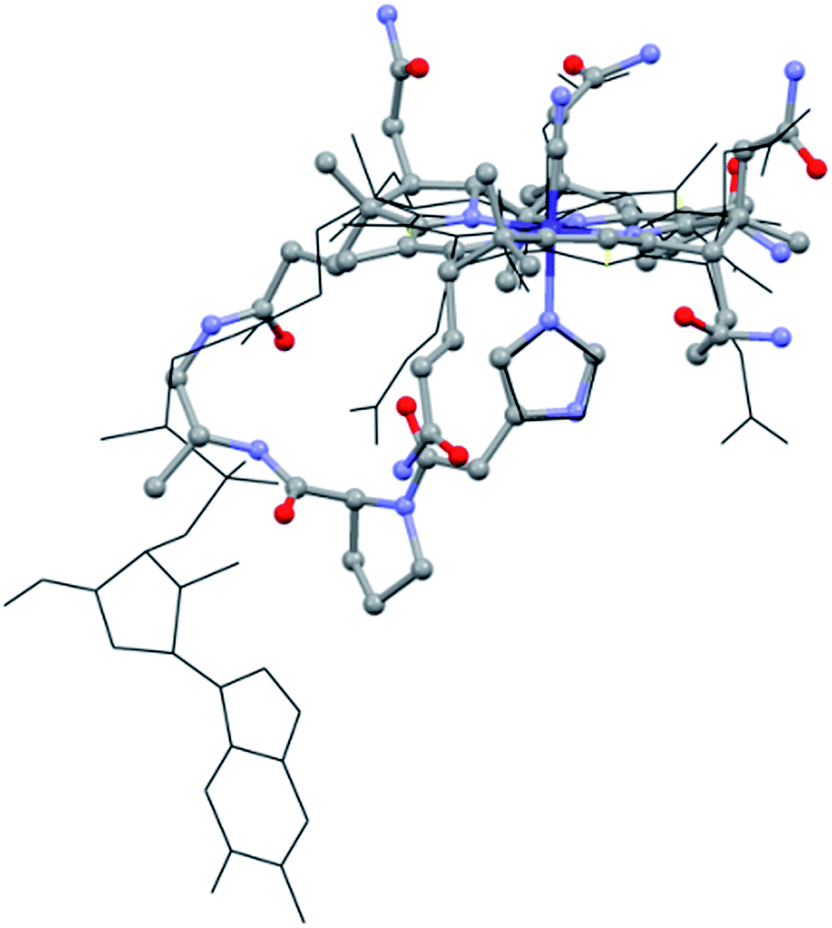 | ||
| Fig. 2 Overlap of the “His-on” configuration in a B12-cofactor protein complex (wireframe; protein: MetH, only MeCbl and His shown, PDB: 1BMT)10 and model of 1-H+ (ball and sticks). | ||
In this compound, the imidazole subunit and the corrin macrocycle are connected via the f-side chain by an ethylenediamine spacer and a proline subunit.50–521-H+ was synthesized as trifluoroacetic acid (TFA) salt in three steps with an overall yield of 19% using common peptide coupling chemistry (ESI, Schemes S1 and S2†). The UV-vis spectrum of 1-H+ resembles that of vitamin B12 (ESI, Fig. S2†), and the high resolution mass spectrum displays a signal at m/z = 610.30054 (calculated: m/z = 610.30032; [(1-H+) + H]2+).
The structural evaluation of the B12@protein model 1-H+ was investigated using 2D-ROESY. Through-space correlation was observed between Him2 (blue on Fig. 3) and the lower side chains 7A and 81 as well as the methyl group 51, all of which are situated at the northern face of the corrin ring. These interactions are complementary to correlations at the southern face of the molecule, between Him5 and the corrin side chains 151, 131, as well as the 1R situated on the peptide backbone (red on Fig. 3). These data thus unambiguously indicate a coordination of the imidazole group through the Nε-nitrogen. These measurements also allowed determining the positioning of the imidazole unit with respect to the macrocycle, and revealed that it follows the C51–C151 axis of the corrin ring (Fig. 3B). Furthermore, comparison with the active site of a “His-on” bound B12-cofactor from crystal structure indicates a good agreement with the structural behavior of 1-H+ (Fig. 2).
