
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic discrimination between formyl groups in regio- and stereoselective intramolecular cross-aldol reactions†
Tomonori
Baba
a,
Junya
Yamamoto
a,
Kazuhiro
Hayashi
a,
Makoto
Sato
b,
Masahiro
Yamanaka
b,
Takeo
Kawabata
a and
Takumi
Furuta
*a
aInstitute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan. E-mail: furuta@fos.kuicr.kyoto-u.ac.jp
bDepartment of Chemistry and Research Center for Smart Molecules, Faculty of Science, Rikkyo University, 3-34-1 Nish-Ikebukuro, Toshima-ku, Tokyo 171-8501, Japan
First published on 22nd February 2016
Abstract
Catalytic discrimination between inequivalent formyl groups was achieved using an aniline-type acid–base catalyst for the regio-, diastereo-, and enantioselective intramolecular cross-aldol reactions of enolizable dials. Although L-proline gave a mixture of the regio- and stereoisomeric products in the presence of an N-containing 1,6-dial, the aniline-type catalyst afforded anti-3,4-disubstituted pyrrolidine in high regio-, and stereoselectivity beyond the background reaction, which led to the regioisomeric 2,3-disubstituted products. The mild reactivity of the aniline-type amine facilitated catalytic discrimination between the inequivalent formyl groups. Kinetic isotope effect studies and reductive amination experiments suggested that the regioselectivity was controlled under the enamine-forming steps.
Introduction
Catalytic discrimination among similarly reactive functional groups is a key to realizing unique and efficient chemo- and regioselective transformations of multi-functionalized molecules;1 however, formidable challenges to such discrimination remain. Intramolecular cross-aldol reactions of enolizable unsymmetric dial 1 require catalytic discrimination between the formyl groups bearing similar reactivities (Fig. 1). Although the reaction provides special versatility toward the production of cyclic β-hydroxy aldehydes potentially found in prostaglandins 2 and nucleic acid medicines 3, control over this reaction is quite challenging due to the production of eight isomers from two regioisomers (from path A and B), including diastereomers (anti/syn), and enantiomers of each isomer (Fig. 1). Reaction selectively may only be achieved by controlling the diastereo- and enantioselectivities, in addition to controlling the regioselectivity of the products (path A vs. path B). In the amine-catalyzed reactions based on an enamine mechanism, a high regioselectivity is expected only under conditions that favor precise discrimination by the amine catalyst between two enolizable formyl groups. Under such conditions, these groups may be individually and selectively converted to the enamine component and the carbonyl component (Fig. 1).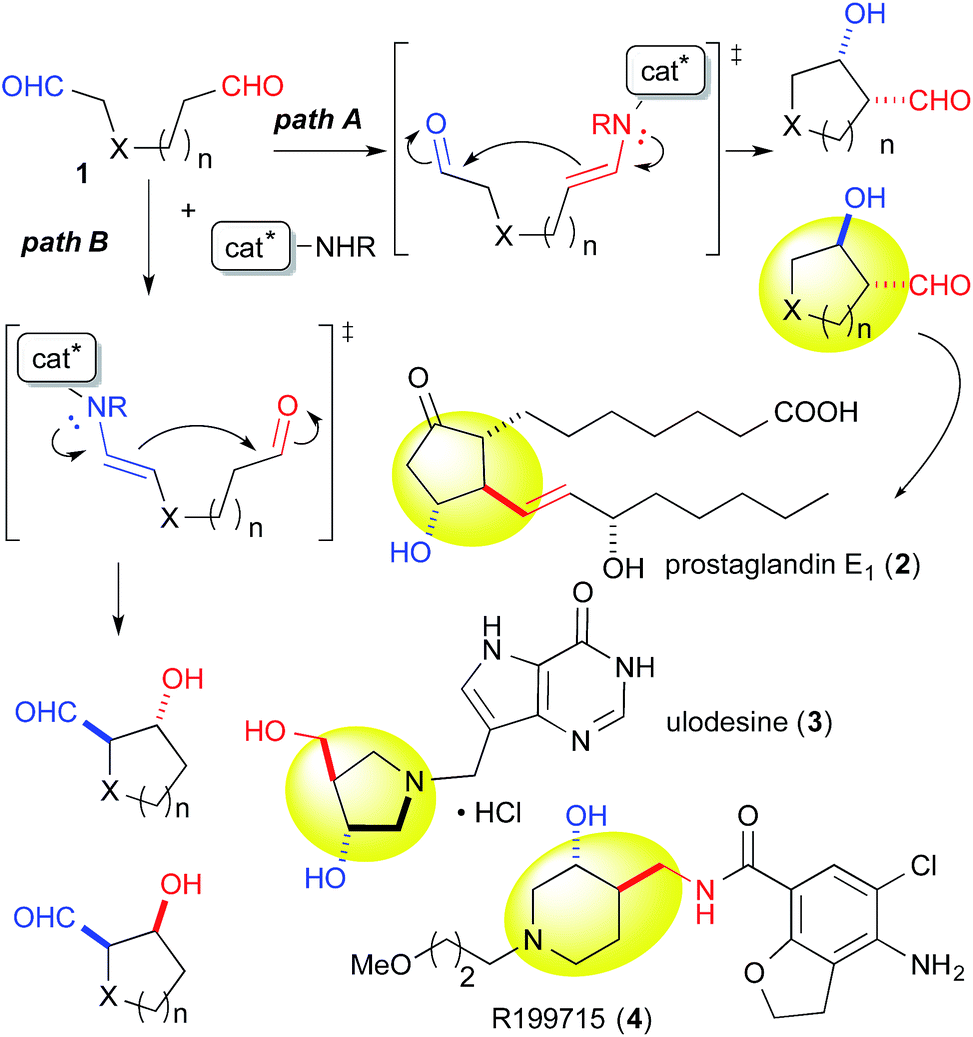 | ||
| Fig. 1 Possible regio- and stereoisomeric products from the intramolecular cross-aldol reaction of an enolizable aliphatic dial, and potentially preparable bioactive compounds bearing carbo- and heterocycles. | ||
In addition to these intramolecular reactions, selective direct intermolecular cross-aldol reactions between enolizable aliphatic aldehydes have been achieved.2,3 The slow addition of a donor or acceptor aldehyde using a syringe pump and/or the addition of excess amounts of one aldehyde to the other are indispensable for obtaining a good yield of the desired cross-aldol product in the case of α-unbranched aldehydes.4 Because these techniques cannot be employed in the intramolecular reaction of 1, the success of the reaction relies purely on the potential of a catalyst to discriminate between formyl groups. For these reasons, intramolecular cross-aldol reactions are highly challenging transformations.
