
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriplet excited state properties in variable gap π-conjugated donor–acceptor–donor chromophores†
Seda
Cekli
a,
Russell W.
Winkel
a,
Erkki
Alarousu
b,
Omar F.
Mohammed
b and
Kirk S.
Schanze
*a
aDepartment of Chemistry and Center for Macromolecular Science and Engineering, University of Florida, Gainesville, Florida 32611-7200, USA. E-mail: kschanze@chem.ufl.edu
bSolar and Photovoltaics Engineering Research Center, Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal 23955-6900, Kingdom of Saudi Arabia
First published on 12th February 2016
Abstract
A series of variable band-gap donor–acceptor–donor (DAD) chromophores capped with platinum(II) acetylide units has been synthesized and fully characterized by electrochemical and photophysical methods, with particular emphasis placed on probing triplet excited state properties. A counter-intuitive trend of increasing fluorescence quantum efficiency and lifetime with decreasing excited state energy (optical gap) is observed across the series of DAD chromophores. Careful study of the excited state dynamics, including triplet yields (as inferred from singlet oxygen sensitization), reveals that the underlying origin of the unusual trend in the fluorescence parameters is that the singlet–triplet intersystem crossing rate and yield decrease with decreasing optical gap. It is concluded that the rate of intersystem crossing decreases as the LUMO is increasingly localized on the acceptor unit in the DAD chromophore, and this result is interpreted as arising because the extent of spin–orbit coupling induced by the platinum heavy metal centers decreases as the LUMO is more localized on the acceptor. In addition to the trend in intersystem crossing, the results show that the triplet decay rates follow the Energy Gap Law correlation over a 1.8 eV range of triplet energy and 1000-fold range of triplet decay rates. Finally, femtosecond transient absorption studies for the DAD chromophores reveals a strong absorption in the near-infrared region which is attributed to the singlet excited state. This spectral band appears to be general for DAD chromophores, and may be a signature of the charge transfer (CT) singlet excited state.
Introduction
The donor–acceptor–donor (DAD) approach has been used extensively for tuning the band gap in π-conjugated systems by controlling the energetics of intramolecular charge transfer from electron rich donor to electron deficient acceptor moieties.1 Numerous studies over the past two decades have investigated DAD chromophores for applications in organic electronics and organic photovoltaic (OPV) devices. For example, in OPVs and light emitting diodes (LEDs), DAD chromophores are widely used to control band gaps for efficient visible absorption and emission at a desired spectral range.2–4 On the other hand, DAD type molecules have also attracted attention for non-linear optical applications due to their high two- and three-photon absorption cross sections.5 As a result of their importance in optoelectronic applications, it is essential to understand the behaviour of the excited states in DAD systems arising from structural and energetic changes in chromophore structure.A great deal of work on the photophysics of π-conjugated DAD oligomers and polymers has been carried out, which mostly focuses on the singlet excited state properties and device performance.6–8 A recent investigation explored singlet fission in oligomers and polymers that feature a DAD electronic structure.9 Even though the long-lived triplet excited state has very interesting properties in organic electronics and photovoltaic applications, to our knowledge, triplet state properties such as T1 energy, S1–T1 splitting energy, intersystem crossing yield and rate in conjugated DAD molecules have not been well studied.10–12 The main reason is that π-conjugated DAD molecules have low intersystem crossing yields13 and they are usually not phosphorescent. Since the non-radiative decay rate of the triplet state increases exponentially with the T1–S0 energy gap due to the Energy Gap Law,14 DAD molecules having a low lying triplet state are not phosphorescent.
Herein we report a systematic study focusing on the correlation between the triplet state properties and energetics in the series of variable band gap π-conjugated DAD molecules shown in Fig. 1. This series consists of organic donor and acceptor moieties with different strengths for controlling the band gap and charge transfer character, as well as two heavy metal (platinum(II)) centers to promote intersystem crossing.15 The spectroscopy and dynamics of the singlet and triplet excited states were probed by using steady-state luminescence spectroscopy and femtosecond–nanosecond transient absorption spectroscopy. In addition to the experimental work, a computational study was conducted to estimate the triplet energies for the series.
 | ||
| Fig. 1 Structures of the DAD molecules with increasing acceptor strength order. | ||
The results presented here for the series of DAD chromophores reveal a counter-intuitive trend in their photophysical properties; in particular we observe an increase in the fluorescence quantum yield and lifetime with decreasing excited state energy. This trend runs counter to the expected Energy Gap Law correlation, where the non-radiative decay rate increases with decreasing excited state energy.14,16–18 Our results demonstrate that the underlying reason for the unusual trend is a correlation between singlet (and triplet) excited state energy and the intersystem crossing rate and quantum efficiency. In addition to this correlation, the study also finds that the triplet decay rates for the series of DAD chromophores follows the usual Energy Gap Law correlation between the non-radiative decay rate (knr) and the triplet energy. Correlation of the results on the present system, along with those from a previous study by Köhler and co-workers19 gives a remarkable linear relation between knr and triplet energy over a 1.8 eV range of triplet energy.
