
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling innate substrate control in site-selective palladium-catalyzed C–H heterocycle functionalization†
Hwanho
Choi
a,
Minsik
Min
bc,
Qian
Peng
*a,
Dahye
Kang
bc,
Robert S.
Paton
*a and
Sungwoo
Hong
*bc
aChemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK. E-mail: qian.peng@chem.ox.ac.uk; robert.paton@chem.ox.ac.uk
bCenter for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon, 34141 Korea. E-mail: hongorg@kaist.ac.kr
cDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 34141 Korea
First published on 8th March 2016
Abstract
Understanding the regioselectivity of C–H activation in the absence of directing groups is an important step towards the design of site-selective C–H functionalizations. The Pd(II)-catalyzed direct arylation of chromones and enaminones provides an intriguing example where a simple substitution leads to a divergence in substrate-controlled site-selectivity. We describe computational and experimental studies which reveal this results from a switch in mechanism and therefore the selectivity-determining step. We present computational results and experimentally measured kinetic isotope effects and labelling studies consistent with this proposal. The C–H activation of these substrates proceeds via a CMD mechanism, which favors more electron rich positions and therefore displays a pronounced kinetic selectivity for the C3-position. However, C2-selective carbopalladation is also a competitive pathway for chromones so that the overall regiochemical outcome depends on which substrate undergoes activation first. Our studies provide insight into the site-selectivity based on the favorability of two competing CMD and carbopalladation processes of the substrates undergoing coupling. This model can be utilized to predict the regioselectivity of coumarins which are proficient substrates for carbopalladation. Furthermore, our model is able to account for the opposite selectivities observed for enaminone and chromone, and explains how a less reactive coupling partner leads to a switch in selectivity.
Introduction
Palladium-catalyzed C–H bond activation is an efficient, atom-economical tool in the synthesis of functionalized organic molecules.1–4 However, the ability to activate a specific ‘inert’ C–H bond in the presence of several others dictates that control must be exerted over the position of activation. This challenge lies at the forefront of developments in synthetic methodology. One such strategy to achieve high site-selectivity is the use of directing groups covalently tethered to the substrate, which coordinate to the transition-metal center and direct C–H activation to a proximal site. Significant developments in terms of ortho,1,2meta3 and para-directing4 groups have been accomplished for aromatic C–H activation. However, an alternative approach – free from directing groups – is to exploit the inherent electronic and steric preferences of both catalyst and substrate to control the regiochemical outcome of C–H functionalizations according to innate reactivities. While controlling the cleavage of a specific C–H bond among several others remains a significant challenge, access to fundamental mechanistic insights into C–H bond activation has been possible from combined computational and experimental investigations.5,6 By adopting such an approach, we now explain contrasting regioselectivities of electron-rich heterocyclic C–H functionalizations, with the intention of providing a rational basis for the design of site-selective catalytic C–H activation.A breakthrough in the regioselective Pd-catalyzed C–H functionalization of indoles was made by Fagnou and coworkers.7 Davies and Macgregor first provided a theoretical basis for understanding C–H activation with Pd(OAc)2 in terms of ambiphilic metal ligand activation (AMLA),8 where the Pd-coordinated acetate acts as proton-abstracting base. Analysis of a wide range of aromatic and heteroaromatic substrates by Fagnou and Gorelsky led to the generalization of this mechanism, termed concerted metalation–deprotonation (CMD) mechanism.9 Since the pioneering work by Fagnou, significant progress has been made to synthesize a variety of bi(hetero)aryl scaffolds via oxidative C–H/C–H cross-coupling approach. Computational studies have been performed to gain considerable insight into Pd-catalyzed C–H functionalization in the presence of directing groups, for both C(sp3)–H and C(sp2)–H activation.10–12 However, mechanistic aspects of C–H bond activation involving non-aromatic substrates remain elusive and predictions of selectivity in the absence of directing groups are difficult. This is exemplified by enolone and enaminone heterocycles: these structures differ only in the identity of a single heteroatom, however, the site of functionalization is completely different under identical conditions, as depicted in Scheme 1.13 The mechanistic basis for this difference has thus far been unexplained.
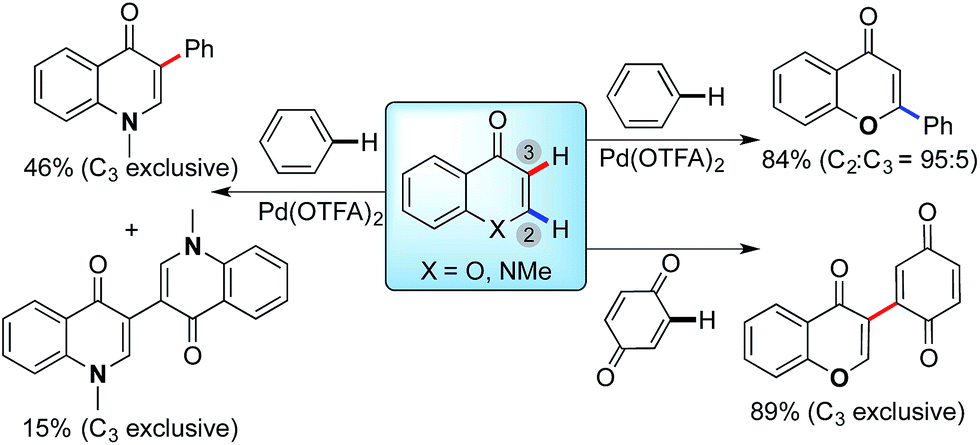 | ||
| Scheme 1 Divergent site-selectivity in the Pd-catalyzed direct arylation of enolone (X = O) and enaminone (X = NMe) substrates with arenes. Standard conditions: Pd(OTFA)2 (20 mol%) and AgOAc (3.0 eq.) in pivalic acid at 100 °C. | ||
We recently described the Pd(II)-catalyzed regioselective arylation of chromones and enaminones with simple arenes via a two fold C–H bond functionalization (Scheme 1).13a,b The pivalate and other bidentate anions have been proposed to act as a catalytic proton shuttle,7 lowering the activation energy of Pd-catalyzed C–H bond cleavage. In our studies, contrasting regioselectivities are observed in the reactions of chromone (X = O) and enaminone (X = NMe) substrates (Scheme 1). The C-2 arylation of chromones proceeded with high regioselectivity (C2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C3 = 95:5).13c Substrates conjugated with a variety of functional groups from electron-donating to electron-withdrawing groups were tested, and the C2-arylated products were obtained with high regioselectivity (∼20:1) in all cases. On the other hand, the C-3 arylation product was exclusively obtained with the enaminone substrate under the same reaction conditions. Furthermore, coupling of chromone with parabenzoquinone also results in a switch to complete selectivity for the C3-position.13d Consequently, we reasoned that the regioselectivity of this oxidative cross-coupling is dramatically affected by the intrinsic properties of the substrates (enaminone vs. enolone). Intrigued by the dramatic change in regioselectivity with subtle alterations (N vs. O) in the substrates, we further investigated to elucidate the origin of the high level of selectivity with density functional theory (DFT) and experimental isotopic labeling studies.
:C3 = 95:5).13c Substrates conjugated with a variety of functional groups from electron-donating to electron-withdrawing groups were tested, and the C2-arylated products were obtained with high regioselectivity (∼20:1) in all cases. On the other hand, the C-3 arylation product was exclusively obtained with the enaminone substrate under the same reaction conditions. Furthermore, coupling of chromone with parabenzoquinone also results in a switch to complete selectivity for the C3-position.13d Consequently, we reasoned that the regioselectivity of this oxidative cross-coupling is dramatically affected by the intrinsic properties of the substrates (enaminone vs. enolone). Intrigued by the dramatic change in regioselectivity with subtle alterations (N vs. O) in the substrates, we further investigated to elucidate the origin of the high level of selectivity with density functional theory (DFT) and experimental isotopic labeling studies.