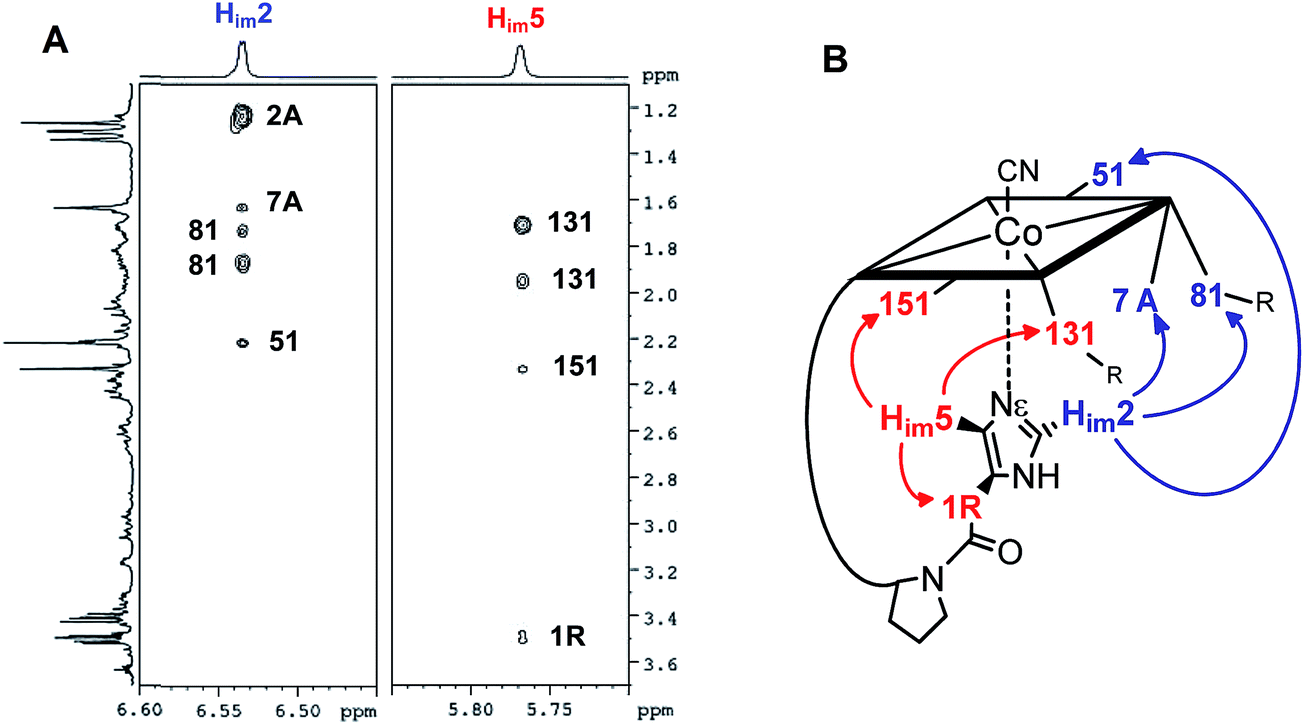 | ||
| Fig. 3 (A): 2D-ROESY coupling of 1-H+ (B): the corresponding correlation in the structure (in D2O, 270 K, 500 MHz) (charge is omitted). | ||
Reversible proton release of 1-H+ (Scheme 1) was then tested by pH titration (from pH 8.5 to pH 12.5) and was detected with UV-vis spectroscopy. It revealed a bathochromic shift of 8 nm for both the α and β bands (ESI, Fig. S3†) which indicates clearly that upon proton release, the imidazole moiety transforms to a stronger σ-donating imidazolate ligand.24,53–55 Additionally, a pKa value of 10.8 was determined for Nδ (ESI, Fig. S3†), which is about 4 pH units lower compared to free imidazole (pKa = 14.5)56 and comparable to the values observed in His-anchored FeIII–13CN protoporphyrin IX@cytochrome c complexes (pKa = 10.1 to 10.6).36
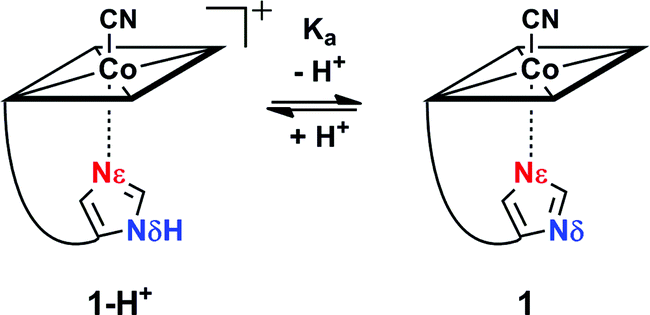 | ||
| Scheme 1 pH equilibrium between 1-H+ and 1. | ||
The electrochemical properties of the cobalt center in 1-H+ and 1 were examined with cyclic voltammetry.57–59 At pH 8.5, a cathodic peak at −920 mV vs. Ag/AgCl was observed for 1-H+ (Table 2, entry 2; Fig. S4†). We assign the cathodic wave to a two electron reduction of the octahedral coordinated Cob(III)alamin in a d6-electron configuration to a CoI-square planar complex in a d8-electron configuration, in agreement with electrochemical studies with B12.57,60 Switching from compound 1-H+ to 1 (pH 12.5) with a CoIII-coordinated imidazolate ligand significantly shifted the cathodic peak by 180 mV to a value of −1100 mV vs. Ag/AgCl (Table 2, entry 1; ESI, Fig. S4†). Deprotonation of 1-H+ to 1 thus stabilizes the CoIII form against reduction.