Results
Structures of the complexes and electrochemistry
The series of seven DAD π-conjugated chromophores shown in Fig. 1 were used to study the effect of energetics and charge transfer character on photophysical properties. Each DAD chromophore features an electron poor arylene unit in the center (acceptor), flanked on either side by electron rich thiophene (Th) or 3,4-ethylenedioxythiophene (EDOT) units (the donors). The π-conjugated DAD systems are “capped” on each end with trans-CC-PtII(PBu3)2(CCPh) units; in previous work we and others have demonstrated that the heavy metal platinum(II) acetylide auxochromes promote intersystem crossing due to enhanced spin–orbital coupling.18,20,21 The design of the series of complexes was based on the concept of systematically varying the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) by using different donor and acceptor groups in the chromophore structure. The change in the primary structural feature is the use of five different acceptor moieties, which increase in acceptor strength in the order TBTz < TPD < TQT < TBT < TPT (in order of decreasing LUMO energy). In addition, in two complexes the donors were replaced with EDOT, the latter of which is slightly more electron rich (stronger donor). Overall, as described in more detail below, across the series of compounds the optical band gap (onset of absorption) varies by ∼0.8 eV.Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were performed in order to obtain the oxidation and reduction potentials for the compounds' redox processes, which allows estimation of the HOMO–LUMO energy gap of the molecules. The HOMO and LUMO levels were calculated from the oxidation and reduction potentials (Eox and Ered, respectively) according to the following equations: EHOMO = −(Eox + 5.1) eV and ELUMO = −(Ered + 5.1).22 A summary of the electrochemical data is provided in Table 1 and the CV and DPV plots are in ESI.†
| E 1/2/V | Frontier orbital energies/eV | HOMO–LUMO gap/eV | |||||
|---|---|---|---|---|---|---|---|
| red | ox1 | ox2 | E LUMO | E HOMO | ΔEgf | ΔEopg | |
| a CV data, conducted in dimethylformamide and referenced to Fc/Fc+ as an internal standard. b CV data, conducted in dichloromethane and referenced to Fc/Fc+ as an internal standard. c DPV data, conducted in dimethylformamide and referenced to Fc/Fc+ as an internal standard. d E LUMO = −(E[red vs. Fc+/Fc] + 5.1) eV. e E HOMO = −(E[ox vs. Fc+/Fc] + 5.1) eV. f Electrochemical gap = Eox − Ered. g Optical gap is found from the onset of absorption spectra. | |||||||
| TBTz | −2.10a | 0.27c | — | −3.00 | −5.37 | 2.37 | 2.37 |
| TPD | −1.84a | 0.45c | — | −3.26 | −5.55 | 2.29 | 2.30 |
| TQT | −1.83b | 0.28b | 0.40b | −3.27 | −5.38 | 2.11 | 2.04 |
| TBT | −1.74b | 0.30b | 0.45b | −3.36 | −5.40 | 2.03 | 1.96 |
| TPT | −1.65b | 0.12b | 0.36b | −3.45 | −5.22 | 1.77 | 1.61 |
| EQE | −1.96b | 0.04b | 0.23b | −3.14 | −5.14 | 2.00 | 1.90 |
| EBE | −1.85b | 0.03b | 0.27b | −3.25 | −5.13 | 1.88 | 1.80 |
All of the molecules exhibit a single, reversible reduction at negative potentials relative to Fc/Fc+. The reduction potentials lie between −2.10 V and −1.65 V and correspondingly the LUMO energies vary between −3.00 eV and −3.45 eV. The variation of the LUMO energy across the series is a direct result of the influence of the acceptor unit; molecular orbital calculations with density functional theory (DFT, see below) indicate that the LUMO is increasingly localized on the core acceptor as the energy decreases.23 For example, the stronger electron-accepting ability of benzothiadiazole results in lower LUMO levels in TBT and EBE compared to TQT and EQE which contain the relatively weaker electron accepting quinoxaline moiety.
In addition, most of the molecules exhibit two oxidation waves at positive potentials. For those that do not exhibit reversible oxidation waves, the peak value from the DPV scan is used to estimate the potential. For the thiophene series, the oxidation potentials vary across a narrow range (+0.12 to +0.45 V) but without a systematic trend. The two EDOT compounds feature reversible waves at ∼+0.04 V, which is generally less than any of the thiophene series, reflecting the stronger donor nature of EDOT.24 Over the entire series, the range of HOMO values is −5.13 to −5.55 eV. There is no trend observed in the HOMO energies of the complexes because the HOMOs are more strongly delocalized throughout the entire molecules.
The HOMO–LUMO gap (ΔEg) is calculated from the oxidation and reduction potentials of the molecules, and the results are listed in Table 1 along with the optical band gap (ΔEopt) estimated from the onset of the long wavelength band in the absorption spectra (Fig. 2). As expected, the increase in acceptor strength causes a systematic decrease in the LUMO levels, resulting in a systematic decrease in the HOMO–LUMO gap across the series. It should be noted that the good agreement between ΔEg and ΔEopt reflects the fact that the long wavelength absorption band (and thus the lowest excited state configuration) is dominated by the HOMO–LUMO transition.
 | ||
| Fig. 2 Normalized absorption (left) and emission (right) spectra of TBTz, TBT and TPT. | ||
Absorption and fluorescence spectroscopy
The optical properties of the molecules were studied by absorption and fluorescence spectroscopy in THF at room temperature, and the spectra of selected examples are illustrated in Fig. 2. In general, all of the molecules feature two primary absorption bands, with the exception of TBTz and TPD. The first band appears in the near-UV and it is assigned to a π–π* transition, while the second band, at lower energy with a maximum in between 500 and 650 nm, is assigned to a charge transfer (CT) transition.25 The unmetallated DAD chromophores (4a–e, ESI S1†) also feature these two primary bands, which suggests that the main absorption bands are mainly due to the transitions localized on the conjugated DAD unit. However, the bands are red-shifted in the corresponding metallated chromophores, indicating that there is extended conjugation due to dπ–pπ orbital overlap with the Pt centers. On the other hand, TBTz and TPD do not exhibit the same two absorption band pattern due to the low acceptor strength of their core arylene units; this results in less charge transfer character in the low energy optical transition.26All of the molecules exhibit fluorescence at room temperature which is Stokes shifted moderately from the long-wavelength absorption band (Fig. 2 and S2†). As can be seen in Fig. 2, the fluorescence spectrum is red-shifted as the acceptor strength of the core arylene unit increases, in a trend that is in accord with the discussion above regarding the HOMO–LUMO gaps. For most of the molecules the fluorescence bands are broad and structure-less, consistent with the charge transfer nature of the underlying optical transitions.27 The TBTz and TPD compounds feature structured emission bands which can be explained by the weaker donor–acceptor interactions found in these chromophores.
To give direct information about the triplet energies for the series, we carried out steady-state emission experiments in a low-temperature glass at 77 K (2-methyltetrahydrofuran solvent) in an effort to observe phosphorescence from the triplet state. The emission scans were done over the 0.70–1.5 μm wavelength range by using a spectrometer equipped with a liquid-nitrogen cooled InGaAs detector. Weak phosphorescence was only observed for TBTz and TPD (ESI, S3†) and from the weak peaks the triplet energies are estimated to be 1.53 eV and 1.59 eV, respectively. The corresponding singlet–triplet splitting for these complexes is 1.09 and 0.88 eV, which is in accord with previous work on related DAD structures.19
Excited state dynamics
Fluorescence quantum yields (ϕf) and decay lifetimes (τf) were determined for the molecules in THF solution and the results are listed in Table 2. The fluorescence quantum yield varies strongly from 0.07 and 0.43 for the thiophene chromophores and 0.29 and 0.42 for the EDOT chromophores. In all cases, the ϕf values are less than 0.5, indicating that a non-radiative decay channel is involved in the deactivation of the singlet excited state. On the basis of previous work with related Pt–acetylide complexes, the non-radiative pathway is considered to be due singlet–triplet intersystem crossing (ISC), which is promoted by the strong spin–orbit coupling induced by the metal complex centers. A remarkable trend is that the fluorescence quantum yield systematically increases with increasing acceptor strength along the thiophene and EDOT series (Fig. 3b).28 As seen in Fig. 3c, a parallel trend is observed for the fluorescence lifetimes (τf) across the series. The fact that there is a parallel variation in ϕf and τf across the series reinforces the conclusion that a systematic variation in a non-radiative decay channel (presumably via ISC) is the source of the observed trends in the emission parameters.