Results and discussion
Isotopic labeling studies
The rate of C–H activation of chromone was investigated experimentally by following the incorporation of deuterium in D2O (Scheme 2). Partial deuteration (40%) takes place in 6 hours, while for enaminone the loss of deuterium is complete within 30 min at the same C3-position. There is no evidence of exchange at the C2-position of either substrate, even though arylation of chromone occurs at C2. This led us to consider the different possible mechanisms to account for these observations.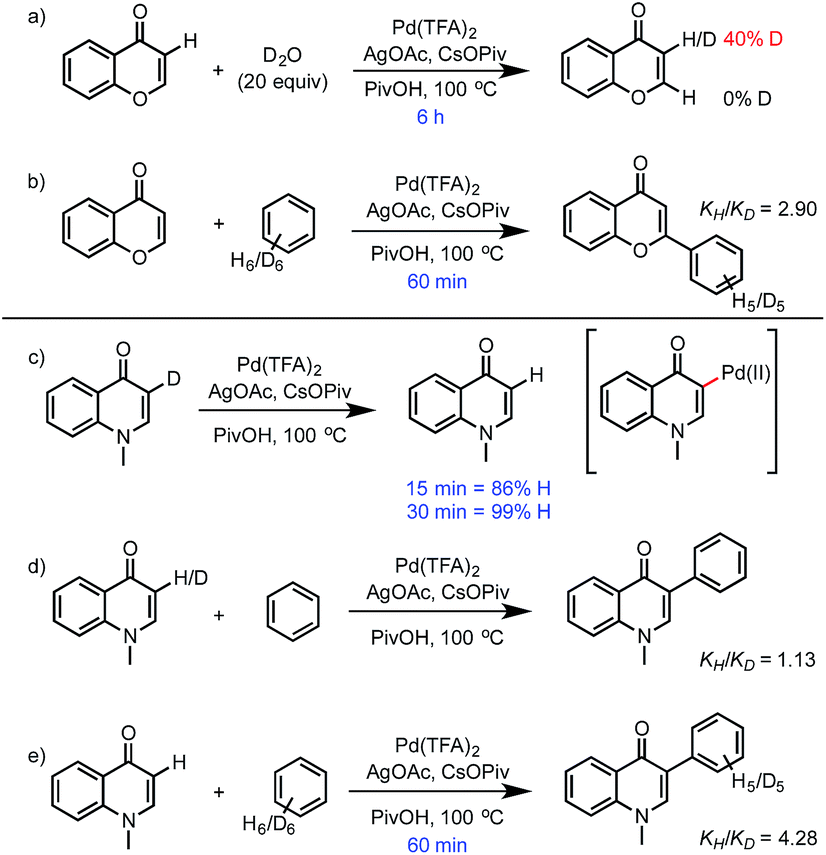 | ||
| Scheme 2 Isotopic labeling studies of chromone and enaminone. | ||
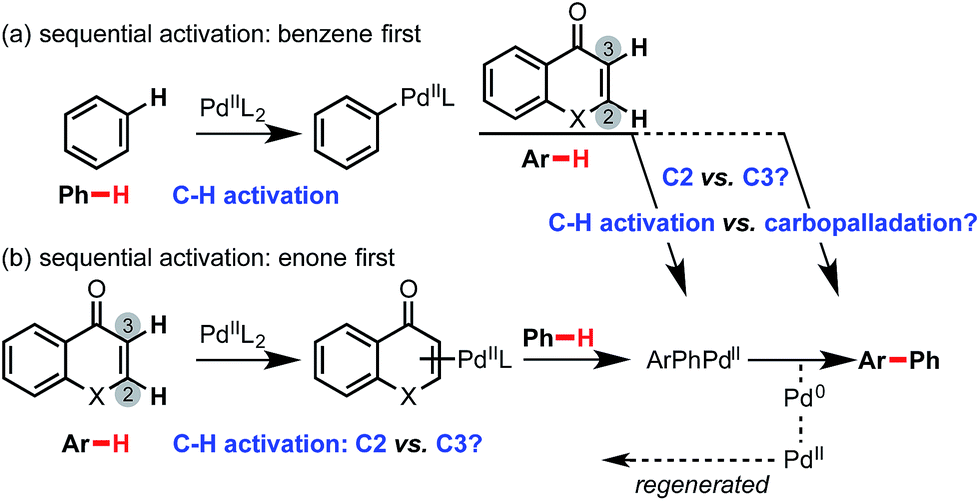 | ||
| Scheme 3 Mechanistic possibilities investigated for Pd-catalyzed oxidative cross-coupling with enolone (X = O) or enaminone (X = NMe) with benzene. | ||
Several potential mechanisms may be conceived for C(sp2)–H activation by a Pd(II)-center (Fig. 1a). We investigated chromone and enaminone substrates undergoing C–H activation by the PdII(OTFA)2 catalyst via electrophilic aromatic substitution (SEAr),12 a termolecular electrophilic substitution (SE3)14 and concerted metalation–deprotonation (CMD)15 pathways. In accord with previous computational results,11b we were unable to locate any stable structures corresponding to the formal Wheland intermediate (INT-A). Attempts to optimize transition structures (TS-B) corresponding to proton abstraction following electrophilic activation by a cationic PdOTFA+ species (SE3 mechanism) were similarly unsuccessful, resulting in the location of CMD transition structures. Mechanisms involving C–H oxidative insertion and σ-bond metathesis have been shown to give dramatically higher activation barriers than CMD and were thus not considered further.
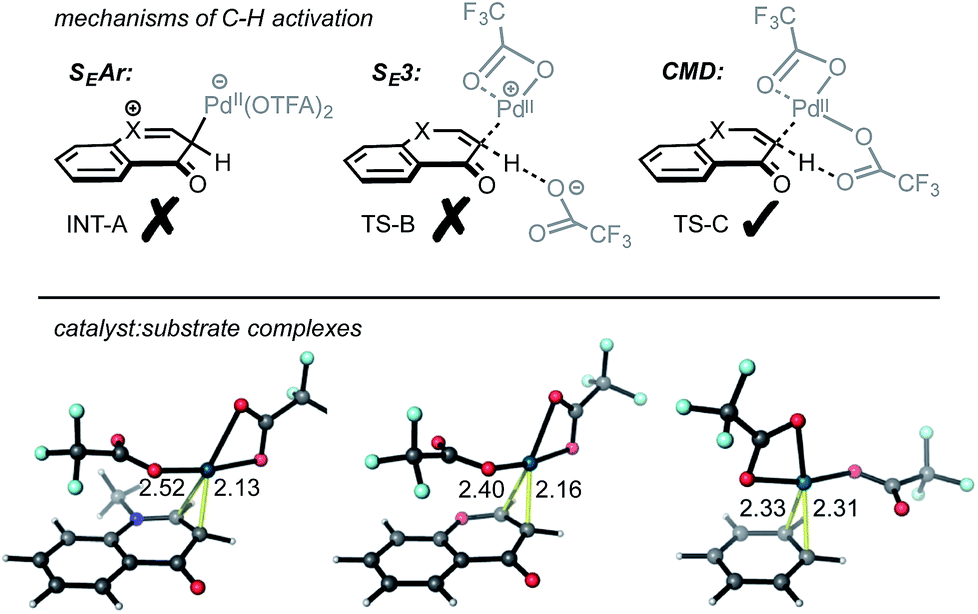 | ||
| Fig. 1 Top: C–H activation mechanisms considered: electrophilic aromatic substitution (SEAr), termolecular electrophilic substitution (SE3) and concerted metalation–deprotonation (CMD). Bottom: catalyst–substrate complexes with distances in Å. | ||
The interaction of Pd(OTFA)2 with the C2/C3 π-bond of both substrate is characterized by a single η2-bound conformation (Fig. 1b): the catalyst is positioned closer to the C3 position due to the greater π-electron density at this position which is more pronounced for enaminone than chromone. In contrast, coordination to benzene is nearly symmetrical. For the CMD pathway, TS-C (at both C2 and C3 positions), was characterized and provides a viable mechanism of C–H activation for all three arenes. The absence of stationary points corresponding to INT-A and TS-B cannot absolutely preclude their existence (absence of evidence is not evidence of absence!), nonetheless, CMD activation barriers and thermochemistry are consistent with experimentally observed hydrogen–deuterium exchange at the C3 position and not at the C2 position (Scheme 2).