| Entry | Compound | E pc (mV) | CN 13C-NMR shiftd (ppm) | λ of α-bandf (nm) |
|---|---|---|---|---|
| a vs. Ag/AgCl, 1.5 mM in 0.1 M KCl, scan rate: 0.005 V s−1. b E pc for the CoIII/CoI reduction. c E pc for the CoII/CoI reduction. d In D2O; 126 MHz. e Values obtained from ref. 63. f In H2O. | ||||
| 1 | 1 | −1100b | 133 | 563 |
| 2 | 1-H+ | −920b | 127 | 556 |
| 3 | B12 | −897b | 124 | 551 |
| 4 | aquaCNCbi | −749c | 112e | 530 |
| 5 | diCNCbi | −1170b | 139e | 580 |
The influence of the axial ligand basicity on the trans-located β-CN moiety was then investigated with NMR spectroscopy. The measurements were performed at two different pH (pH 7.0 and 12.5), with an isotopically labeled 13CN ligand on the β-side of the cobalt. The 13C-NMR of 1-H+ revealed a broad signal at 127 ppm (13CN moiety) which shifts downfield (133 ppm) upon deprotonation (1) (ESI, Fig. S5†). This behavior is explained by the coordination of a stronger σ-donor (imidazolate, 1), which results in a decreased polarization of the trans-located cyanide π-electron density.61,62 The broadening of the 13CN lines of 1 and 1-H+ (375 vs. 150 Hz) is due to the distinct nuclear quadrupolar properties of 59Co, which significantly increase the t1 and t2 relaxation rates of the directly bound carbon. These effects also prevent the observation of resolved 13C-multiplets due to scalar coupling between 13C and 59Co. Other broadening mechanisms like anisotropic effects or paramagnetic impurities can be excluded, because all remaining 13C resonances of the measured compounds are sharp (ESI, Fig. S5†).
Control experiments (CV, UV-vis, 13C-NMR) were performed with a ‘blocked’ model at pH 7.0 and pH 12.5 in the same manner described for 1-H+ and 1 (ESI, Fig. S6†). B12 was used for this purpose, since it contains an α-coordinating Dmbz ligand resembling the imidazole unit, for which reversible protonation of both nitrogens is impossible (Fig. 1, left). In effect, all control experiments with B12 exhibited no significant shifts upon pH changes, thus proving that all results obtained with 1-H+ and 1 originated from the deprotonation of the imidazole ligand.