Indeed, we encountered difficulties in our efforts to apply the intramolecular cross-aldol reaction. An examination of the reaction of N-Ts dial 1a in the presence of L-proline (5 mol%) toward an efficient synthesis of the chiral pyrrolidine (Table 2, entry 6) revealed that the reaction yielded an undesirable mixture of products. After NaBH4 reduction, the reaction mixture afforded nearly all possible regio- and stereoisomeric products, [anti-5a (9%, 6% ee), syn-6 (31%, >99% ee), dehydrated 9 (2%)] and [anti-7 (5%, 42% ee), syn-8 (17%, 53% ee)], from the enamines of the C(6)- and C(1)-formyl groups, as well as the diol 10 (20%), which corresponded to residual starting material. The regioselectivity of the reaction (5a + 6 + 9)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(7 + 8), was found to be 1.8:1. This undesirable result indicated that L-proline could not discriminate between the formyl groups of 1a.
:(7 + 8), was found to be 1.8:1. This undesirable result indicated that L-proline could not discriminate between the formyl groups of 1a.
Herein, we describe the catalytic discrimination between formyl groups by aniline-type acid–base catalysts based on their distinct mild reactivities. This approach yielded the first examples of regio-, diastereo-, and enantioselective intramolecular cross-aldol reactions of enolizable unsymmetric dials.5
Results and discussion
We assumed that L-proline was too reactive to discriminate between formyl groups bearing similar reactivities (Fig. 2A). Therefore, we hypothesized that an acid–base catalyst bearing a mildly reactive amine could be advantageous in discriminating between the different formyl groups.6 Our focus centered on an aniline-type amine as an amine with one of the lowest reactivities (Fig. 2B). We were particularly interested in aniline-type axially chiral amino acids,7 and therefore we prepared (R)-11 and (R)-12, 13, 14, which possessed tetrazole and sulfonamide groups as acidic moieties, respectively (Fig. 2C).8 Although aniline derivatives have been frequently employed as organocatalysts toward iminium activation,9 their application toward enamine catalysis has not received significant attention due to their weak basic and nucleophilic properties.10 Therefore, we initially evaluated the catalytic activities of these aniline catalysts by performing intramolecular enolexo-intramolecular aldol reaction of 1,6-hexanedial (15) (Table 1).11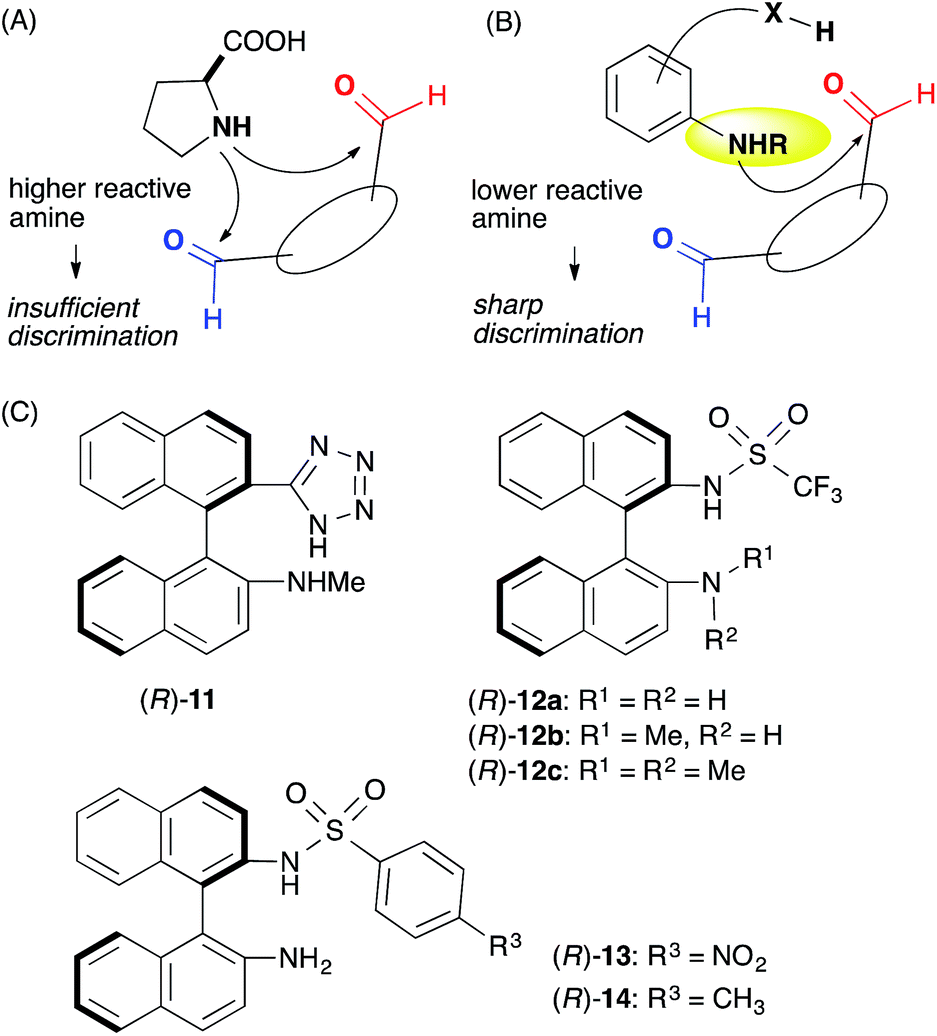 | ||
| Fig. 2 Working hypothesis underlying formyl group discrimination. (A) Aliphatic amino acid with a high reactivity. (B) Aniline-type acid–base catalyst with a low reactivity. (C) Axially chiral anilines bearing an acidic moiety. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time (h) | Yielda (%) 18b, 19 | d.r. (ee%) 18 (ee%):19e (ee%) |