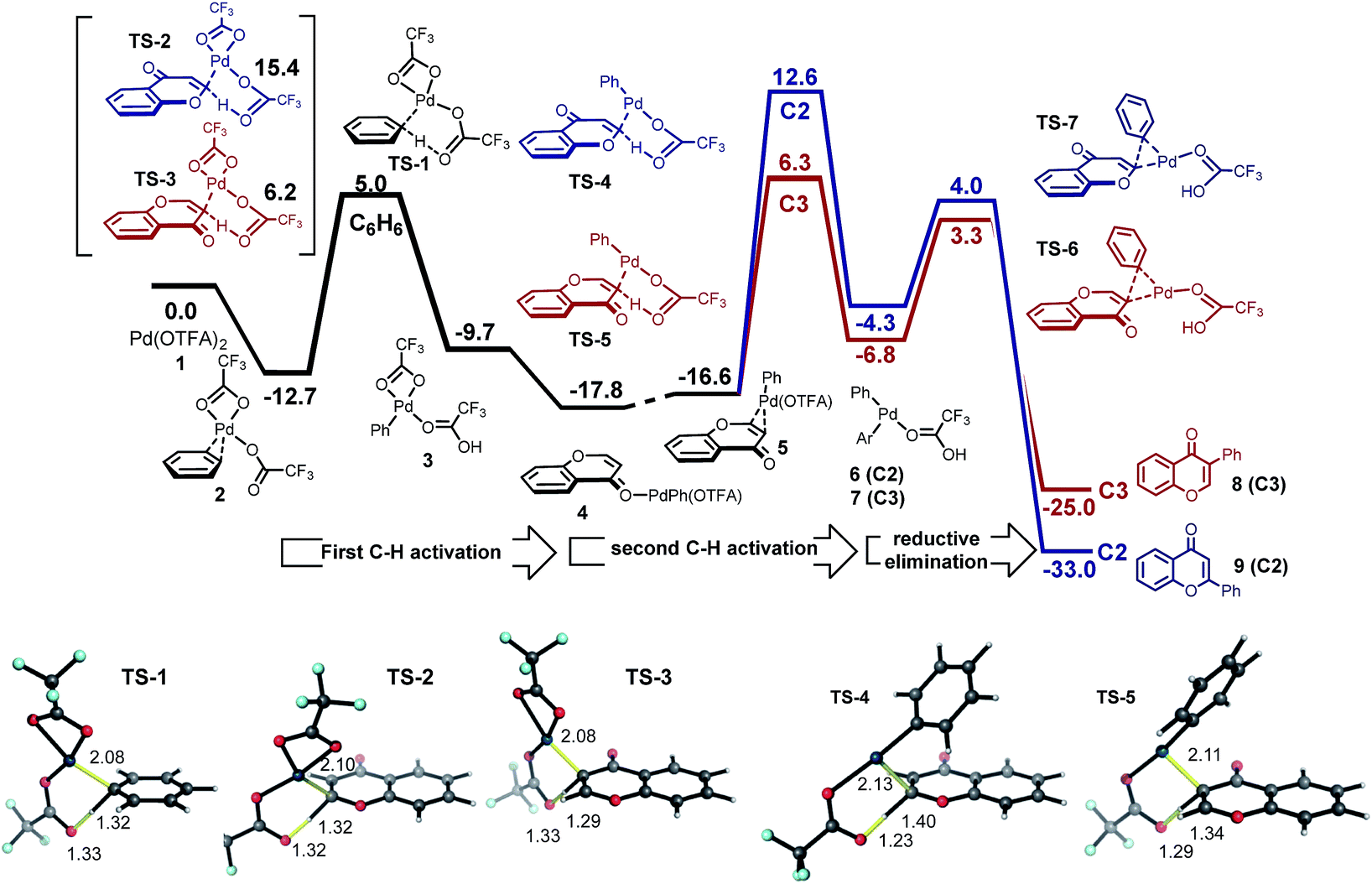 | ||
| Fig. 2 Coupling of chromone and benzene via sequential C–H activation. Palladation of benzene (40-fold excess) with Pd(OTFA)2 is marginally faster than for the C3-position of chromone; palladation of chromone by PhPd(OTFA) and subsequent reductive elimination: the C3-regioselectivity afforded by this mechanism is inconsistent with experiment. Structures are colored as follows: Pd (dark blue), F (light blue), O (red), C (grey), H (white). | ||
In fact, the C–H activation of benzene via CMD TS-1, is computed to occur more quickly than for either position of chromone – the free energy barrier is 1.2 kcal mol−1 lower than for the C3-activation. This activation of benzene benefits from the statistical effect of a 40-fold excess of benzene use experimentally, and also from the presence of six equivalent C–H bonds. In addition to a lower barrier, the thermochemistry of benzene activation is also more favorable, forming arylpalladium species (intermediate 3) which is more stable than either position of chromone. Given the preferential reactivity of benzene towards C–H activation and the greater stability computed for the arylpalladium intermediate formed, we investigated the subsequent reaction of the phenylpalladium intermediate with chromone. Firstly, we considered the potential for a second CMD processes in which the phenylpalladium species activates the C2 and C3 positions of chromone. Followed by (irreversible) reductive elimination, this process leads to the coupled heterobiaryl product (Fig. 2 second C–H activation and reductive elimination). As with the CMD TSs involving Pd(OTFA)2, a sizable difference in reactivity is computed in favor of CMD activation at the C3 position (viaTS-4 and TS-5). This preference is a result of the greater π-electron density at the C3-position. This C–H activation step has a higher computed barrier than the initial activation of benzene and lies above the subsequent reductive elimination step. This mechanism cannot account for the high level of C2-arylation selectivity, since this isomer is kinetically disfavored by a much high barrier for the second CMD step. Nor can it account for the observed primary KIE when benzene-d6 is used (Scheme 2b), and so this mechanism was rejected.
We investigated an alternative to sequential CMD steps: a migratory insertion/carbopalladation of the phenylpalladium intermediate across the isolated π-bond of chromone (Fig. 3). Arylpalladium species are known experimentally to add across the π-bonds of enones, which have been studied computationally.17 Prior to locating the TS structures for this process, we carried out an exhaustive 2D-scan of the potential energy surface (see ESI†) in which the distances between the phenyl ipso-carbon and C2 and C3 positions of chromone defined the search coordinates. This established the existence of two carbopalladation TSs and also confirmed that there is no feasible means of interconversion between the two adducts. Regioisomeric structures TS-8 and TS-9 were found to lie much lower in free energy than those structures previously obtained for the CMD-reductive elimination pathway (Fig. 3, migratory insertion). While caution should be exercised in comparing different mechanisms, the difference of more than 7 kcal mol−1 in favor of carbopalladation lends rather strong support for this mechanism.
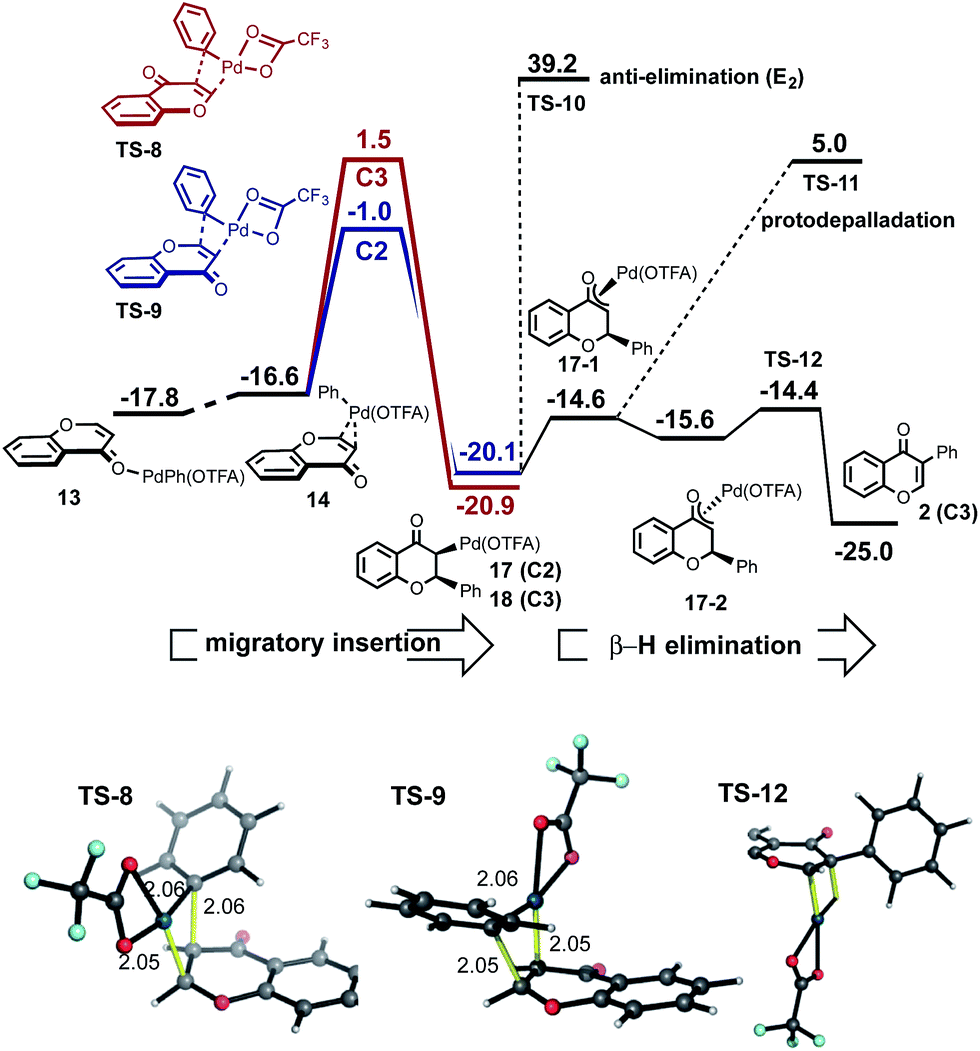 | ||
| Fig. 3 Migratory insertion/carbopalladation of PhPd(OTFA) across the π-bond of chromone is favored relative to CMD; the correct C2-regioselectivity is computed from this step, which is irreversible. | ||
Additionally, the relative stability of carbopalladation TSs is consistent with the observed level of C2-selectivity for chromone arylation: the experimental value of 95:5 compares very favorably with our computed selectivity of 97:3 based on a ΔΔG‡ of 2.5 kcal mol−1 between TS-8 and TS-9. Our calculations support the view that the subsequent steps following carbopalladation are relatively facile, making carbopalladation itself irreversible and therefore the regiodetermining step in chromone arylation (Fig. 3, β-H elimination).