Plotting the position of the cathodic peak Epc against the upper cyanide 13C-NMR shift for the ‘blocked’ model B12, the biomimetic compounds 1 and 1-H+ as well as two other compounds which present distinct lower axial ligand (aquacyanocobinamide, aquaCNCbi; Fig. 4, right; and dicyanocobinamide, diCNCbi; Fig. 4, right) gives a more extensive picture on the influence of lower ligand modulation (Fig. 4).13,24,64,65 The linear correlation suggests that the relative σ-donating capacities of the lower ligand similarly influence both the cathodic peak (Epc) and the upper CN 13C shift. Furthermore, by considering that aquaCNCbi represents a model for base-off B12, important insights are extracted on how Cbls and Cbl@protein complexes may alter their electrochemical properties in biological systems. Indeed, when considering B12 alone, the tuning of the redox properties is reached only through the “base-on/base-off” switch, where the α-Dmbz ligand is replaced by the weaker electron donating H2O molecule. However, this modification only leads to the destabilization of the cobalt center, as shown by the less negative Epc value for the Co(II)/Co(I) reduction of aquaCNCbi (Epc = −749 mV vs. Ag/AgCl, Table 2, entry 4). The contrary effect, a stabilization of the metal ion seems not to be possible in this coordination mode. Switching the α-Dmbz ligand of B12 to an imidazole moiety, as encountered in 1-H+, exhibits only a small effect on the CoIII/CoI reduction (ΔV = −23 mV, Table 2, entry 2 and 3). However, deprotonation to an imidazolate moiety (1) is now possible and leads to a substantial cathodic shift of −203 mV (Table 2, entry 1–3). The lower ligand modulation of 1-H+ thus leads to a doubling of the available redox range (Fig. 4, −749 to −1100 mV vs. Ag/AgCl) compared to B12 alone (Fig. 4, −749 to −897 mV vs. Ag/AgCl). This behavior underscores strikingly that (partial) deprotonation of the coordinated His-residue offers B12-dependent proteins an elegant tool for fine-tuning their electron donating properties on demand. The lower ligand modulation is also reflected in a bathochromic shift of the α-band (Table 2) and the β-band wavelengths (data not shown). Furthermore, linear correlations are observed between the Epc and the α-band (Fig. 4) or the β-band (data not shown). This behavior is in line with recent spectroscopic and theoretical studies, which show that the position of the α-band reflects the dz2 contribution of the lower coordinating ligand to the HOMO orbital.24,53–55