| a Determined by the integration of the 1H NMR signals in the presence of dibenzyl ether as an internal standard. b The combined yield of the E/Z isomers. c The absolute configurations of the major enantiomers of anti-18 for entry 1 were determined to be (1S,2R). d The absolute configurations of the major enantiomers of anti-18 for entries 2–7 were determined to be (1R,2S). e The absolute configuration of the major enantiomer of syn-19 was determined to be (1S,2S). | |||||
| 1 | (R)-11 | DMSO | 96 | 40, 52 | 1 (50c):1.3 (93) |
| 2 | (R)-12a | DMSO | 4 | 58, 9 | 6.4 (87d):1 (13) |
| 3 | (R)-12a | DMF | 4 | 53, 12 | 4.4 (95d):1 (37) |
| 4 | (R)-12a | Acetone | 24 | 62, 8 | 7.8 (95d):1 (34) |
| 5 | (R)-12a | THF | 36 | 74, 5 | 15 (97d):1 (12) |
| 6 | (R)-13 | DMSO | 192 | 80, 5 | 16 (87d):1 (29) |
| 7 | L-Proline | DMSO | 6 | 13, 59 | 1 (67d):4.5 (19) |
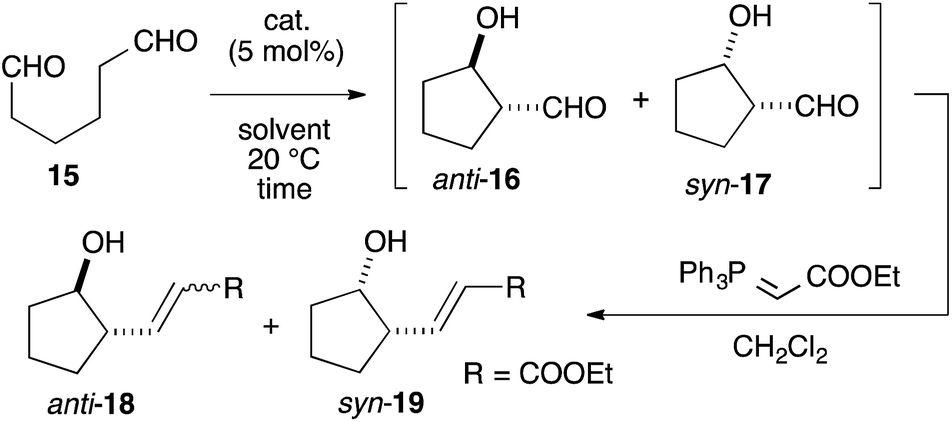
To our delight, cat. (R)-11 exhibited sufficient catalytic activity toward the reactions.12 In the presence of 5 mol% cat. (R)-11, the 5-membered ring-forming enolexo-intramolecular aldolization of 15 proceeded smoothly to afford anti-16 and syn-17 in DMSO. A subsequent Wittig olefination gave the corresponding anti-18 and syn-19 with a high enantioselectivity toward the syn isomer (93% ee), although the diastereoselectivity of the products was moderate (anti-18:syn-19 = 1:1.3) (Table 1, entry 1). A survey of catalysts and solvents (see ESI†) identified the primary amine catalyst (R)-12a, which possessed a triflic amide, as capable of affording anti-18 as the major product in a 58% yield with a high diastereo- (anti:syn = 6.4:1) and enantioselectivity (87% ee) (entry 2). DMF and acetone were good solvents for this reaction and improved the ee of anti-18 (entries 3 and 4). Changing the solvent to THF increased the diastereo- and enantioselectivities as well as the chemical yield of anti-18, giving an anti:syn ratio of 15:1, an ee of 97%, and a 74% yield after 36 h (entry 5). The catalyst (R)-13, which bore a p-Ns group, was also active in this reaction and yielded anti-18 in an 80% yield with an 87% ee in a high diastereoselectivity (anti:syn = 16:1) (entry 6). The opposite diastereo- and moderate enantioselectivities obtained in the presence of L-proline13 revealed that these axially chiral anilines provided a unique chiral environment suitable for the 1,6-dial (entry 7). The tertiary amine (R)-12c did not promote the reaction, suggesting that the aniline-type catalysts promoted the reaction via enamine catalysis. The catalytic activities listed in Table 1 verified that the anilines are useful organocatalysts, even in enamine catalysis.