The greater π-electron density at the C3-position of chromone favors the delivery of the electrophilic Pd(OTFA) center to this position in TS-9, which leads to the C2-delivery of the phenyl group in the carbopalladation step. Our prediction of a preference for chromone towards carbopalladation rather than concerted metalation–deprotonation can be understood in terms of the inherent reactivity of the α,β-unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, which although conjugated is not part of an aromatic ring system. In contrast, (hetero)aromatic systems would be expected to resist the loss of aromaticity which necessarily results from carbopalladation and will favor the CMD pathway where this is less severe. The KIE value, kH/kD = 2.90 observed for benzene-h6/d6 suggests turnover-limiting C–H cleavage of benzene (Scheme 2b), consistent with our calculations in which the initial C–H activation of benzene has a higher barrier than the subsequent carbopalladation step.
C bond, which although conjugated is not part of an aromatic ring system. In contrast, (hetero)aromatic systems would be expected to resist the loss of aromaticity which necessarily results from carbopalladation and will favor the CMD pathway where this is less severe. The KIE value, kH/kD = 2.90 observed for benzene-h6/d6 suggests turnover-limiting C–H cleavage of benzene (Scheme 2b), consistent with our calculations in which the initial C–H activation of benzene has a higher barrier than the subsequent carbopalladation step.
Conversion of the intermediate following carbopalladation into the arylated product could occur via an (E2) anti-elimination TS-10 or alternatively, epimerization via a Pd-enolate and subsequent syn β-hydride elimination TS-12 (Fig. 3). Alternatively, protonolysis of the intermediate would lead to the product of conjugate addition of the phenyl group to the enone (TS-11). Although this is not observed in our experiments, different solvents have been shown to lead to a switch between arylation and conjugate addition products.18 The computed barrier to anti-elimination is highly uncertain, since the identity of the external base is unknown, however with an anionic trifluoroacetate the barrier to this step is prohibitively high. In contrast, syn β-hydride elimination is facile, provided the intermediate is able to interconvert to the epimeric form (from π-coordinated 10-1 to 10-2via free rotation in the O-bound enolate) in which the metal is now on the same side of the ring as the β-hydrogen.
The regioselectivity of chromone does not arise from the site-selectivity of C–H activation, since in this respect the C3-position is kinetically favored. Rather, with benzene undergoing C–H activation first, carbopalladation of chromone becomes the selectivity-determining step. The computed regioselectivity of migratory insertion by 2.5 kcal mol−1 accounts quantitatively for the experimentally observed preference for C2-arylation. Sequential C–H activation at a single Pd(II)-center (in either order), followed by reductive elimination is uncompetitive, with activation barriers over 7 kcal mol−1 higher than the favored carbopalladation route and predicts the incorrect regioselectivity. Based on this new understanding, we realized that C3-selective arylation of chromone should result whenever C–H activation becomes the selectivity-determining step. For example, by coupling with a partner which is much less prone to undergo C–H activation, but is itself a proficient substrate for carbopalladation, such as p-benzoquinone (Fig. 4) this should be possible. Indeed, our calculations confirm that p-benzoquinone is much less reactive towards C–H activation (CMD TS-21 lies significantly higher in free energy than reaction at the C3 position of chromone, by 8.7 kcal mol−1). Therefore, for this coupling the C–H activation of chromone occurs first. Conversely (unlike benzene), p-benzoquinone is a good substrate towards carbopalladation, and so the intrinsic reactivity towards C–H activation at the C3-position of chromone ensures selectivity for this regioisomer. The very high levels of selectivity (ΔΔG‡ of 9.2 kcal mol−1) predicted by this model supports our previous experimental observations of the coupling(s) of chromone with p-benzoquinone13d and alkenes,19 which occur exclusively at the C3-position.