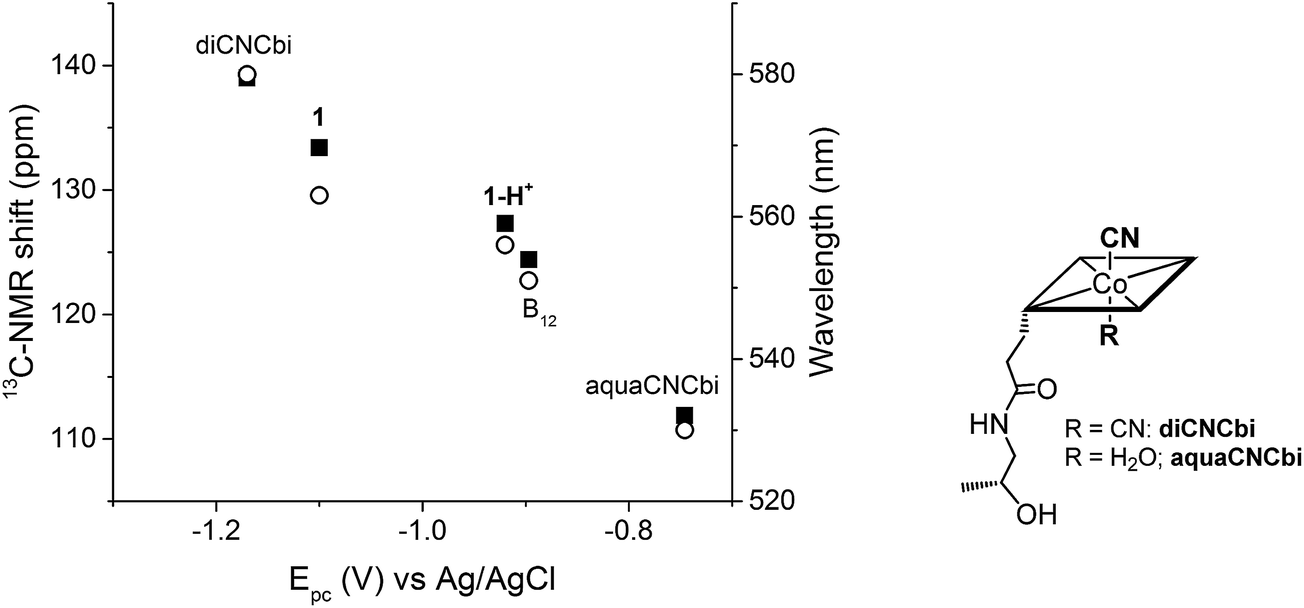 | ||
| Fig. 4 Left: Correlation between Epc (CoIII/CoI reduction for all compounds, except aquaCNCbi: CoII/CoI reduction) and the 13C-NMR shift of CN (left axis, ■) and the α-band wavelength (right axis, ○); B12 data correspond to both pH 7.0 and pH 12.5. Right: Structure of aquaCNCbi and diCNCbi (charges are omitted).66 | ||
In His-on Cbl complexes, the deprotonation of the His residue is achieved through an H-bonded chain in between the ligand triad. However, under aqueous conditions, deprotonation was only achieved at pH values above the pKa of 1-H+ (10.8). For mimicking the biological situation with the model 1-H+ more closely, we thus decided to investigate the interactions between the imidazole of 1-H+ and an exogenous carboxylate as H-bonding acceptor. For this purpose, we applied first benzoic acid in dioxane–H2O mixtures of 1-H+,67 which did not result in any spectroscopic changes. Immobilized 1-H+ (1-H+SP) on C18 silica material behaved differently and allowed studying proton release with diffuse reflectance spectroscopy, an approach recently introduced by our group.14
1-H+SP remains base-on upon immobilization, as indicated by the characteristic reflectance spectrum of a “base-on” Cbl (ESI, Fig. S7†). In agreement with studies under homogenous conditions, treatment of 1-H+SP with a strong base (1 M NaOH) led to a bathochromic shift (6 nm for the α-band) and indicated imidazole deprotonation (Fig. 5; ESI, Fig. S7†). However, the addition of pure toluene, which lacks any H-bond acceptor, to 1-H+SP did not lead to any changes. In contrast, the further addition of benzoate (tetrabutylammonium salt) to this solvent led to a slight, but characteristic bathochromic shift of the α-band (4 nm) and β-band (2 nm) (Fig. 5; ESI, Fig. S8†), which suggests hydrogen-bonding between benzoate and the Nδ hydrogen of 1-H+SP, similar to the situation proposed for this class of proteins.10,13
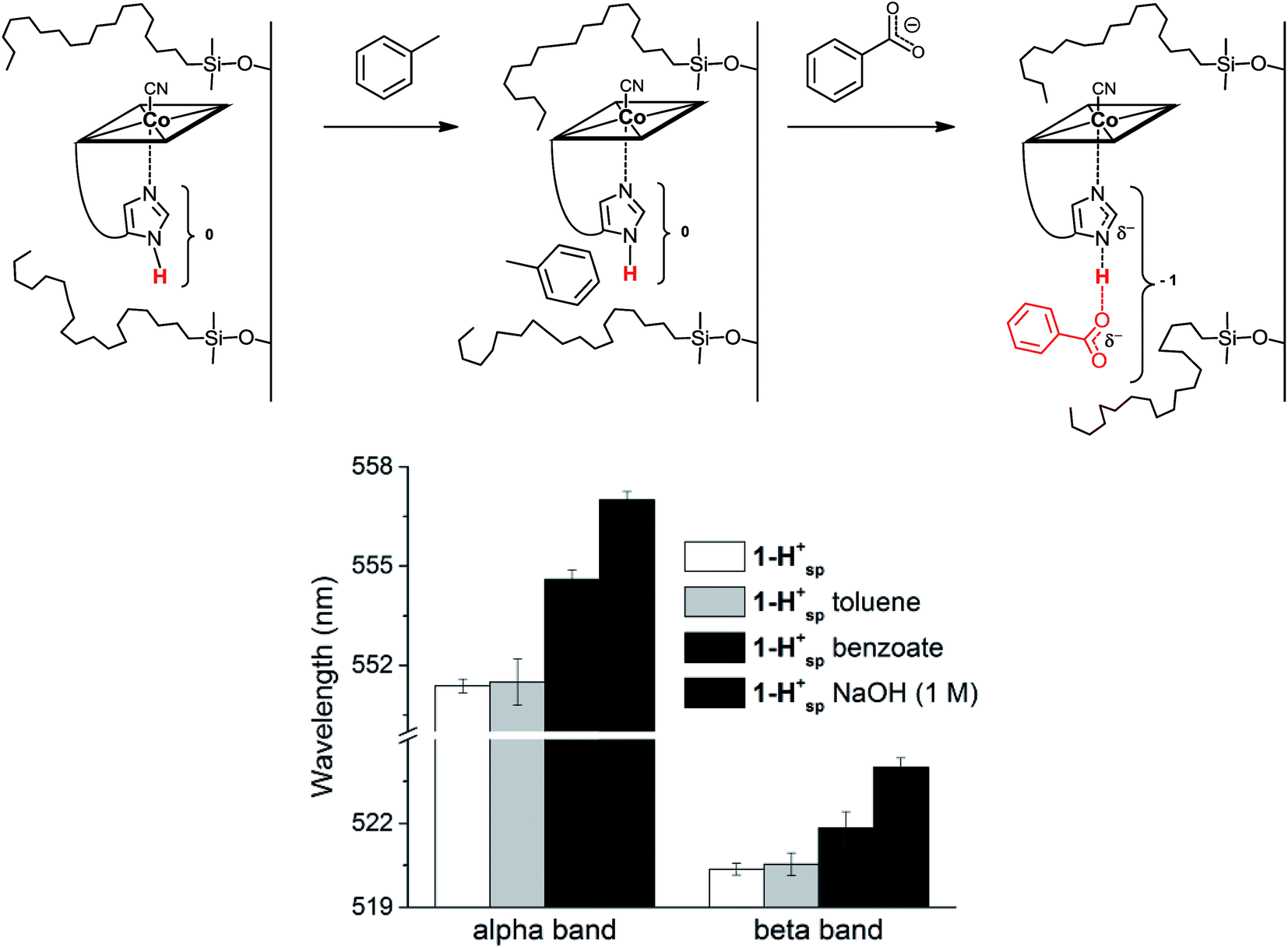 | ||
| Fig. 5 Top: Proposed mechanism for the reaction of 1-H+SP immobilized on C18 silica (left) with toluene (middle) and with benzoate (tetrabutylammonium salt; right). Bottom: Absorbance for the α and β bands of immobilized 1-H+SP (white), 1-H+SP after the addition of toluene (light grey), 1-H+SP after the addition of a solution of benzoate in toluene (dark grey) and 1-H+SP after the addition of 1 M NaOH (black) (n = 4) (charges are omitted). | ||