λ
abs, nma (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) ε) |
λ f nm | ϕ f | τ f ns | τ T μs | ϕ Δ | τ S ns | k ISC (109) s−1 | E T eV | ΔESTk eV | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Measured in THF at room temperature. b Measured in THF using rhodamine B (ϕf = 0.69)29 as an actinometer. c Measured in THF using tetraphenylporphyrin (ϕf = 0.12)30 as an actinometer. d Measured by TCSPC in dry THF. e Measured by nanosecond transient absorption spectroscopy, in deoxygenated THF. f TPT is not a singlet oxygen sensitizer. g Measured by femtosecond transient absorption spectroscopy in THF. The fitting parameters and kinetics plots are shown in ESI (Fig. S8). h Calculated from kISC = ϕISC/τS where it is assumed that ϕISC ≈ ϕΔ. i T1 energy computed by DFT. j Measured in MeTHF at 77 K. k Energy difference between the experimental S1 energy found from the emission spectrum and the T1 energy calculated by DFT. l Estimated from the linear relationship presented in Fig. 6A and B. | ||||||||||
| TBTz | 463 (4.95) | 515 | 0.07b | 0.17 | 3.9 | 0.83 | 0.20 | 4.19 | 1.38 (1.53)j | 1.09 |
| TPD | 475 (4.84) | 542 | 0.14b | 0.50 | 6.2 | 0.57 | 0.52 | 1.10 | 1.49 (1.59)j | 0.88 |
| TQT | 370, 518 (4.84, 4.63) | 660 | 0.38b | 3.89 | 2.62 | 0.24 | 2.14 | 0.112 | 1.22 | 0.88 |
| TBT | 370, 546 (4.71, 4.54) | 685 | 0.43b | 4.84 | 1.81 | 0.15 | 3.16 | 0.048 | 1.08 | 0.93 |
| TPT | 406, 638 (4.81, 4.48) | 835 | 0.009c | 0.28 | 0.18 | NAf | 0.22 | 0.005l | 0.71 | 0.85 |
| EQE | 386, 559 (4.76, 4.53) | 709 | 0.29c | 2.25 | 1.78 | 0.36 | 2.38 | 0.15 | 1.20 | 0.75 |
| EBE | 389, 596 (4.78, 4.57) | 758 | 0.42c | 3.65 | 1.37 | 0.32 | 5.76 | 0.056 | 1.01 | 0.83 |
 | ||
| Fig. 3 Photophysical properties of the molecules (thiophene series are in orange, EDOT series are in blue). (a) Optical gap, (b) fluorescence quantum yield, (c) fluorescence lifetime, (d) singlet oxygen quantum yield. | ||
In order to quantify the intersystem crossing yield, we used the indirect approach of determining the singlet oxygen yield (ϕΔ) for the series of molecules. Previous studies indicate that there is a correlation between the singlet oxygen and triplet yields,13 although it is prudent to realize that the singlet oxygen yield is a lower limit for the triplet yield. The singlet oxygen quantum yields were determined in benzene-d6 by monitoring the emission at 1260 nm. With the exception of TPT,31 all of the molecules sensitize singlet oxygen. As seen from Table 2, the singlet oxygen quantum yields vary from 0.83 for TBTz to 0.15 for TBT and 0.36 for EQE and 0.32 to EBE. As seen in Fig. 3, there is a noteworthy inverse correlation in the singlet oxygen yields compared to the fluorescence quantum yields and lifetimes. This is strong evidence supporting the hypothesis that the triplet yield systematically decreases as the acceptor strength increases.
Parallel photophysical measurements were also performed on the unmetallated donor–acceptor–donor chromophores, and the results are collated in Fig. S5† along with the experimental details. Interestingly, the inverse correlation between ϕf and ϕΔ is also present in the series of unmetallated chromophores, suggesting that the trend is not only limited to chromophores that contain heavy metal centers. This supports the hypothesis that the effects of structure and energetics on intersystem crossing dynamics and efficiency in donor–acceptor systems are general, and may apply to other π-conjugated chromophores and polymers as discussed below.
Transient absorption spectroscopy
To provide information regarding the spectroscopy and dynamics of the excited states of the DAD molecules, nanosecond and femtosecond transient absorption (TA) spectroscopy studies were conducted. First, the nanosecond TA difference spectra of the molecules were collected following 355 nm pulsed laser excitation in deoxygenated THF. The nanosecond TA spectrum of the selected molecules (TBTz, TBT and TPT) are illustrated in Fig. 4, and the decay lifetimes of the transients, which are assigned to the triplet states, are listed in Table 2 (τT). The TA spectra of the remaining molecules are in Fig. S5.† In every case, the transients were completely quenched by oxygen for air-saturated solutions, leading to assignment of the transients to the triplet–triplet absorption. For all of the complexes, a broad triplet–triplet (T1 → Tn) absorption band was observed at longer wavelength compared to the CT band, in addition to a negative feature corresponding to the ground state absorption (bleach). In contrast to the Pt-complexes, the unmetallated DAD chromophores exhibit very weak or no triplet absorption, which indicates that the Pt centers are necessary to promote intersystem crossing (ISC). The triplet–triplet absorption lifetimes of the complexes vary between 6.2 and 0.18 μs for the thiophene series, and 1.78–1.37 μs for the EDOT series. It is observed that, in general, the triplet lifetimes of the molecules decrease with the increasing acceptor strength for both series, and as discussed below, this trend is in accord with the Energy Gap Law. | ||
| Fig. 4 (a) Normalized steady-state absorption spectra of selected molecules in THF at room temperature. (b) Normalized nanosecond transient absorption difference spectra, following nanosecond-pulsed 355 nm laser excitation pulse (4 mJ per pulse) in argon-purged THF. | ||
In order to probe the dynamics of the singlet state, femtosecond TA was carried out in the visible and near-IR regions (375–1300 nm). TA spectra for the selected complexes, as well as the kinetics traces of the excited state absorption are shown in Fig. 5. Focusing first on the visible region, the spectra are characterized by a bleach and broad excited-state absorption in the red that appears promptly after the 100 fs excitation and then evolves over the first few nanoseconds. The spectral evolution is assigned to triplet–triplet absorption resulting from singlet–triplet intersystem crossing, and the final spectra (∼3 ns delay) are in good agreement with the triplet state spectra observed in the nanosecond timescale (Fig. 4). Turning to the near-IR region (Fig. 5), it is quite interesting that a distinct transient absorption band was observed for all of the molecules in the 850–1300 nm region. This near-IR absorption decays on the same timescale as the spectral evolution observed in the visible region, and on this basis it is assigned to the singlet excited state. A similar spectral feature was reported in a femtosecond transient absorption study of a related unmetallated DAD chromophore and it was assigned to the singlet state.32 To our knowledge, there are few studies32–34 that have reported singlet excited state absorption in the near-IR region for intramolecular charge transfer chromophores. This intense S1 → Sn absorption band is one of the initial examples of near-IR singlet excited state transitions in DAD chromophores and illustrates a potentially important spectroscopic signature in these chromophore systems.