DFT calculations using the model catalyst 20 and the dial 15 also supported the enamine mechanism, including the rate-determining iminium-to-enamine transformation (see ESI†). The most stable transition state (TS) for the stereo-determining C–C bond-forming step explained the stereochemistry of anti-16via C–C bond formation between the Si faces of the enamine and the formyl group (Fig. 3). The structural and electronic factors in the TS played a crucial role in controlling the stereoselectivity. The structurally favored conformation of the enamine and the C–C bond-forming moieties caused strong hydrogen bonds to form between the sulfonamide NH and the formyl carbonyl group, thereby facilitating a fit into the chiral space and stabilizing the TS for (1R,2R)-16.
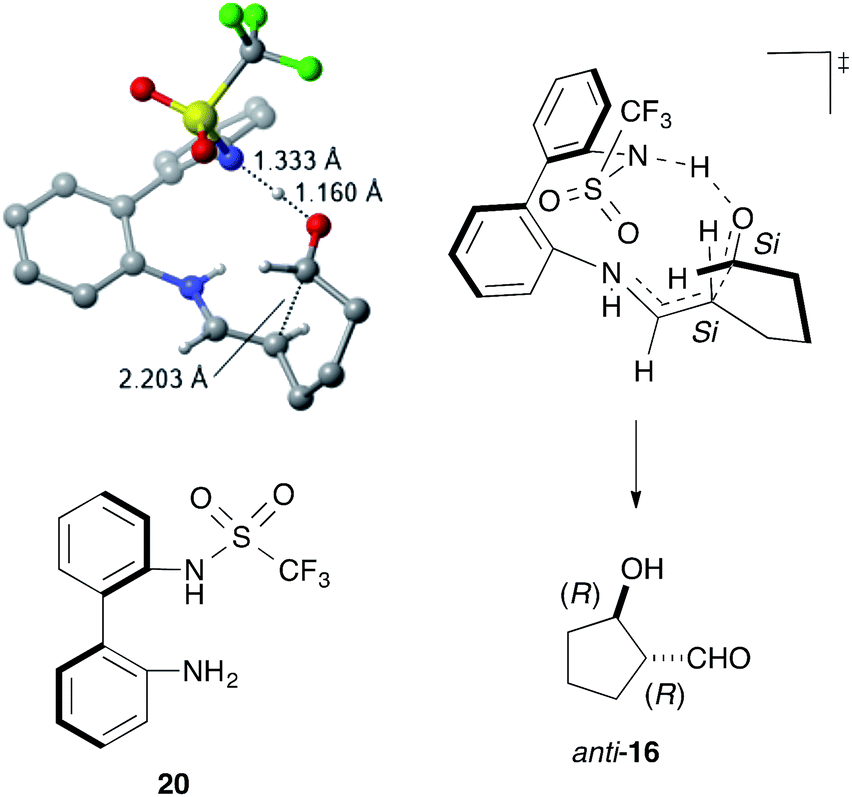 | ||
| Fig. 3 The most stable transition state for the stereo-determining C–C bond formation. Unimportant hydrogen atoms were omitted for clarify. | ||
With effective catalysts in hand, we moved again to examine the cross-aldol reaction of 1a (Table 2). Although the reaction in the presence of cat. (R)-12a gave dehydrated 9 as the major product (see ESI†), the milder acidic (R)-13 afforded 3,4-disubstituted anti-5a (59%) as the major product in 89% ee with a high diastereoselectivity (anti-5a:syn-6 = 12:1) and the concomitant formation of the regioisomer, 2,3-disubstituted anti-7 (8%) (Table 2, entry 1). The regioselectivity of the reaction, (5a + 6 + 9):(7 + 8), was found to be 8.0:1. This regioselectivity contrasts significantly with the corresponding value associated with the L-proline catalyzed reaction (entry 6). This selectivity indicated that cat. (R)-13 discriminated between the different formyl groups, which could not be distinguished by L-proline, and converted the C(6)-formyl group into the enamine component and the C(1)-formyl group into the carbonyl component, as shown in Fig. 1 (path A). The reaction in DMF improved the diastereoselectivity to 18:1 and the enantioselectivity to 93% ee, although the regioselectivity decreased slightly (entry 2). During this survey, we found that 1a was labile in DMSO and gave the opposite regioisomeric adducts anti-7 and syn-8 in the absence of a catalyst (regioselectivity, (5a + 6 + 9):(7 + 8) = 1:8.8) (entry 7). These results revealed that cat. (R)-13 overcame the background reaction to predominantly yield anti-5a. We also tested the primary alkyl amine catalyst, L-isoleucine, which was successfully employed in the intermolecular cross-aldol reaction with an α-branched substrate (entry 5).3j This catalyst predominantly gave anti-7 and syn-8; however, no significant ee value was obtained in syn-8, and a diastereoselectivity (7:8 = 1:1.2) similar to that of the background reaction (7:8 = 1:1.1) (entry 7) suggested that this catalyst did not overcome the background reaction. The combination of aniline (21) and p-Ns aniline (22) also afforded anti-7 and 8 as the major products (entry 4). These results indicated that cooperative activation of the substrate by acidic and basic moieties in biaryl framework was required for regioselectivity toward anti-5a.