In summary, we present an unprecedented supramolecular assembly mimicking the regulatory ligand triad of “His-on” metal-cofactor@protein complexes. The data presented herein give evidence that deprotonation of an axially coordinating imidazole to form imidazolate stabilizes significantly (ΔV = −203 mV) the CoIII form of the corrinoid. However, such behavior cannot be achieved with B12 in its base-on form. Modulating the corrinoid redox properties with this elegant trick represents therefore a sophisticated strategy for tailoring the electronic properties of Cbls in biological systems.
Acknowledgements
The authors thank L. Bigler for the recording of the HR-ESI-MS spectra, C. Männel-Croisé for help with the DRUV-Vis measurements and R. Alberto for support. A generous gift of vitamin B12 from DSM Nutritional Products AG (Basel/Switzerland) is acknowledged. This work was supported by the Swiss National Science Foundation (Grant No. 200020-149108).References
- K. Gruber, B. Puffer and B. Kräutler, Chem. Soc. Rev., 2011, 40, 4346–4363 RSC.
- R. Banerjee, Chemistry and Biochemistry of B12, Wiley-Interscience, New York, 1st edn, 1999 Search PubMed.
- F. Zelder, Chem. Commun., 2015, 51, 14004–14017 RSC.
- P. Friedrich, U. Baisch, R. W. Harrington, F. Lyatuu, K. Zhou, F. Zelder, W. McFarlane, W. Buckel and B. T. Golding, Chem.–Eur. J., 2012, 18, 16114–16122 CrossRef CAS PubMed.
- A. J. Brooks, C. C. Fox, E. N. G. Marsh, M. Vlasie, R. Banerjee and T. C. Brunold, Biochemistry, 2005, 44, 15167–15181 CrossRef CAS PubMed.
- A. J. Brooks, M. Vlasie, R. Banerjee and T. C. Brunold, J. Am. Chem. Soc., 2004, 126, 8167–8180 CrossRef CAS PubMed.
- D. Bucher, G. M. Sandala, B. Durbeej, L. Radom and D. M. Smith, J. Am. Chem. Soc., 2012, 134, 1591–1599 CrossRef CAS PubMed.
- J. Masuda, N. Shibata, Y. Morimoto, T. Toraya and N. Yasuoka, Structure, 2000, 8, 775–788 CrossRef CAS PubMed.
- A. M. Calafat and L. G. Marzilli, J. Am. Chem. Soc., 1993, 115, 9182–9190 CrossRef CAS.
- C. L. Drennan, S. Huang, J. T. Drummond, R. G. Matthews and M. L. Ludwig, Science, 1994, 266, 1669–1674 CAS.
- J. T. Jarrett, C. Y. Choi and R. G. Matthews, Biochemistry, 1997, 36, 15739–15748 CrossRef CAS PubMed.
- J. M. Puckett, M. B. Mitchell, S. Hirota and L. G. Marzilli, Inorg. Chem., 1996, 35, 4656–4662 CrossRef CAS.
- K. S. Conrad, C. D. Jordan, K. L. Brown and T. C. Brunold, Inorg. Chem., 2015, 54, 3736–3747 CrossRef CAS PubMed.