 | ||
| Fig. 5 Femtosecond transient absorption difference spectra of TBTz, TBT and TPT at indicated delay times following 355 nm laser excitation pulse (1.5 μJ per pulse) in THF and the temporal evolution of transient absorption at selected wavelengths. | ||
Kinetics obtained at selected visible and near-IR wavelengths are shown in the right side of Fig. 5; while it is clear that the dynamics occur on the same timescale, the fits to the near-IR band decay (which is assigned to the singlet state) are the most robust due to the lack of spectral overlap, and these are used to determine the singlet lifetimes. These values are listed in Table 2 as τS, and it is seen that they are in excellent agreement with the fluorescence lifetimes (τf). The τS listed in Table 2 are used to calculate the intersystem crossing rate (kISC) for the molecules from the equation, kISC = ϕISC/τS35 where ϕISC is estimated from the singlet oxygen quantum yield36 (i.e., we assume that ϕISC ≈ ϕΔ).37 In general, it is seen that the rate of intersystem crossing decreases as the acceptor strength increases.
Computational results
A key aspect of this work is to correlate the observed properties to the energies of the singlet and triplet excited states. Due to the fact that phosphorescence was not observed in most of the molecules, it was necessary to apply density functional theory (DFT) calculations to estimate the triplet energies. In addition, the computations were also carried out on the singlet ground state and they provide insight regarding the energies and electronic distributions for the frontier molecular orbitals. Results concerning the HOMO and LUMO orbitals are included in the ESI;† here we focus on the use of the DFT calculations to estimate the triplet energies.The previously reported ΔSCF method was used due to its ability to accurately predict the triplet energies of platinum acetylide chromophores.38 The lowest energy triplet conformation was used for the energetics calculations (see ESI† for optimized singlet and triplet geometries).39 The calculated triplet energy, T1, is determined by taking the difference in energy of the optimized triplet and optimized singlet (ground state) geometries (adiabatic energy). These computed triplet energies (T1) are listed in Table 2. As a point of reference for the computations, it is satisfying that the computed T1 energies for TBTz′ and TPD′ are in good agreement with the experimental phosphorescence energies (see Table 2, spectroscopic values are in parentheses). Additionally, when these data are plotted alongside experimental data from Köhler,19 a clear linear correlation is observed, and this will be discussed below.
Discussion
Charge transfer extent and the dynamics of intersystem crossing
A key outcome of this work is the correlation between the excited state energy (which correlates with acceptor strength) and the efficiency and dynamics of intersystem crossing. Qualitatively it is found that as the singlet excited state energy decreases (optical bandgap decreases, acceptor strength increases), the triplet yield decreases (Fig. 3 and 6). This finding is quite significant, because given the widespread use of donor–acceptor–donor type chromophores in organic electronics, the finding is relevant to the performance of this class of materials in applications. In particular, triplet state formation via intersystem crossing has an impact on the efficiency of OLEDs and organic solar cells.40–42 In this section we explore the basis for this effect, by considering several correlations, along with the theory of spin–orbit coupling in the context of charge-transfer excited states. | ||
| Fig. 6 The natural log of intersystem crossing rate vs. (a) singlet energy, (b) triplet energy. Black dashed lines are given as guides to the eye.37 | ||
First, we review the pertinent experimental results. As seen in Table 2 and Fig. 3 (with the exception of TPT)28 the fluorescence quantum yields (ϕfl) and lifetimes (τf) increase as the optical gap decreases (TBTz < TPD < TQT < TBT, EQE < EBE). In addition, the singlet oxygen quantum yields (ϕΔ), which reflect a lower limit for the triplet yield, decrease as the optical gap decreases. Taken together, these three trends indicate that the rate (kISC) and efficiency of intersystem crossing decrease as the acceptor strength increases. This prediction is borne out when kISC is computed from the experimental parameters (Table 2). The significance of this result is that it signals that the dynamics of ISC is correlated with the excited state energy, and at the molecular level it may be determined by the strength of the charge transfer interaction the DAD chromophore systems.
In order to understand the trends in ISC dynamics in more detail, we considered how kISC varies with several molecular energetic terms for the DAD chromophores. First, we explored the relationship between kISC and the energy gap between the S1 and T1 states (singlet–triplet splitting, ΔEST) in order to determine whether kISC exhibits a Marcus normal or inverted region dependence on ΔEST. Indeed, there are several literature reports which suggest that for specific chromophores kISC exhibits a Marcus normal region dependence on ΔEST due to increased Franck–Condon overlap of the singlet state with the triplet state as the gap increases.43–46 However, in the present case we find that there is no clear correlation observed between kISC and ΔEST (see ESI Fig. S9†). This finding means that a factor other than the energy gap between the lowest singlet and triplet excited states determines the rate of the transition.
Second, we turned to consider how the dynamics of ISC correlate with excited state energies across the series. Interestingly, in this case when ln(kISC) is plotted vs. the energy of the S1 and T1 states (Fig. 6A and B), excellent correlations are observed: ln(kISC) decreases almost linearly with decreasing excited state energy. As noted above, the S1 and T1 energies decrease with increasing charge transfer character in the DAD chromophore series as a result of the decrease in HOMO–LUMO gap. Thus, we conclude that kISC decreases as the extent of the charge transfer interaction increases in the DAD chromophores.