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | t (h) | Yielda (%) 5ab, 6c, 7c, 8c, 9, 10 | Regioselectivity (5a + 6 + 9):(7 + 8) |
d.r. 5a:6 |
d.r. 7:8 |
ee (%) 5a | ee (%) 8 |
| a Determined by the integration of the 1H NMR signals in the presence of dibenzyl ether as an internal standard. b The absolute configurations of the major enantiomers of anti-5a for entries 1–3 were determined to be (3S,4S). c The relative stereochemistry of all isomers was determined. The absolute configurations are tentatively based on the assumption that both products were generated from the same enamine geometry for anti-5a. d 74% ee was observed. e 6% ee, >99% ee, 42% ee, were observed for 5a, 6, and 7, respectively. f The absolute configurations of the major enantiomers was determined to be (2R,3R). | |||||||||
| 1 | (R)-13 | DMSO-d6 | 72 | 59, 5, 8d, 0, 0, 25 | 8.0:1 |
12:1 |
>99:1 |
89 | — |
| 2 | (R)-13 | DMF | 72 | 54, 3, 10, 0, 0, 28 | 5.7:1 |
18:1 |
>99:1 |
93 | — |
| 3 | (R)-13 | THF | 72 | 18, 4, 6, 0, 0, 50 | 3.7:1 |
4.5:1 |
>99:1 |
82 | — |
| 4 | 21 + 22 | DMSO-d6 | 48 | 5, 6, 22, 34, 0, 14 | 1:5.1 |
1:1.2 |
1:1.9 |
— | — |
| 5 | L-Isoleucine | DMSO-d6 | 56 | 5, 6, 27, 31, 4, 15 | 1:3.8 |
1:1.3 |
1:1.2 |
n.d. | 6 |
| 6 | L-Proline | DMSO-d6 | 24 | 9e, 31e, 5e, 17, 2, 20 | 1.8:1 |
1:3.4 |
1:3.4 |
6 | 53f |
| 7 | — | DMSO-d6 | 72 | 3, 3, 37, 42, 0, 3 | 1:8.8 |
1:1.0 |
1:1.1 |
— | — |
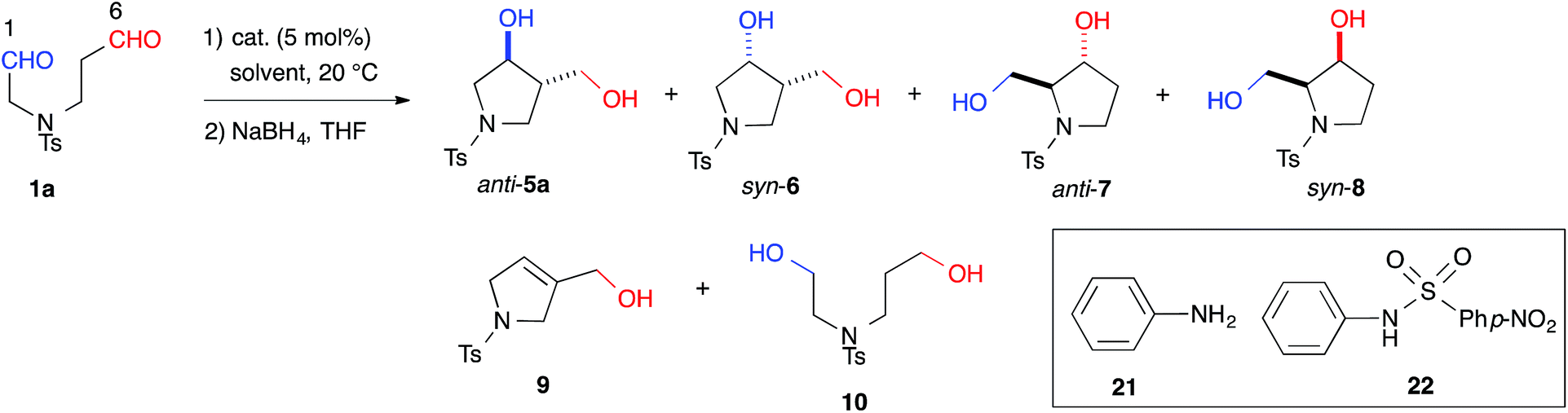
The yield of anti-5a was improved to 75% through elongation of the reaction time without decreasing the enantioselectivity (Table 3). The N-Alloc-, N-Cbz-, and N-Boc-protected dials 1b–1d were applicable in this reaction and afforded anti-5b–5d in 90% ee (Table 3). It should be noted that 5d was isolated as the sole product in 95% yield, suggesting that the formyl groups of 1d were perfectly distinguished by cat. (R)-13. Anti-5d is an intermediate to the phosphorylase inhibitor, ulodesine (3, Fig. 1).14
|
|
|---|
| a Determined by the integration of the 1H NMR signals in the presence of dibenzyl ether as an internal standard. b Determined by the integration of the 1H NMR signals in the presence of 1,3-dinitrobenzene as an internal standard. c The absolute configurations of anti-(3S,4S)-5b–5d were determined by transforming into anti-5a. |
|
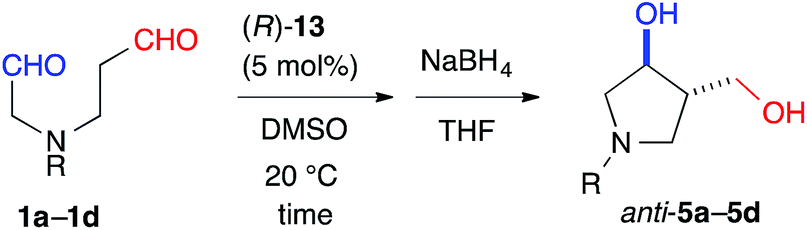