- C. Männel-Croisé and F. Zelder, Chem. Commun., 2011, 47, 11249–11251 RSC.
- W. Buckel and B. T. Golding, Chem. Soc. Rev., 1996, 25, 329–337 RSC.
- S. Dong, R. Padmakumar, N. Maiti, R. Banerjee and T. G. Spiro, J. Am. Chem. Soc., 1998, 120, 9947–9948 CrossRef CAS.
- K. L. Brown, Chem. Rev., 2005, 105, 2075–2149 CrossRef CAS PubMed.
- R. G. Matthews, M. Koutmos and S. Datta, Curr. Opin. Struct. Biol., 2008, 18, 658–666 CrossRef CAS PubMed.
- R. Banerjee and S. Chowdhury, in Chemistry and Biochemistry of B12, ed. R. Banerjee, Wiley-Interscience, New York, 1st edn, 1999, pp. 707–730 Search PubMed.
- F. Mancia, N. H. Keep, A. Nakagawa, P. F. Leadlay, S. McSweeney, B. Rasmussen, O. Diat and P. R. Evans, Structure, 1996, 4, 339–350 CrossRef CAS PubMed.
- W. Buckel, G. Bröker, H. Bothe, A. J. Pierik and B. T. Golding, in Chemistry and Biochemistry of B12, ed. R. Banerjee, Wiley-Interscience, New York, 1st edn, 1999, pp. 757–782 Search PubMed.
- R. Reitzer, K. Gruber, G. Jogl, U. G. Wagner, H. Bothe, W. Buckel and C. Kratky, Structure, 1999, 7, 891–902 CrossRef CAS PubMed.
- J. T. Jarrett, M. Amaratunga, C. L. Drennan, J. D. Scholten, R. H. Sands, M. L. Ludwig and R. G. Matthews, Biochemistry, 1996, 35, 2464–2475 CrossRef CAS PubMed.
- T. A. Stich, A. J. Brooks, N. R. Buan and T. C. Brunold, J. Am. Chem. Soc., 2003, 125, 5897–5914 CrossRef CAS PubMed.
- U. Ermler, R. A. Siddiqui, R. Cramm and B. Friedrich, EMBO J., 1995, 14, 6067–6077 CAS.
- J. D. Satterlee and J. E. Erman, Biochemistry, 1991, 30, 4398–4405 CrossRef CAS PubMed.
- J. H. Dawson, Science, 1988, 240, 433–439 CAS.
- T. Spiro, ACS Chem. Biol., 2008, 3, 673–675 CrossRef CAS PubMed.
- D. B. Goodin and D. E. McRee, Biochemistry, 1993, 32, 3313–3324 CrossRef CAS PubMed.
- D. Nonaka, H. Wariishi, K. G. Welinder and H. Fujii, Biochemistry, 2010, 49, 49–57 CrossRef CAS PubMed.
- T. G. Spiro, G. Smulevich and C. Su, Biochemistry, 1990, 29, 4497–4508 CrossRef CAS PubMed.
- V. P. Chacko and G. N. La Mar, J. Am. Chem. Soc., 1982, 104, 7002–7007 CrossRef CAS.
- L. Banci, I. Bertini, P. Turano, M. Tien and T. K. Kirk, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 6956–6960 CrossRef CAS.
- H. Fujii, J. Am. Chem. Soc., 2002, 124, 5936–5937 CrossRef CAS PubMed.
- H. Fujii and T. Yoshida, Inorg. Chem., 2006, 45, 6816–6827 CrossRef CAS PubMed.
- S. E. Bowman and K. L. Bren, Inorg. Chem., 2010, 49, 7890–7897 CrossRef CAS PubMed.
- L. Banci, A. Rosato and P. Turano, J. Biol. Inorg. Chem., 1996, 1, 364–367 CrossRef CAS.
- C. Olea Jr, J. Kuriyan and M. A. Marletta, J. Am. Chem. Soc., 2010, 132, 12794–12795 CrossRef PubMed.
- S. Hirota, S. M. Polson, J. M. Puckett, S. J. Moore, M. B. Mitchell and L. G. Marzilli, Inorg. Chem., 1996, 35, 5646–5653 CrossRef CAS PubMed.
- S. J. Moore, A. Kutikov, R. J. Lachicotte and L. G. Marzilli, Inorg. Chem., 1999, 38, 768–776 CrossRef CAS.