Given the observed trend between kISC and the strength of the donor–acceptor interaction for the series of DAD chromophores, it is logical to consider that the effect has its origin in the molecular-electronic structures of the chromophores. In order to explore the origin of the effect in more depth, we consider Fermi's golden rule approximation for the rate of intersystem crossing,47
 | (1) |
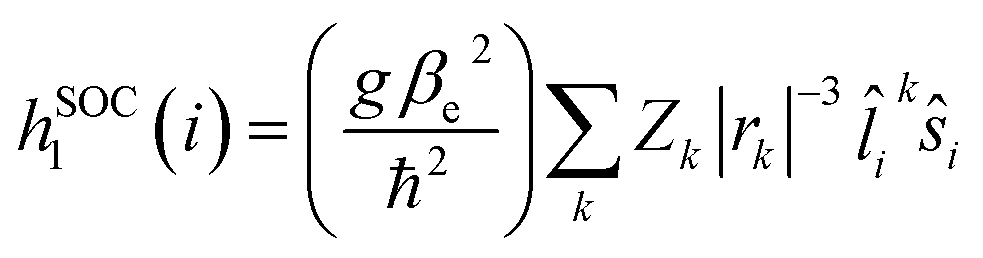 | (2) |
 | (3) |
Consideration of eqn (2) reveals that it is dominated by the heavy atom (platinum in the present systems) due to its considerably larger Z value compared to the light elements. Thus, as shown in the first term, the spin orbit coupling (and thus kISC) will inversely scale with the third power of the distance between the electron and platinum, (|rPt|−3). The important consequence of this relationship in the context of the results on the DAD chromophores is the following. As acceptor strength increases, the LUMO is increasingly localized on the acceptor unit which is in the core of the chromophore. As a result, with increasing acceptor strength, the excited electron is more localized on the acceptor (LUMO), which results in a generally larger |rPt|, and consequently reduced SOC due to the contribution of the (|rPt|−3) to the coupling operator.
Despite the convoluted nature of the arguments presented above, several clear conclusions can be drawn. The experimental results show that there is a distinct correlation between the excited state energies, which are controlled by the donor–acceptor interaction, and the dynamics and efficiency of intersystem crossing. Conventional wisdom would suggest that this effect is determined by energetics (i.e., ΔEST and the Franck–Condon factors); however, such correlations do not emerge. Rather, we hypothesize based on the arguments above, that the effect is dominated by the electronic term, namely the degree of spin orbit coupling, which to a first order approximation is determined by the distance between the excited electron and the heavy atom (in the present systems platinum). This average distance is increased when the LUMO is more localized on the acceptor, and this gives rise to the decreased spin orbit coupling as the acceptor strength increases.
Triplet decay rates and the Energy Gap Law
As noted above, a second clear trend that emerges from the present study is an Energy Gap Law correlation between the triplet energies of the DAD chromophores and the triplet decay rate. This is illustrated in Fig. 7, where the complexes are ordered with respect to decreasing triplet lifetime (τT), in order to emphasize the correlation. This observation shows that the smaller HOMO–LUMO gap in DAD complexes decreases the triplet energy, resulting in an increase in the triplet decay rate, kT. | ||
| Fig. 7 Triplet state properties of the molecules (thiophene series are in orange, EDOT series are in blue) (a) triplet lifetime, (b) calculated triplet energy. | ||
In order to understand the quantitative relationship between kT and energetics, we consider an Energy Gap Law correlation. The Energy Gap Law is an established theory which states that the non-radiative rate for excited state decay decreases exponentially with increasing energy difference between the states involved, and its mechanism is controlled by the Franck–Condon overlap of their wave-functions.14,16–19 The Energy Gap Law is expressed by eqn (4), where F is the Franck–Condon factor, γ consists of the molecular parameters, and ω is the high frequency vibrational mode that is coupled to the triplet decay. This equation suggests a linear relationship between the log of the non-radiative decay rate and the singlet–triplet energy gap.
 | (4) |
Fig. 8 illustrates an Energy Gap Law correlation for the present series of DAD chromophores, as ln(kT) vs. the triplet energy (ET).50 The plot also includes the results for triplet decay rates for a series of structurally-related π-conjugated platinum acetylide chromophores reported by Köhler and co-workers.19 Several points are of note. First, a clear linear trend is evident in the correlation, showing that for the present series of DAD chromophores triplet decay rates follow the Energy Gap Law. When one includes the results from Köhler and co-workers, the linear correlation holds over more than a 1.8 eV range of excited state energy (0.7–2.5 eV), and over 1000-fold range of decay rates (103 to 106 s−1). To our knowledge, this is the largest range of energy explored for a single set of structurally-related chromophores in an Energy Law correlation. Finally, the fact that the correlation holds across a series of chromophores that have very little charge transfer character (Köhler and co-workers)19 to those with considerable charge transfer (e.g., TPT and EBE) indicates that the triplet decay dynamics are determined by the Franck–Condon factors, and not strongly affected by the electronic terms (e.g., spin–orbit coupling).
 | ||
| Fig. 8 The natural log of the triplet decay rate (kT) of the studied DAD molecules (black squares) and the natural log of the non-radiative decay rate of triplet state from Köhler et al.19 work (red circles) are plotted against the triplet energy. The straight line corresponds to the linear fitting. | ||
Are the effects general for donor–acceptor systems?
In the first section of the discussion part it was shown that the ISC efficiency is controlled by the distance between the heavy metal (platinum) center and the acceptor unit, and it decreases as acceptor strength increases because the LUMO localizes on the acceptor unit, which is spatially separated from the heavy atom. This result might suggest that the effects are restricted only to the special case of organometallic π-conjugated donor–acceptor systems. However, as noted above, the unmetallated donor–acceptor–donor chromophores also exhibit a similar (yet not as well defined) inverse relationship between excited state energy (acceptor strength) and ISC efficiency (Fig. S5†). In particular, in the series of in unmetallated donor–acceptor–donor chromophores, the fluorescence quantum yield and lifetime increase and the singlet oxygen yield decreases with increasing acceptor strength. Indeed, the present study was motivated on the basis of observations made previously on structurally related donor–acceptor–donor chromophores, where it was observed that the triplet yield was apparently much lower in donor–acceptor–donor chromophores compared to similarly structured oligomers that lacked a strong donor–acceptor interaction.13,51,52 In addition, a very recent study of a series of DAD bithiophene chromophores highlights a trend of increasing fluorescence quantum yield trend with decreasing HOMO–LUMO gap.53 In this report, the authors give a complicated explanation for the trend in terms of reverse intersystem crossing from an upper triplet state,53 but we posit that the trend could be the result of the same effect uncovered in the present study. Specifically, the rate and efficiency of ISC decrease as the acceptor strength increases. Taken together, these results suggest that the findings reported here may be not just limited to the specific case of heavy metal substituted donor–acceptor chromophores. If this is correct, then our results could be more broadly significant, as they may pertain more widely to donor–acceptor chromophores (and polymers) of interest in a number of applications, including organic solar cells and light emitting diodes.One may question how the relationship described above and in eqn (1) and (2) could apply in chromophores that lack a heavy metal center. In this regard it is important to recognize that most π-conjugated DAD-type chromophores contain thiophene-based donor units, and ISC in thiophene-based chromophores has been attributed to spin–orbit coupling induced by sulfur (S) as a heavy atom.54 Thus, the arguments made above which are focused on the spin–orbit coupling effect of platinum and the distance between the heavy atom and the acceptor unit (LUMO), may also apply to the S atoms which are located in the thiophene donor units, and are spatially distant from the acceptor moiety.