The 6-membered ring-forming cross-aldol reactions were further examined under the conditions used for the 5-membered ring formation (Table 4). As expected, 3,4-anti disubstituted piperidine (24) was obtained from the reaction of N-Boc 1,7-dial (23) in the presence of 5 mol% cat. (R)-13, followed by successive NaBH4 reduction with high regio- and diastereoselectivities, although the enantioselectivity was low (entry 1). Unlike the five-membered ring formation, cat. (R)-12a did not promote dehydration and gave 24 as the major product, with a slightly better enantioselectivity (entry 2). Changing the solvent to THF and lowering the temperature to 0 °C in the presence of cat. (R)-12a improved the enantioselectivity to 86% ee with excellent regioselectivity (entry 3). The high regioselectivity indicated that the formyl groups of 23 were distinguished by the catalyst, leading to a reaction from the enamine of the C(7)-formyl group. This reaction provided a promising tool for constructing a 3,4-anti disubstituted chiral piperidine, such as R199715 (4, Fig. 1), previously prepared from 24.15
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp (°C) | Time (h) | Yieldb (%) | ee (%) |
| a The absolute configurations of anti-24 was determined to be (3R,4R). b Determined by the integration of the 1H NMR signals in the presence of 1,3-dinitrobenzene as an internal standard. | ||||||
| 1 | (R)-13 | DMSO | 20 | 120 | 82 | 30 |
| 2 | (R)-12a | DMSO | 20 | 1.5 | 91 | 40 |
| 3 | (R)-12a | THF | 0 | 36 | 96 | 86 |
The formyl groups of 25 without a heteroatom were also discriminated well by cat. (R)-13 to afford cyclopentane, anti-26, regioselectively in 74% yield with 82% ee (Scheme 1).
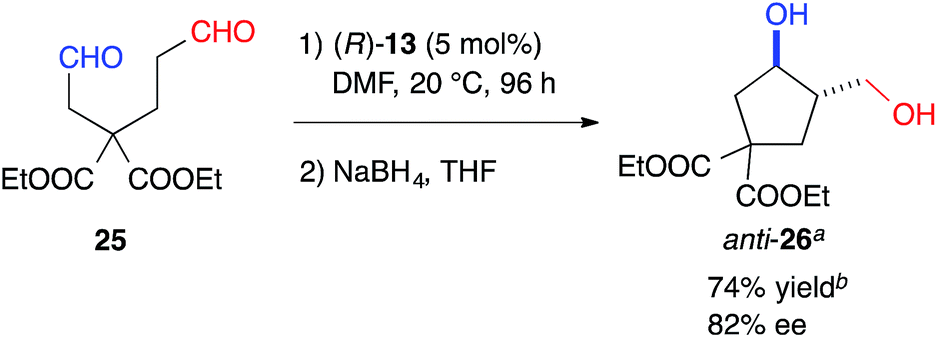 | ||
| Scheme 1 Intramolecular cross-aldol reaction to a chiral cyclopentane. aThe absolute configuration of anti-26 was determined to be (3R,4S). bDetermined by the integration of the 1H NMR signals in the presence of 1,3-dinitrobenzene as an internal standard. | ||
The origin of the regioselectivity was explored by treating a mixture of the aldol-adducts anti-7′ and syn-8′, the minor regioisomers, with cat. 13. The absence of a conversion to anti-5a′ suggested that the regioselectivity was governed by kinetic factors (Fig. 4A). The rate-determining step of the reaction was revealed by the kinetic isotope effect (KIE) using the α-deuterated substrate 1a-D. The apparent primary KIE (kH/kD = 3.4) indicated that the rate-determining step was associated with the enamine-forming step from the iminium intermediate, which involved C–H bond cleavage (Fig. 4B).16 Therefore, the regioselectivity of the reaction was defined by the enamine-forming steps rather than by the C–C bond-forming step. Furthermore, the regioselectivity of the reaction decreased to 1.2:1 for (5a-D + 6-D):(7-D + 8-D) from 8.0:1 for (5a + 6 + 9):(7 + 8) (Table 2, entry 1). This result indicated that the kinetics associated with the iminium-to-enamine intermediate affected the regioselectivity.