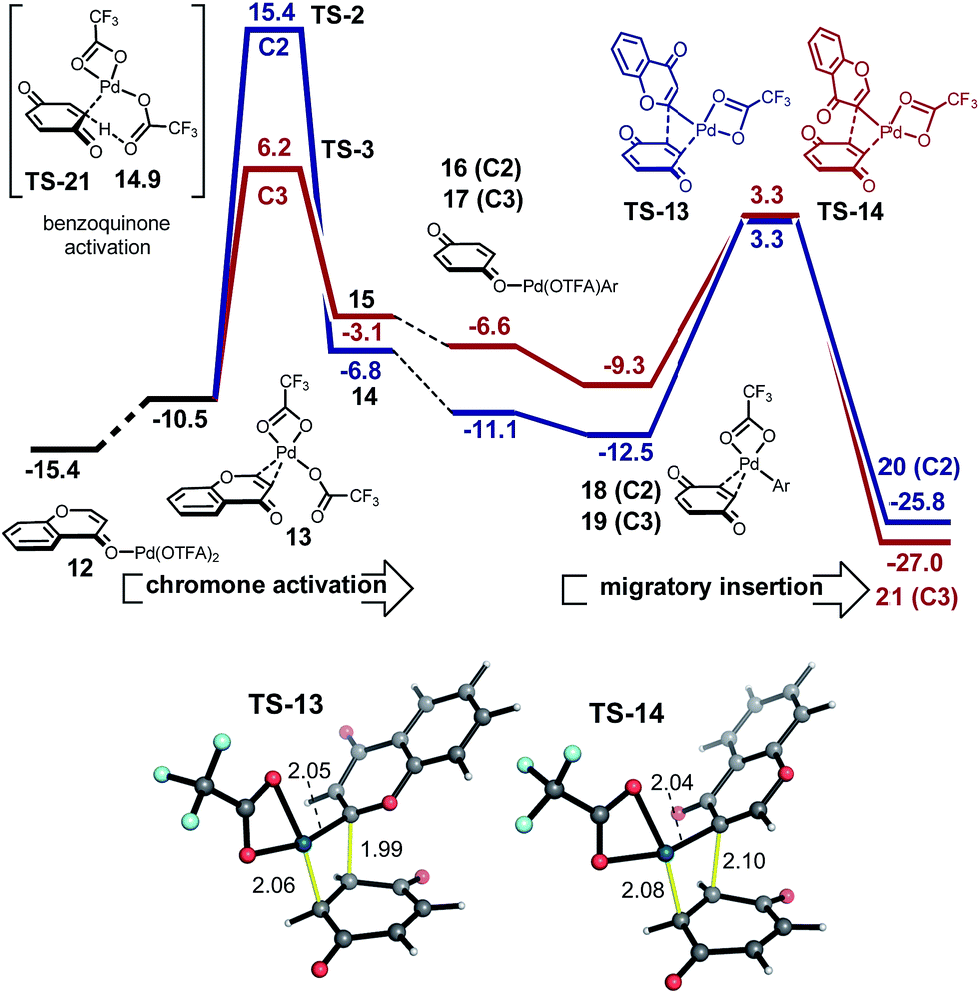 | ||
| Fig. 4 Coupling of chromone and p-benzoquinone via a C–H activation–carbopalladation sequence; C3-regiochemistry is predicted and observed experimentally. | ||
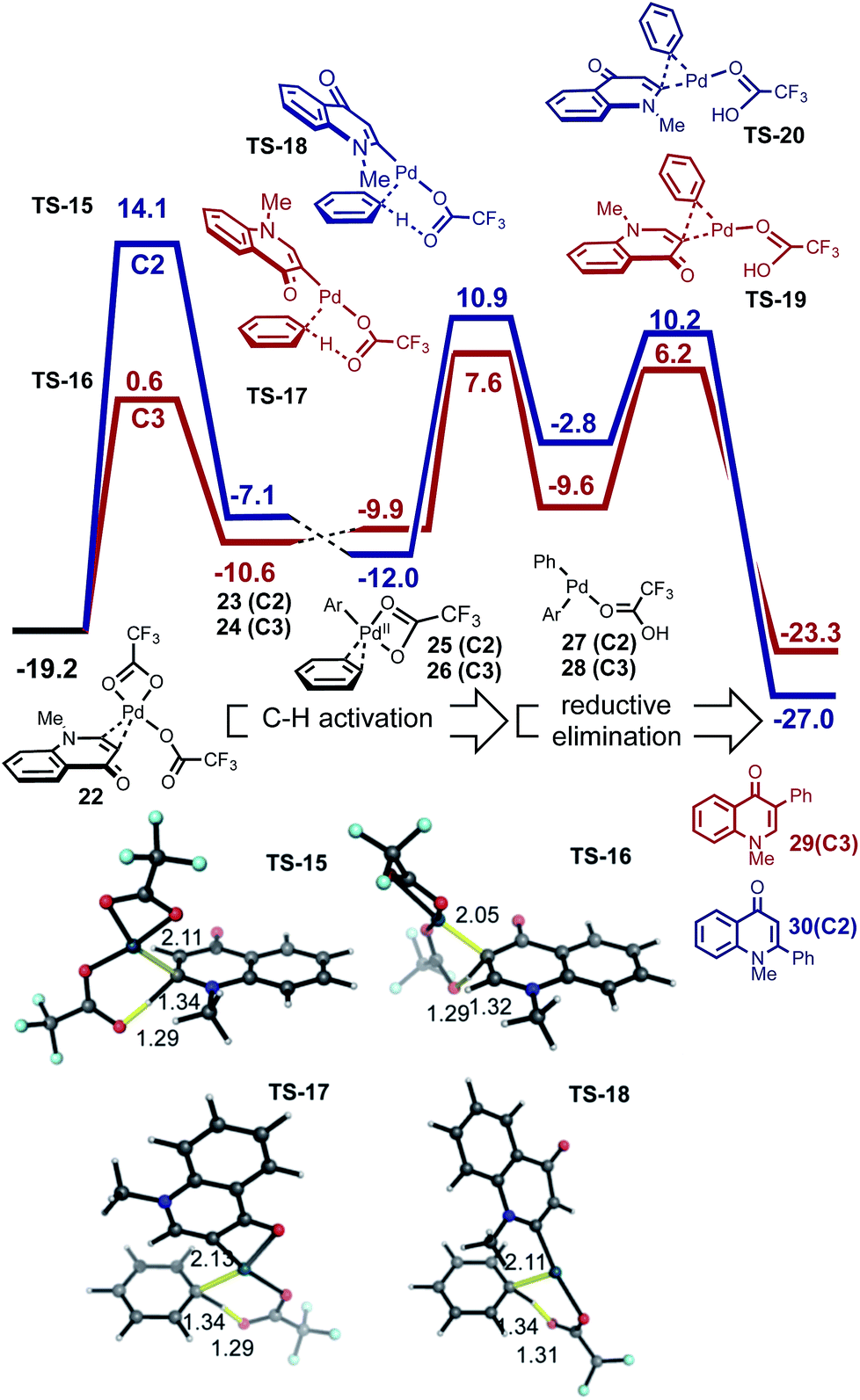 | ||
| Fig. 5 Sequential C–H activation of enaminone and benzene followed by reductive elimination. The C3-product is kinetically favored. | ||
The ease and reversibility of C–H activation of enaminone at the C3-position suggested by the computed reaction coordinate in Fig. 5 was confirmed experimentally (Scheme 2c). Exposure of C3-deuterated enaminone substrate to the catalytic reaction conditions in the absence of benzene, led to 99% conversion to the C3-protio form within 30 minutes. Reaction times required for benzene heterocoupling take much longer (on the order of 8 hours), which suggests that enaminone C–H activation cannot be turnover-limiting in this process. Attempts to obtain primary kinetic isotope effect (KIE) measurements for C3-deutero-enaminone are complicated by scrambling of the D atoms in the substrate: based on conversion after 60 min a kH/kD value of 1.13 was obtained. In contrast, a primary KIE value, kH/kD = 4.28, was observed based on parallel experiments of benzene and benzene-d6 with enaminone (Scheme 2e). This suggests turnover-limiting C–H cleavage of benzene,20 although large intrinsic isotope effects have also been observed in arene oxidative coupling where transmetalation of two bimetallic intermediates is the limiting step.21,22
Based on our computational and experimental evidence for facile C3–H activation of enaminone and the comparatively slow C–H activation of benzene, we reason that palladation of enaminone will occur first. C–H activation at the C2 position of enaminone is unlikely to occur due to a significantly higher free energy barrier (33.3 kcal mol−1). Additionally, homocoupling only shows C–C formation at the C3-position of enaminone.23 Computed mechanism of CMD activation of benzene and subsequent reductive elimination is shown in Fig. 5. The ΔΔG‡ between the regioisomeric pathways amounts to 6.5 kcal mol−1 (defined by the difference between turnover-limiting TS-15 for C2 and TS-17 for C3 positions) in favor of the C3-product. The passage viaTS-16 in step 1, followed by TS-17 accounts for the C3-selectivity since both CMD TSs along the C2-pathway are less stable. The observed primary KIE with benzene-d6 is consistent with our computed free energy profile since the turnover-limiting TS is the second CMD activation step in which benzene undergoes C–H activation in TS-17. We also computed the analogous mechanism for the homocoupling of enaminone at the C3-position, which is formed in 15% yield. The second CMD-step of homocoupling has a TS of similar stability to TS-17, which is 1.3 kcal mol−1 lower in free energy. Thus this process is competitive with benzene heterocoupling as found experimentally, although since this mechanism is bimolecular in enaminone it becomes less competitive during the reaction, as substrate is consumed and benzene remains in a large excess. These results show that the turnover limiting step in the arylation of both enolone and enaminone substrates is the C–H activation of benzene, although the catalytic species involved is prediction to be different (Pd(OTFA)2vs. PdAr(OTFA)2). The computed free energy span24 is 26.8 kcal mol−1 for the C3-arylation of enaminone and 20.4 kcal mol−1 for the C2-arylation of chromone. This is consistent with the lower reactivity observed for enaminone (as judged by lower conversion obtained experimentally for the latter).
C bond localization in the substrates studied here enables a competitive carbopalladation pathway to occur. The inherent polarization of the CC bond in chromone and enaminone, and the sites of HOMO/LUMO coefficients act to favor C2-arylation (Scheme 4c) since the metal is delivered to the more electron-rich position in concert with C–C formation.