Conclusions
This study provides detailed insight regarding the relationship between molecular/electronic structure and photophysics in a series of donor–acceptor–donor π-conjugated chromophores. The results demonstrate a clear correlation showing that the rate of intersystem crossing decreases as the extent of charge transfer in the chromophore increases. This distinct correlation gives rise to counter-intuitive effects such as increasing fluorescence quantum yield and excited state lifetime with decreasing excited state energy. While this effect is demonstrated by using organometallic auxochromes to enhance spin–orbit coupling, previous results on related all-organic DAD chromophores13,51,55 suggest that it is a general effect and can be used to aid the design of chromophores where fluorescence quantum yields need to be optimized and/or intersystem crossing minimized. In addition, the study also demonstrated that the Energy Gap Law for non-radiative decay is quantitatively followed for a series of organometallic chromophores over an unprecedented range of energy (1.8 eV) and rates (1000-fold). Taken together, these results provide considerable insight into excited state dynamics in donor–acceptor–donor π-conjugated chromophores, which are the essential building blocks in organic and polymer solar cells.Experimental methods
Synthesis and characterization
Complete details concerning the synthesis and characterization of all complexes are provided in the ESI.† The synthesis and characterization of TBT is reported elsewhere.561H (300 or 500 MHz) and 13C NMR (75.4 or 125.7 MHz) spectra were recorded on either a Varian Mercury 300 or a Varian Inova 500 spectrometer. 31P (121.4 MHz) spectra were recorded on the Varian Mercury 300 spectrometer. High-resolution mass spectrometry was collected with either an Agilent 6200 ESI-TOF or an AB Sciex 5800 MALDI TOF/TOF in the Chemistry Department at the University of Florida.Photophysical and electrochemical methods
Steady-state absorption spectra were recorded on a Shimadzu UV-1800 dual beam spectrophotometer. Corrected steady-state emission measurements were performed on a Photon Technology International (PTI) spectrophotometer. Low-temperature luminescence measurements and near-IR emission measurements were conducted on a PTI Quantamaster near-IR spectrophotometer equipped with an InGaAs photodiode detector. For low temperature measurements, samples were prepared in methyl-tetrahydrofuran and degassed by five repeated cycles of freeze–pump–thaw on a high-vacuum line and cooled in a LN2-cooled Oxford Instruments Optistat DN-1704 optical cryostat connected to an Omega CYC3200 temperature controller.Fluorescence lifetimes were collected in anhydrous THF on a Picoquant FluoTime 100 time-correlated single photon counting (TCSPC) instrument and analyzed with FluoFit Software. Fluorescence quantum yields were reported relative to known standards and include ±10% error. The optical density of solutions at the excitation wavelength was 0.03–0.07 and corrected by the difference in the refractive index of standard and sample solvents. Singlet oxygen quantum yield measurements were conducted in deuterated benzene after 10 min of purging with oxygen. Singlet oxygen quantum yield values were reported relative to terthiophene (ϕΔ = 0.84)57 and are estimated to have ±15% error.
Nanosecond transient absorption spectroscopy measurements were performed on an in-house apparatus that is described in detail elsewhere.58 The third harmonic of a Continuum Surelite series Nd:YAG laser (λ = 355 nm, 10 ns FWHM, 10 mJ per pulse) was used as the excitation source. Probe light was produced by a xenon flash lamp and the transient absorption signal was detected with a gated-intensified CCD mounted on a 0.18 M spectrograph (Princeton PiMax/Acton Pro 180). The optical density of the solutions was adjusted to ∼0.7 at the excitation wavelength. Samples were measured in a cell that holds a total volume of 10 mL and the content was continuously recirculated through the pump–probe region of the cell. Samples were prepared in anhydrous THF and degassed by bubbling argon for 45 min before the acquisition. The transient absorption (TA) spectrum was collected from 350 nm to 850 nm with a 60 ns initial camera delay and with different subsequent delay time increments depending on the triplet lifetime of the molecule. Fifty averages were obtained at each delay time.
Ultrafast pump–probe experiments were performed with femtosecond (fs) transient absorption spectroscopy with broadband capabilities. Detailed information of the experimental setup can be found elsewhere.59 Briefly, an Ultrafast Systems Helios femtosecond transient absorption spectrometer equipped with UV-visible and near-infrared detectors was used to measure the samples in this study. The white light continuum probe pulse was generated in a thick sapphire plate (800–1300 nm) and in a CaF2 crystal (350–700 nm spectral range) using a few μJ pulse energy of the fundamental output of a Ti:sapphire fs regenerative amplifier operating at 800 nm with 35 fs pulses and a repetition rate of 1 kHz. The pump pulses at 355 nm were created from fs pulses generated in an optical parametric amplifier (Newport Spectra-Physics). The sample solution was constantly stirred to avoid photodegradation in scanned volume. The pump and probe beams were overlapped both spatially and temporally on the sample solution, and the transmitted probe light from the samples was collected on the broad-band UV-visible-near-IR detectors to record the time-resolved excitation-induced difference spectra.