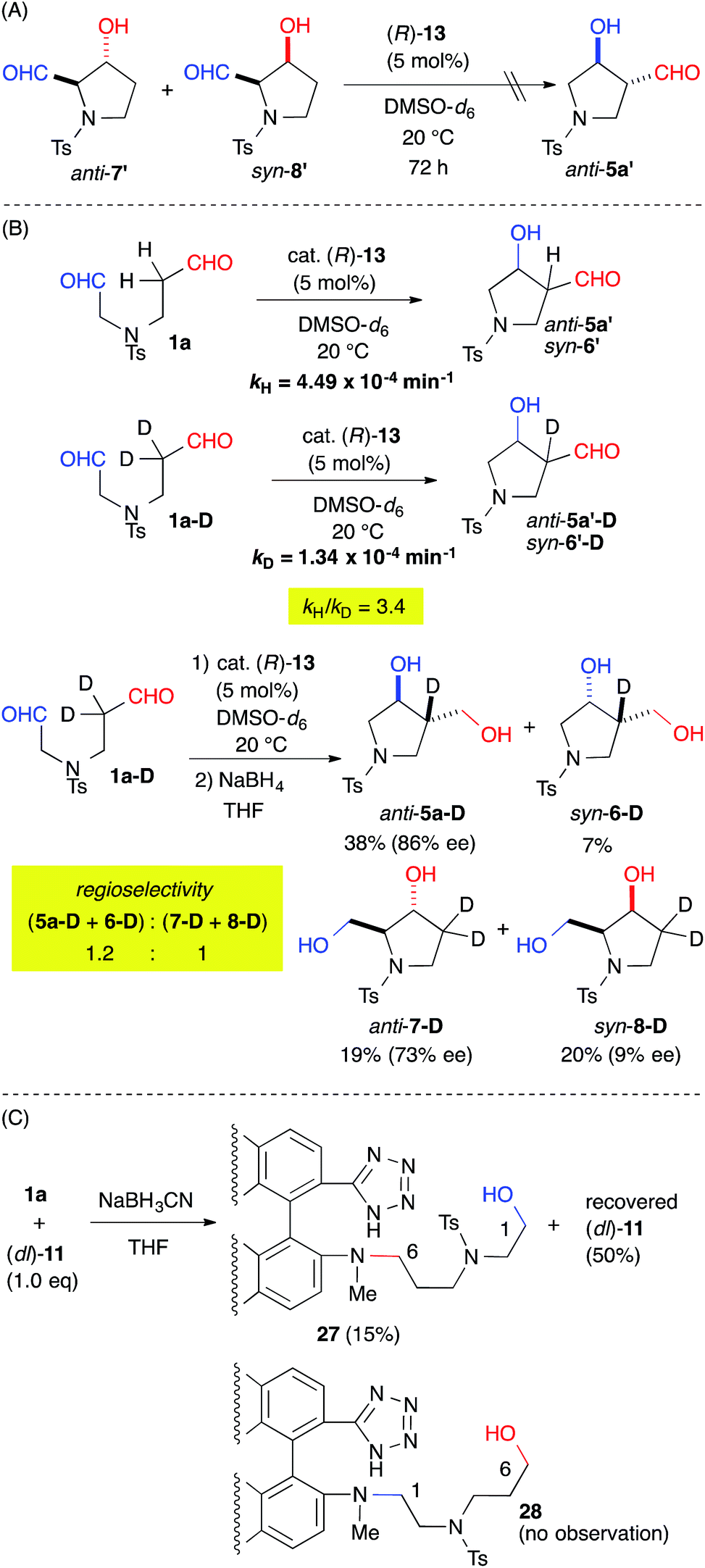 | ||
| Fig. 4 Mechanistic investigation. | ||
A reductive amination of dial 1a with the catalyst was also carried out to evaluate the regioselective iminium formation prior to the rate-determining step (Fig. 4C). The NaBH3CN reduction of an equimolar mixture of 1a and cat. (dl)-11, which gave a regioselectivity {(5a + 6 + 9):(7 + 8): = 7.4:1} similar to that obtained from cat. (R)-13 (see ESI†), gave 27 in a 15% yield with a 50% recovery of (dl)-11 without any observable appearance of 28. This result suggested predominant iminium formation at the C(6)-formyl group of 1a.