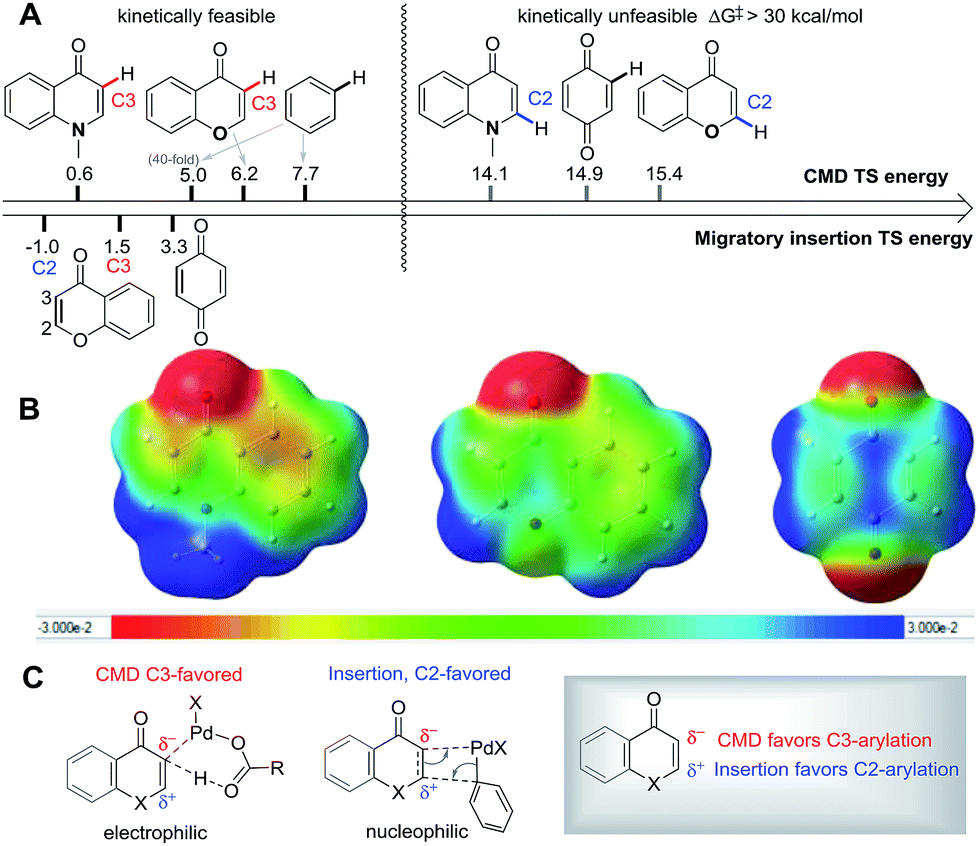 | ||
| Scheme 4 (A) Energy of key transition states for site-selectivity; (B) electrostatic potential map (ESP) with iso-value = 0.001 C: favorable site-selectivity in the reaction. | ||
The scope of our newly developed mechanistic model is applicable to other substrates and Pd-catalyzed functionalizations, so that we can predict regioselectivity in the arylation and alkenylation for chromone and other heteroamatic substrates, such as coumarins (Scheme 5). Based on the inherent polarization of chromone and coumarin substrates we predict that C–H activation of both substrates will occur preferentially at the enone α-position, and that carbopalladation will deliver the aryl group preferentially to the β-position. Alkenylation is thus expected to result in α-selectivity since heterocyclic CMD-activation dictates selectivity: indeed, experimentally this is the sole regioisomer for each substrate.19a,b Conversely, arylation is expected to result in β-selectivity as carbopalladation by the arylpalladium intermediate dictates selectivity, which is seen experimentally for both substrate.13a,b,19c Our model is consistent with experimental isotopic labeling studies, which show the C3 (α)-position of coumarin to be most susceptible towards palladation. A significant level of deuterium incorporation (41% D after 12 h) is observed at this position in the presence of D2O (20 equiv.).19b The calculated MOs and ESP map of coumarin (see ESI†) confirm that the polarization is in a similar sense as the other enolones considered, and can thus be used to make tangible predictions of regioselectivity for real catalytic reactions.
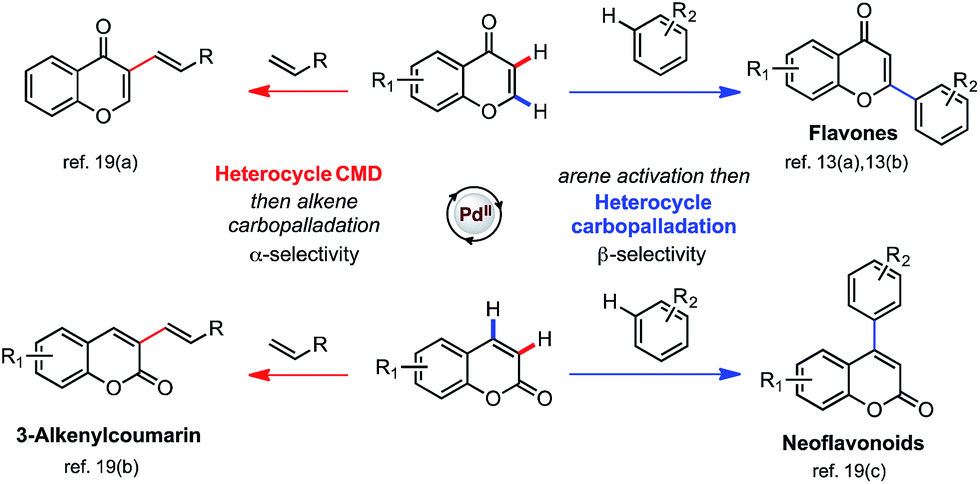 | ||
| Scheme 5 Experimental regioselectivities of the arylation and alkenylation of chromone and coumarin substrates are in accord with the computational model. | ||
Our work illustrates that the site-selectivity of Pd(II)-catalyzed C–H functionalizations will not always correspond to the position which undergoes metalation most quickly. This may seem relatively trivial, although it does establish that universal predictions of site-selectivity that focus on substrate and catalyst alone are impossible. For heterocyclic systems such as we have considered here, where there is a sizable difference in the π-Lewis acidities at different positions, large and opposite selectivities will be attainable depending on whether C–H activation by a CMD mechanism or carbopalladation occurs.
In each case the metal favors delivery to the more electron-rich position. In fact, we observe a strong linear correlation between the activation barriers of the CMD transition states with Pd(OTFA)2 and two geometric parameters in those structures: the C–Pd distance (R2 = 0.92) and the distortion of the C–H bond out of the ring-plane (R2 = 0.99) as shown in Fig. 6. The linear correlation of other structural parameters with reactivity are less quantitatively useful (see ESI†) but provide further support for the electrophilic nature of the C–H activation step. For example, a longer C2C3 bond is present in the more stable CMD TS structures (R2 = 0.83) due to the resonance contribution from heteroatom lone-pair donation. TS structures which exhibit more agostic character,25 as judged by a shorter Pd–H distance, are disfavored (R2 = 0.78). While metalation and deprotonation remain concerted in these TS structures, those substrates and positions of activation that are more electron rich benefit from more advanced C–Pd formation in the TS and an ensuing increase in C–H acidity due to the distortion out-of-plane of the ipso C–H bond.