The cyclic voltammetry (CV) and differential pulse voltammetry (DPV) experiments were performed on a Bioanalytical Systems CV50W electrochemical analyzer at a sweep rate of 100 mV s−1 and 20 mV s−1 respectively, by using a platinum button as a working electrode, a platinum wire as a counter electrode, and a silver wire as a pseudo-reference electrode. Solutions of samples were prepared in dichloromethane or dimethylformamide with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as a supporting electrolyte. The electrochemical potentials were internally calibrated against the standard ferrocene/ferrocenium redox couple (Fc/Fc+). The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) levels for each complex were reported with respect to the potential of a Fc/Fc+ redox couple (−5.1 eV vs. vacuum).22
Computational details
All calculations were carried out using DFT as implemented in Gaussian 09 Rev. C.01.60 Geometries were optimized using the B3LYP functional along with the 6-31G(d) basis set for C, H, O, N, the 6-31+G(d) basis set for P, S, and the SDD basis set for Pt. To minimize computational cost, the PBu3 ligands were replaced with PMe3. These molecules are designated by the addition of a prime (′) to their name, e.g., the acronym for TBTz with PMe3 ligands is TBTz′. All singlet optimizations were started from idealized geometries and computed without symmetry constraints. Two conformations were run for each complex, as the DAD cores are capable of multiple geometries. These starting conformations differ only in the conformational orientation of the acceptor unit with respect to the thiophene donors. Triplet optimizations were run on both conformations and were initialized from the optimized singlet geometries using the unrestricted formalism. All optimized structures were characterized by vibrational frequency calculations and were shown to be minima by the absence of imaginary frequencies. Structures were visualized using Chemcraft Version 1.7.Acknowledgements
This work was supported by the National Science Foundation (Grant No. CHE-115164 and CHE-1504727). The authors acknowledge the University of Florida Research Computing (http://researchcomputing.ufl.edu) for providing computational resources and support that have contributed to the research results reported in this publication.Notes and references
- S. Hellstrom, F. Zhang, O. Inganas and M. R. Andersson, Dalton Trans., 2009, 45, 10032–10039 RSC.
- P. Beaujuge, S. Ellinger and J. Reynolds, Nat. Mater., 2008, 7, 795–799 CrossRef CAS PubMed.
- A. P. Kulkarni, X. Kong and S. A. Jenekhe, Adv. Funct. Mater., 2006, 16, 1057–1066 CrossRef CAS.
- Z. Zhu, D. Waller, R. Gaudiana, M. Morana, D. Mühlbacher, M. Scharber and C. Brabec, Macromolecules, 2007, 40, 1981–1986 CrossRef CAS.
- S. Yao, H.-Y. Ahn, X. Wang, J. Fu, E. W. van Stryland, D. J. Hagan and K. D. Belfield, J. Org. Chem., 2010, 75, 3965–3974 CrossRef CAS PubMed.
- K. Colladet, S. Fourier, T. J. Cleij, L. Lutsen, J. Gelan, D. Vanderzande, L. Huong Nguyen, H. Neugebauer, S. Sariciftci and A. Aguirre, Macromolecules, 2007, 40, 65–72 CrossRef CAS.
- T. T. Steckler, X. Zhang, J. Hwang, R. Honeyager, S. Ohira, X.-H. Zhang, A. Grant, S. Ellinger, S. A. Odom and D. Sweat, J. Am. Chem. Soc., 2009, 131, 2824–2826 CrossRef CAS PubMed.
- M. Wang, X. Hu, P. Liu, W. Li, X. Gong, F. Huang and Y. Cao, J. Am. Chem. Soc., 2011, 133, 9638–9641 CrossRef CAS PubMed.
- E. Busby, J. Xia, Q. Wu, J. Z. Low, R. Song, J. R. Miller, X. Zhu, L. M. Campos and M. Y. Sfeir, Nat. Mater., 2015, 14, 426–433 CrossRef CAS PubMed.
- M. Albota, D. Beljonne, J.-L. Brédas, J. E. Ehrlich, J.-Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin and S. R. Marder, Science, 1998, 281, 1653–1656 CrossRef CAS PubMed.
- M. Kirkus, L. Wang, S. Mothy, D. Beljonne, J. Cornil, R. A. Janssen and S. C. Meskers, J. Phys. Chem. A, 2012, 116, 7927–7936 CrossRef CAS PubMed.
- D. Veldman, S. C. Meskers and R. A. Janssen, Adv. Funct. Mater., 2009, 19, 1939–1948 CrossRef CAS.
- A. Parthasarathy, S. Goswami, T. S. Corbitt, E. Ji, D. Dascier, D. G. Whitten and K. S. Schanze, ACS Appl. Mater. Interfaces, 2013, 5, 4516–4520 CAS.
- W. Siebrand, J. Chem. Phys., 1967, 46, 440–447 CrossRef CAS.
- N. Turro, Modern Molecular Photochemistry, Menlo Park, CA, 1978 Search PubMed.
- J. V. Caspar, B. P. Sullivan, E. M. Kober and T. J. Meyer, Chem. Phys. Lett., 1982, 91, 91–95 CrossRef CAS.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- C. E. Whittle, J. A. Weinstein, M. W. George and K. S. Schanze, Inorg. Chem., 2001, 40, 4053–4062 CrossRef CAS PubMed.
- J. S. Wilson, N. Chawdhury, M. R. A. Al-Mandhary, M. Younus, M. S. Khan, P. R. Raithby, A. Köhler and R. H. Friend, J. Am. Chem. Soc., 2001, 123, 9412–9417 CrossRef CAS PubMed.
- F. N. Castellano, I. E. Pomestchenko, E. Shikhova, F. Hua, M. L. Muro and N. Rajapakse, Coord. Chem. Rev., 2006, 250, 1819–1828 CrossRef CAS.
- Y. Liu, S. Jiang, K. Glusac, D. H. Powell, D. F. Anderson and K. S. Schanze, J. Am. Chem. Soc., 2002, 124, 12412–12413 CrossRef CAS PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- A. P. Kulkarni, P.-T. Wu, T. W. Kwon and S. A. Jenekhe, J. Phys. Chem. B, 2005, 109, 19584–19594 CrossRef CAS PubMed.
- J. Roncali, P. Blanchard and P. Frère, J. Mater. Chem., 2005, 15, 1589–1610 RSC.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396–1407 CrossRef CAS PubMed.
- S. C. Price, A. C. Stuart, L. Q. Yang, H. X. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625–4631 CrossRef CAS PubMed.
- G. Heimel, M. Daghofer, J. Gierschner, E. J. List, A. C. Grimsdale, K. Müllen, D. Beljonne, J.-L. Brédas and E. Zojer, J. Chem. Phys., 2005, 122, 054501 CrossRef PubMed.
- We note that TPT displays anomalous behavior, with an unusually low fluorescence yield and lifetime. This behavior is not clearly understood, but it is believed to be due to an additional non-radiative decay channel that operates in this compound.