- M. Fasching, W. Schmidt, B. Krautler, E. Stupperich, A. Schmidt and C. Kratky, Helv. Chim. Acta, 2000, 83, 2295–2316 CrossRef CAS.
- B. Kräutler, Öster. Chem. Zeit., 1989, 2–9 Search PubMed.
- M. Kumar and P. M. Kozlowski, Chem. Commun., 2012, 48, 4456–4458 RSC.
- T. Hayashi, Y. Morita, E. Mizohata, K. Oohora, J. Ohbayashi, T. Inoue and Y. Hisaeda, Chem. Commun., 2014, 50, 12560–12563 RSC.
- M. Bröring, Angew. Chem., Int. Ed., 2007, 46, 6222–6224 CrossRef PubMed.
- J. P. Collman, N. K. Devaraj, R. A. Decréau, Y. Yang, Y.-L. Yan, W. Ebina, T. A. Eberspacher and C. E. D. Chidsey, Science, 2007, 315, 1565–1568 CrossRef CAS PubMed.
- T. Itoh, K. Yano, T. Kajino, Y. Inada and Y. Fukushima, Biotechnol. Bioeng., 2006, 93, 476–484 CrossRef CAS PubMed.
- A. Hosseini, C. J. Barile, A. Devadoss, T. A. Eberspacher, R. A. Decreau and J. P. Collman, J. Am. Chem. Soc., 2011, 133, 11100–11102 CrossRef CAS PubMed.
- Y. Morita, K. Oohora, A. Sawada, K. Doitomi, J. Ohbayashi, T. Kamachi, K. Yoshizawa, Y. Hisaeda and T. Hayashi, Dalton Trans., 2016, 45, 3277–3284 RSC.
- K. Zhou and F. Zelder, Angew. Chem., Int. Ed., 2010, 49, 5178–5180 CrossRef CAS PubMed.
- F. Zelder, K. Zhou and M. Sonnay, Dalton Trans., 2013, 42, 854–862 RSC.
- K. ó. Proinsias, M. Giedyk and D. Gryko, Chem. Soc. Rev., 2013, 42, 6605–6619 RSC.
- J. M. Pratt, in Inorganic Chemistry of Vitamin B12, ed. J. M. Pratt, Academic Press, New York, 1972, p. 44 Search PubMed.
- T. Andruniow, P. M. Kozlowski and M. Z. Zgierski, J. Chem. Phys., 2001, 115, 7522–7533 CrossRef CAS.
- R. A. Firth, H. A. O. Hill, J. M. Pratt, R. J. P. Williams and W. R. Jackson, Biochemistry, 1967, 6, 2178–2189 CrossRef CAS PubMed.
- T. Eicher and S. Hauptmann, The Chemistry of Heterocycles: Structures, Reactions, Synthesis, and Applications 2nd, John Wiley & Sons, 2003 Search PubMed.
- D. Lexa and J. M. Saveant, Acc. Chem. Res., 1983, 16, 235–243 CrossRef CAS.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, Chem. Sci., 2013, 4, 819–823 RSC.
- I. A. Dereven'kov, D. S. Salnikov, R. Silaghi-Dumitrescu, S. V. Makarov and O. I. Koifman, Coord. Chem. Rev., 2016, 309, 68–83 CrossRef.
- D. Lexa, J. M. Sayeant and J. Zickler, J. Am. Chem. Soc., 1980, 102, 2654–2663 CrossRef CAS.
- P. L. Gaus and A. L. Crumbliss, Inorg. Chem., 1976, 15, 739–741 CrossRef CAS.
- R. A. Firth, H. A. O. Hill, J. M. Pratt, R. G. Thorp and R. J. P. Williams, J. Chem. Soc. A, 1968, 2428–2433 RSC.
- K. L. Brown, in Chemistry and Biochemistry of B12, ed. R. Banerjee, Wiley-Interscience, New York, 1st edn, 1999, pp. 197–238 Search PubMed.
- A. J. Reig, K. S. Conrad and T. C. Brunold, Inorg. Chem., 2012, 51, 2867–2879 CrossRef CAS PubMed.
- K. L. Brown, Dalton Trans., 2006, 1123–1133 RSC.
- G. L. Nelsestuen, T. H. Zytkovicz and J. B. Howard, J. Biol. Chem., 1974, 249, 6347–6350 CAS.
- M. Komiyama, M. L. Bender, M. Utaka and A. Takeda, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 2634–2638 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04396d |
| This journal is © The Royal Society of Chemistry 2016 |