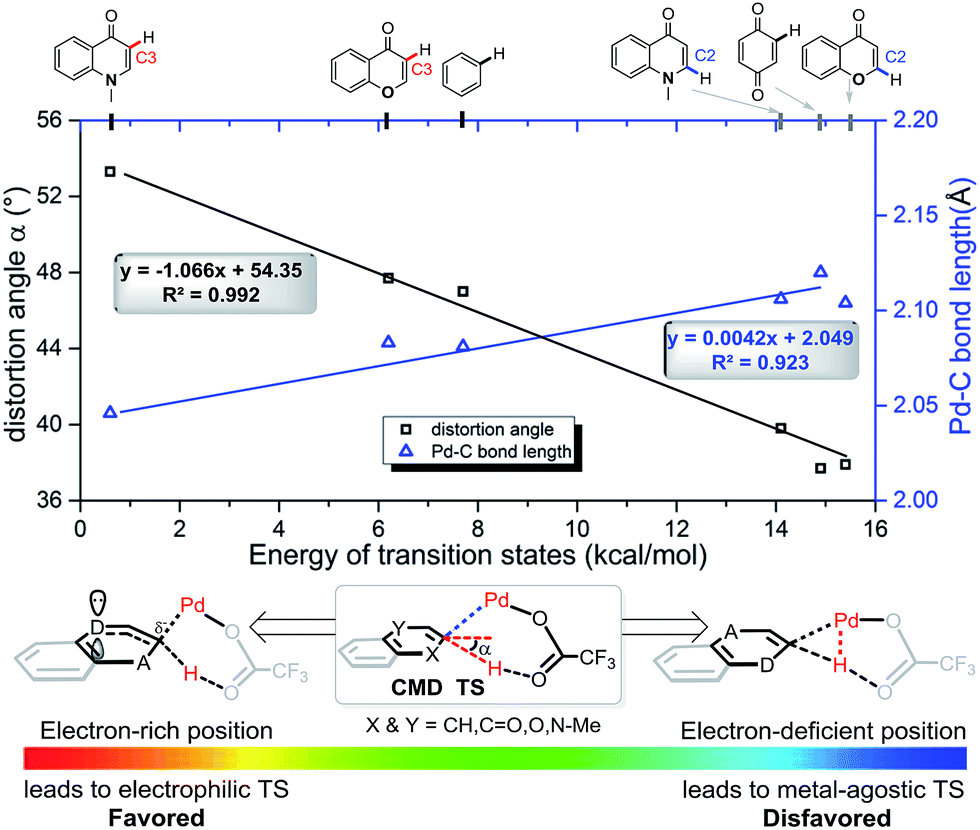 | ||
| Fig. 6 For all CMD TSs the Pd–C distance and out-of-plane deformation of the C–H bond correlate strongly with the TS energy. | ||
Conclusions
In summary, we have performed a detailed mechanistic study incorporating computational and experimental approaches to account for the divergent site-selectivity in Pd(II)-catalyzed direct arylations of enolones and enaminones. The computed mechanism for the two substrates is consistent with observed regioselectivities and the measured kinetic isotope effects which show benzene activation to be turnover-limiting. C–H activation of these substrates by a CMD mechanism favors more π-electron rich positions and displays a pronounced selectivity for the C3-position of both chromone and enaminone. Exclusive C3-selectivity in enaminone arylation occurs since the C–H activation barrier at C2 is prohibitively high. The carbopalladation of chromone favors the delivery of the aryl group to the C2-position since the metal interacts more strongly at the neighboring C3-site. The divergence in selectivity results from a switch in mechanism between these two competing pathways (CMD vs. carbopalladation), and it is possible to reason which will occur based on the inherent reactivities of the substrates towards C–H activation. We have shown that the favorability of CMD and carbopalladation steps can be understood based on the innate electronics of the reacting species, which forms the basis for rational predictions of site-selectivity. The regioselectivity in C–H functionalization of coumarin substrates can be accurately rationalized by this model.Methods
Experimental details
To a capped sealed tube were added Pd(TFA)2 (20 mol%), AgOAc (3.0 equiv.) and CsOPiv (3.0 equiv.). Subsequently, 1-methylquinolin-4(1H)-one-3 (or deuterated at the C3-position) (0.1 mmol), benzene (or benzene-d6) (0.5 mL) and pivalic acid (0.5 mL) were added. The reactions were stirred at 100 °C for 60 min. The reaction mixture was cooled to room temperature, and then diluted with CH2Cl2 and NaHCO3. After stirring for 10 min, the mixture was washed sequentially with aqueous NaHCO3 and NH4Cl. The combined organic layers were dried over MgSO4. The filtrate was concentrated in vacuo and the yield was analyzed by 1H NMR.Computational methods
All stationary points were fully optimized at the density functional theory level in Gaussian 09 (rev. D.01),26 using the dispersion-corrected ω-B97XD functional27 without symmetry constraints. The effective core potentials (ECPs) of Hay and Wadt with a double-ζ basis set (LanL2DZ)28 were used for Pd, and the 6-31G(d) basis set was used for H, C, N, O and F(BS1). The energies were further evaluated using a larger basis set (6-311+G(d,p) basis set for H, C, N, O and F) and a triple-ζ basis set and associated ESP (LanL2TZ(f))29 for Pd(BS2) by single-point calculations. Single point calculations with the B3LYP functional30 with a D3-dispersion correction31 with zero-damping at short rangei with BS2 were used to corroborate these results – large differences between C2 and C3 C–H activation are also observed with this method and reaffirm the differences in selectivity presented in the manuscript. The effects of solvation were described with an implicit description of acetic acid using the CPCM treatment,32 where the United Atom Topological Model (UAHF) was used to define the solute cavity. All optimized species were verified as either minima or transition structures by the presence of zero or a single imaginary vibrational frequency. Gibbs free energies were evaluated at the reaction temperature using vibrational frequencies: the rigid rotor harmonic oscillator description was used above 100 cm−1 while the free rotor description is used below this value and a damping function switches between the two expressions about this point, as previously described by Grimme.33 Saddle points were connected to minima in the usual way with intrinsic reaction coordinate (IRC) calculations.34 Computed structures are displayed with PyMol.35The accurate computation of free energy changes for associative processes in solution remains a significant challenge, particularly since entropic terms associated with translation and rotation are hard to evaluate accurately in the condensed phase. A number of post hoc corrections have been employed in the literature, however, several have been criticized for their arbitrariness, and introducing unphysical assumptions.36 We have treated the translational entropy according to the model developed by Shakhnovich, where the accessible free space associated with the solvent of interest is used instead of that which would be accessible to an ideal gas – this model reduces the magnitude of the Strans relative to the gas phase, however, the dependence on temperature remains unchanged.37 This was performed for acetic acid as solvent, to model pivalic acid, using a molarity for AcOH of 17.4 mol L−1 and a solvent volume (computed with B3LYP/6-31G*) of 86.1 Å3 per molecule. This correction is justified in terms of the physical basis of this model, along with a correction to a 1 M standard state for all species except for benzene, which is present in a 40-fold excess, causing a corresponding increase in its chemical potential by RTln(40). Nevertheless, we focus our analysis of computational results in terms of relative, rather than absolute predictions of reactivity. For example, the assessing the relative feasibility of initial possibilities for C–H activation relies on the comparison of TS-1, TS-2, TS-3, TS-16 and TS-17 which are structurally similar and identical in terms of the number of reacting species.
Acknowledgements
This research was supported financially by the Royal Society (RG110617), the European Community (FP7-PEOPLE-2012-IIF under grant agreement 330364 to QP), and Institute for Basic Science (IBS-R010-G1). We acknowledge the use of the EPSRC UK National Service for Computational Chemistry Software (CHEM773) in carry-ing out this work.Notes and references
- For selected reviews on o-C–H activation, see: (a) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (b) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (e) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (f) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (g) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (h) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (i) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (j) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (k) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed; (l) M. S. Sigman and E. W. Werner, Acc. Chem. Res., 2012, 45, 874 CrossRef CAS PubMed; (m) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed; (n) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC; (o) J. J. Topczewski and M. S. Sanford, Chem. Sci., 2015, 6, 70 RSC.
- (a) G. Brasche, J. García-Fortanet and S. L. Buchwald, Org. Lett., 2008, 10, 2207 CrossRef CAS PubMed; (b) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350 CrossRef CAS PubMed; (c) A. Deb, S. Bag, R. Kancherla and D. Maiti, J. Am. Chem. Soc., 2014, 136, 13602 CrossRef CAS PubMed.
- For examples of Pd catalyzed m-C–H activation by chelation assistance, see: (a) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518 CrossRef CAS PubMed; (b) S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778 CrossRef CAS PubMed; (c) H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567 CrossRef CAS PubMed; (d) R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215 CrossRef CAS PubMed; (e) M. Bera, A. Modak, T. Patra, A. Maji and D. Maiti, Org. Lett., 2014, 16, 5760 CrossRef CAS PubMed; (f) Y. Deng and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 888 CrossRef CAS PubMed; (g) M. Bera, A. Maji, S. K. Sahoo and D. Maiti, Angew. Chem., Int. Ed., 2015, 54, 8515 CrossRef CAS PubMed; (h) S. Li, H. Ji, L. Cai and G. Li, Chem. Sci., 2015, 6, 5595 RSC.
- For examples of p-C–H activation by chelation assistance: S. Bag, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888 CrossRef CAS PubMed.
- For selected reviews on computational studies of C–H activation: (a) P. E. M. Siegbahn and M. R. A. Blomberg, Dalton Trans., 2009, 5832 RSC; (b) D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749 CrossRef CAS PubMed; (c) S. I. Gorelsky, Coord. Chem. Rev., 2013, 257, 153 CrossRef CAS; (d) Y. Boutadla, D. L. Davies, S. A. Macgregor and A. I. Poblador-Bahamonde, Dalton Trans., 2009, 5820 RSC.
- For reviews see: (a) G.-J. Cheng, X. Zhang, L. W. Chung, L. Xu and Y.-D. Wu, J. Am. Chem. Soc., 2015, 137, 1706 CrossRef CAS PubMed; (b) T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532 CrossRef CAS PubMed and references therein; specific examples: (c) I. A. Sanhueza, A. M. Wagner, M. S. Sanford and F. Schoenebeck, Chem. Sci., 2013, 4, 2767 RSC.
- With pivalate: (a) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496 CrossRef CAS PubMed; with phosphate: (b) G. Jindal and R. B. Sunoj, J. Am. Chem. Soc., 2014, 136, 15998 CrossRef CAS PubMed; (c) G. Jindal and R. B. Sunoj, Org. Lett., 2015, 17, 2874 CrossRef CAS PubMed.