- C. A. Parker and W. T. Rees, Analyst, 1960, 85, 587–600 RSC.
- A. B. Ormond and H. S. Freeman, Dyes Pigm., 2013, 96, 440–448 CrossRef CAS.
- TPT does not sensitize singlet oxygen likely due to its triplet excited state (T1) being too low in energy (<0.98 eV).61.
- B. P. Karsten, R. K. M. Bouwer, J. C. Hummelen, R. M. Williams and R. A. J. Janssen, J. Phys. Chem. B, 2010, 114, 14149–14156 CrossRef CAS PubMed.
- I. V. Rubtsov, K. Susumu, G. I. Rubtsov and M. J. Therien, J. Am. Chem. Soc., 2003, 125, 2687–2696 CrossRef CAS PubMed.
- K. Susumu, T. V. Duncan and M. J. Therien, J. Am. Chem. Soc., 2005, 127, 5186–5195 CrossRef CAS PubMed.
- C. C. Byeon, M. M. McKerns, W. Sun, T. M. Nordlund, C. M. Lawson and G. M. Gray, Appl. Phys. Lett., 2004, 84, 5174–5176 CrossRef CAS.
- The exact relationship between the triplet and singlet oxygen yields is ϕΔ = (ϕISCϕen) where ϕISC is the intersystem crossing (triplet) yield and ϕen is the efficiency of energy transfer from the excited sensitizer and triplet oxygen. In the text we assume that ϕISC ∼ ϕΔ, which is based on the assumption that ϕen = 1. Given that the triplet lifetimes for most of the chromophores are >1 μs, the latter assumption is reasonable. However, it is important to keep in mind that in fact the ϕISC calculated represents a lower limit for the actual value.
- TPT cannot be calculated from kISC = ϕISC/τS as other complexes since its ϕISC is unknown. The kISC of TPT is estimated (∼5.4 × 106 s−1) from the linear relationship presented in Fig. 6A and B, which will be discussed later.
- T. M. Cooper, D. M. Krein, A. R. Burke, D. G. McLean, J. E. Rogers and J. E. Slagle, J. Phys. Chem. A, 2006, 110, 13370–13378 CrossRef CAS PubMed.
- Note that the conformation of the central acceptor unit, relative to the attached thiophenes, changes throughout the series.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- F. Guo, Y.-G. Kim, J. R. Reynolds and K. S. Schanze, Chem. Commun., 2006, 17, 1887–1889 RSC.
- Y. Shao and Y. Yang, Adv. Mater., 2005, 17, 2841–2844 CrossRef CAS.
- J. J. Cavaleri, K. Prater and R. M. Bowman, Chem. Phys. Lett., 1996, 259, 495–502 CrossRef CAS.
- M. Etinski and C. M. Marian, Phys. Chem. Chem. Phys., 2010, 12, 15665–15671 RSC.
- J. G. Langan, E. V. Sitzmann and K. B. Eisenthal, Chem. Phys. Lett., 1984, 110, 521–527 CrossRef CAS.
- C. M. Marian, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 187–203 CrossRef CAS.
- D. Peceli, H. H. Hu, D. A. Fishman, S. Webster, O. V. Przhonska, V. V. Kurdyukov, Y. L. Slominsky, A. I. Tolmachev, A. D. Kachkovski, A. O. Gerasov, A. E. Masunov, D. J. Hagan and E. W. Van Stryland, J. Phys. Chem. A, 2013, 117, 2333–2346 CrossRef CAS PubMed.
- J. Michl, J. Am. Chem. Soc., 1996, 118, 3568–3579 CrossRef CAS.
- R. McWeeny and B. T. Sutcliffe, Methods of molecular quantum mechanics, Academic pressLondon, 1969 Search PubMed.
- Note that the triplet energy values used in this correlation were obtained from the DFT calculations.
- D. G. Patel, F. Feng, Y.-y. Ohnishi, K. A. Abboud, S. Hirata, K. S. Schanze and J. R. Reynolds, J. Am. Chem. Soc., 2012, 134, 2599–2612 CrossRef CAS PubMed.
- Y. Yang, R. T. Farley, T. T. Steckler, S.-H. Eom, J. R. Reynolds, K. S. Schanze and J. Xue, Appl. Phys. Lett., 2008, 93, 163305 CrossRef.
- L. Ren, F. Liu, X. Shen, C. Zhang, Y. Yi and X. Zhu, J. Am. Chem. Soc., 2015, 137, 11294–11302 CrossRef CAS PubMed.
- D. Beljonne, Z. Shuai, G. Pourtois and J.-L. Brédas, J. Phys. Chem. A, 2001, 105, 3899–3907 CrossRef CAS.
- J. Pina, J. S. Seixas de Melo, H. D. Burrows, R. M. Batista, S. P. Costa and M. M. M. Raposo, J. Phys. Chem. A, 2007, 111, 8574–8578 CrossRef CAS PubMed.
- J. Mei, K. Ogawa, Y. G. Kim, N. C. Heston, D. J. Arenas, Z. Nasrollahi, T. D. McCarley, D. B. Tanner, J. R. Reynolds and K. S. Schanze, ACS Appl. Mater. Interfaces, 2009, 1, 150–161 CAS.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1993, 22, 113–262 CrossRef CAS.
- R. T. Farley, PhD. thesis, University of Florida, 2007.
- A. O. El-Ballouli, E. Alarousu, M. Bernardi, S. M. Aly, A. P. Lagrow, O. M. Bakr and O. F. Mohammed, J. Am. Chem. Soc., 2014, 136, 6952–6959 CrossRef CAS PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. Hratchian, A. Izmaylov, J. Bloino, G. Zheng, J. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. Peralta, F. Ogliaro, M. Bearpark, J. Heyd, E. Brothers, K. Kudin, V. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. Burant, S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. Millam, M. Klene, J. Knox, J. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. Stratmann, O. Yazyev, A. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. Martin, K. Morokuma, V. Zakrzewski, G. Voth, P. Salvador, J. Dannenberg, S. Dapprich, A. Daniels, O. Farkas, J. Foresman, J. Ortiz, J. Cioslowski and D. Fox, Gaussian 09 (Revision C.01), Gaussian, Inc., Wallingford, CT, 2011 Search PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including synthetic procedures, NMR spectra, additional photophysical data and spectra. See DOI: 10.1039/c5sc04578a |
| This journal is © The Royal Society of Chemistry 2016 |