- D. L. Davies, S. Donald and S. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754 CrossRef CAS PubMed.
- (a) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496 CrossRef CAS PubMed; (b) M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754 CrossRef CAS PubMed; (c) D. García-Cuadrado, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066 CrossRef PubMed; (d) D. Garcia-Cuadrado, P. de Mendoza, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2007, 129, 6880 CrossRef CAS PubMed; (e) S. I. Gorelsky, D. Laporte and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848 CrossRef CAS PubMed; (f) S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Org. Chem., 2012, 77, 658 CrossRef CAS PubMed; and for an overview of mechanistic work: (g) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef.
- (a) D. G. Musaev, A. Kaledin, B.-F. Shi and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 1690 CrossRef CAS PubMed; (b) R. Giri, Y. Lan, P. Liu, K. N. Houk and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 14118 CrossRef CAS PubMed; (c) M. Travis, T. M. Figg, M. Wasa, J.-Q. Yu and D. G. Musaev, J. Am. Chem. Soc., 2013, 135, 14206 CrossRef PubMed; (d) A. P. Smalley and M. J. Gaunt, J. Am. Chem. Soc., 2015, 137, 10632 CAS.
- (a) G.-J. Cheng, Y.-F. Yang, P. Liu, P. Chen, T.-Y. Sun, G. Li, X. Zhang, K. N. Houk, J.-Q. Yu and Y.-D. Wu, J. Am. Chem. Soc., 2014, 136, 894 CrossRef CAS PubMed; (b) Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344 CAS.
- (a) M. Catellani and G. P. Chiusoli, J. Organomet. Chem., 1992, 425, 151 CrossRef CAS; (b) C. C. Hughes and D. Trauner, Angew. Chem., Int. Ed., 2002, 41, 1569 CrossRef CAS; (c) E. J. Hennessy and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 12084 CrossRef CAS PubMed; (d) B. S. Lane, M. A. Brown and D. Sames, J. Am. Chem. Soc., 2005, 127, 8050 CrossRef CAS PubMed.
- (a) M. Min, H. Choe and S. Hong, Asian J. Org. Chem., 2012, 1, 47 CrossRef CAS; (b) Y. Moon, D. Kwon and S. Hong, Angew. Chem., 2012, 124, 11495 CrossRef; (c) Experimentally, in the coupling of chromone with benzene we observe that Pd(OPiv)2 gives 94:6 selectivity for C2-arylation ; (d) Y. Moon and S. Hong, Chem. Commun., 2012, 48, 7191 RSC.
- B. Glover, K. A. Harvey, B. Lui, M. J. Sharp and M. F. Tymoschenko, Org. Lett., 2003, 5, 301 CrossRef CAS PubMed.
- (a) V. I. Sokolov, L. L. Troitskaya and O. A. Reutov, J. Organomet. Chem., 1979, 182, 537 CrossRef CAS; (b) A. D. Ryabov, I. K. Sakodinskaya and A. K. Yatsimirsky, J. Chem. Soc., Dalton Trans., 1985, 2629 RSC; (c) M. Gomez, J. Granell and M. Martinez, Organometallics, 1997, 16, 2539 CrossRef CAS; (d) M. Gomez, J. Granell and M. Martinez, J. Chem. Soc., Dalton Trans., 1998, 37 RSC; (e) B. Biswas, M. Sugimoto and S. Sakaki, Organometallics, 2000, 19, 3895 CrossRef CAS.
- A. Petit, J. Flygare, A. T. Miller, G. Winkel and D. H. Ess, Org. Lett., 2012, 14, 3680 CrossRef CAS PubMed.
- (a) J. C. Holder, L. Zou, A. N. Marziale, P. Liu, Y. Lan, M. Gatti, K. Kikushima, K. N. Houk and B. M. Stoltz, J. Am. Chem. Soc., 2013, 135, 14996 CrossRef CAS PubMed; (b) S. E. Walker, J. A. Jordan-Hore, D. G. Johnson, S. A. Macgregor and A.-L. Lee, Angew. Chem., Int. Ed., 2014, 53, 13876 CrossRef CAS PubMed; (c) C. L. Boeser, J. C. Holder, B. L. H. Taylor, K. N. Houk, B. M. Stoltz and R. N. Zare, Chem. Sci., 2015, 6, 1917 RSC.
- S. E. Walker, J. Boehnke, P. E. Glen, S. Levey, L. Patrick, J. A. Jor-dan-Hore and A.-L. Lee, Org. Lett., 2013, 15, 1886 CrossRef CAS PubMed.
- (a) D. Kim and S. Hong, Org. Lett., 2011, 13, 4466 CrossRef CAS PubMed; (b) M. Min, Y. Kim and S. Hong, Chem. Commun., 2013, 49, 196 RSC; (c) M. Min and S. Hong, Chem. Commun., 2012, 48, 9613 RSC.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed.
- D. Wang, Y. Izawa and S. S. Stahl, J. Am. Chem. Soc., 2014, 136, 9914 CrossRef CAS PubMed.
- (a) T. A. Dwight, N. R. Rue, D. Charyk, R. Josselyn and B. DeBoef, Org. Lett., 2007, 9, 3137 CrossRef CAS PubMed; (b) N. A. B. Juwaini, J. K. P. Ng and J. Seayad, ACS Catal., 2012, 2, 1787 CrossRef CAS; (c) S. Potavathri, K. C. Pereira, S. I. Gorelsky, A. Pike, A. P. LeBris and B. DeBoef, J. Am. Chem. Soc., 2010, 132, 14676 CrossRef CAS PubMed; (d) H. Li, J. Liu, C.-L. Sun, B.-J. Li and Z.-J. Shi, Org. Lett., 2011, 13, 276 CrossRef CAS PubMed; (e) A. N. Campbell, E. B. Meyer and S. S. Stahl, Chem. Commun., 2011, 47, 10257 RSC; (f) C.-Y. He, Q.-Q. Min and X. Zhang, Organometallics, 2012, 31, 1335 CrossRef CAS; (g) Z. Li, L. Ma, J. Xu, L. Kong, X. Wu and H. Yao, Chem. Commun., 2012, 48, 3763 RSC; (h) G. Wu, J. Zhou, M. Zhang, P. Hu and W. Su, Chem. Commun., 2012, 48, 8964 RSC; (i) F. Chen, Z. Feng, C.-Y. He, H.-Y. Wang, Y.-L. Guo and X. Zhang, Org. Lett., 2012, 14, 1176 CrossRef CAS PubMed; (j) B. Liu, Y. Huang, J. Lan, F. Song and J. You, Chem. Sci., 2013, 4, 2163 RSC; (k) X. Chen, X. Huang, Q. He, Y. Xie and C. Yang, Chem. Commun., 2014, 50, 3996 RSC.
- (a) H. Ge, M. J. Niphakis and G. I. Georg, J. Am. Chem. Soc., 2008, 130, 3708 CrossRef CAS PubMed; (b) Y. W. Kim, M. J. Niphakis and G. I. Georg, J. Org. Chem., 2012, 77, 9496 CrossRef CAS PubMed; (c) Y.-Y. Yu, L. Bi and G. I. Georg, J. Org. Chem., 2013, 78, 6163 CrossRef CAS PubMed.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2009, 42, 1074 CrossRef PubMed.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS; (c) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS; (d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS PubMed.
- (a) S. Grimme, S. Ehrlich and L. Georigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed; (b) S. Grimme, Chem.–Eur. J., 2012, 18, 9955 CrossRef CAS PubMed.
- (a) H. P. Hratchian and H. B. Schlegel, J. Chem. Phys., 2004, 120, 9918 CrossRef CAS PubMed; (b) H. P. Hratchian and H. B. Schlegel, J. Chem. Theory Comput., 2005, 1, 61 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 1.5.0.4, Schrödinger, LLC Search PubMed.
- R. E. Plata and D. A. Singleton, J. Am. Chem. Soc., 2015, 137, 3811 CrossRef CAS PubMed.
- M. Mammen, E. I. Shakhnovich, J. M. Deutch and G. M. Whitesides, J. Org. Chem., 1998, 63, 3821 CrossRef CAS.
- Calculations for a model N–H enaminone substrate show a similarly high preference for C–H activation at the C3-position over C2 (NMe ΔΔG = 13.5 kcal mol−1; N–H ΔΔG = 14.6 kcal mol−1) indicating that steric interactions due to the N-methyl group are not decisive.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04590h |
| This journal is © The Royal Society of Chemistry 2016 |