These experiments suggested that the regioselectivity was controlled by the kinetics of the enamine formation. Selective iminium formation at the C(6)-formyl group ([29] > [30]) of 1a-1d may have affected the kinetics associated primarily with the production of the major regioisomers (Fig. 5). The thermodynamic stability of 29 due to the iminium cation located two carbons away from the electron-withdrawing NR group may have contributed to the predominant formation of 29 relative to 30 during the equilibration step. The steric factor of the catalyst may also play a role in the preferential formation of the sterically less congested 29 than 30. The regioselective formation of anti-26 from dial 25 bearing gem-diester group instead of the NR group might be mainly governed by the steric factor (Scheme 1). The mild reactivity of the aniline-type catalyst may have facilitated the discrimination between tiny electronic differences and/or steric circumstances of the C(1)- and C(6)-formyl groups.17
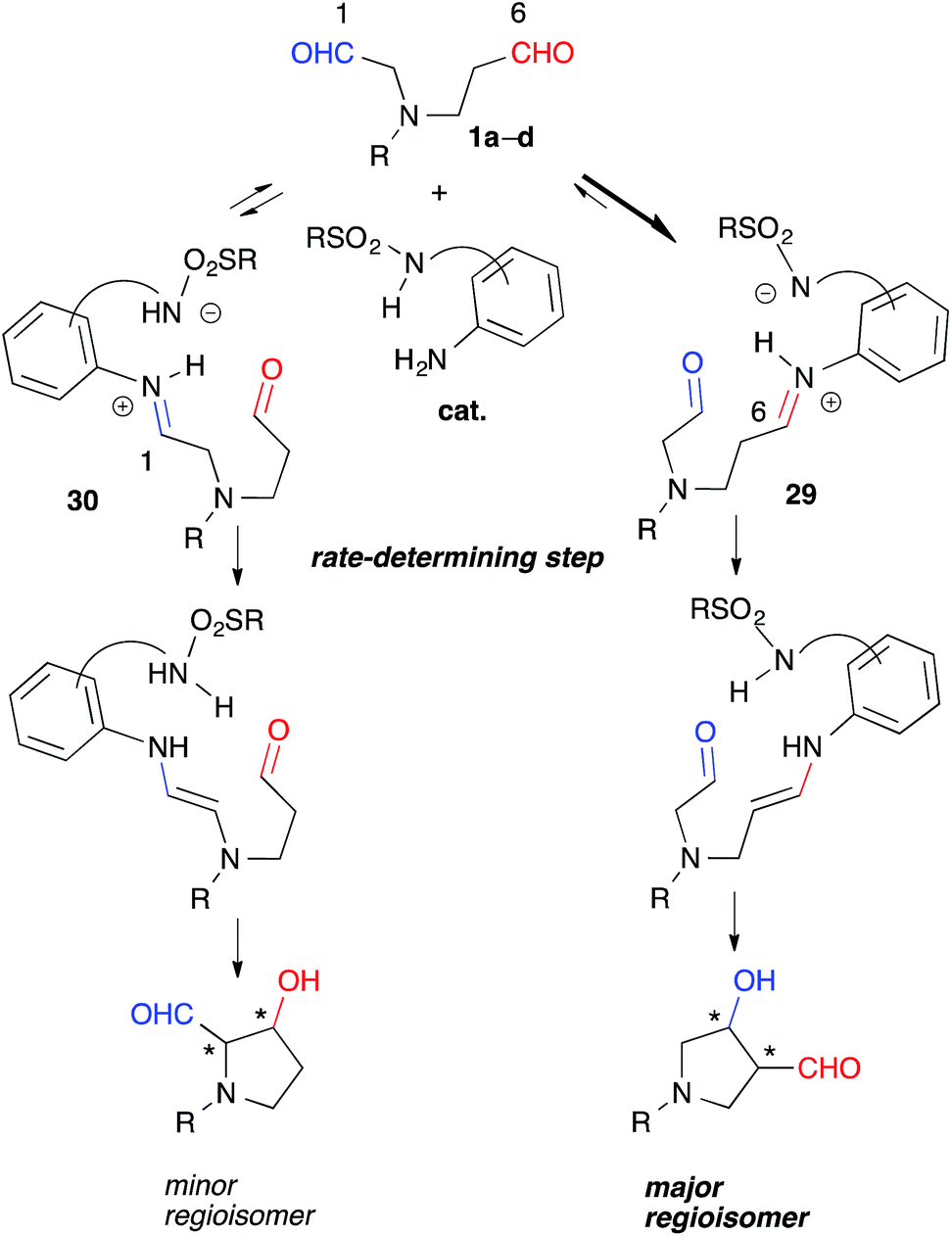 | ||
| Fig. 5 Possible explanation for regioselectivity. | ||
Conclusions
In summary, catalytic discrimination among formyl groups was achieved in the highly regio-, diastereo-, and enantioselective intramolecular cross-aldol reactions of enolizable 1,6- and 1,7-dials. The key to realizing formyl group discrimination was the mild reactivity of the aniline-type acid–base catalysts, which led to excellent regioselectivity. Mechanistic investigations including kinetic isotope effect studies and reductive amination experiments revealed that the regioselectivity was controlled under the enamine-forming steps. The high accessibility to the chiral pyrrolidines and piperidines provided a prominent feature of this cross-aldol reaction. Further mechanistic investigations are currently underway.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, a Grant-in-Aid for Scientific Research on Innovative Areas “Advanced Molecular Transformations by Organocatalysts”, the Collaborative Research Program of Institute for Chemical Research, Kyoto University (grant# 2015-72), and The Naito Foundation. The authors thank KYOWA HAKKO BIO CO., LTD. for generously providing cis-3-hydroxy-L-proline.Notes and references
- (a) N. A. Afagh and A. K. Yudin, Angew. Chem., Int. Ed., 2010, 49, 262 CrossRef CAS PubMed; (b) D. Lee and M. S. Taylor, Synthesis, 2012, 44, 3421 CrossRef CAS; (c) J. Mahatthananchai, A. M. Dumas and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 10954 CrossRef CAS PubMed.
- For the reviews, see (a) B. Alcaide and P. Almendros, Angew. Chem., Int. Ed., 2003, 42, 858 CrossRef CAS PubMed; (b) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600 RSC; (c) U. Scheffler and R. Mahrwald, Synlett, 2011, 1660 CAS.
- For examples, see (a) A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 6798 CrossRef CAS PubMed; (b) A. B. Northrup, I. K. Mangion, F. Hettche and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2004, 43, 2152 CrossRef CAS PubMed; (c) R. I. Storer and D. W. C. MacMillan, Tetrahedron, 2004, 60, 7705 CrossRef CAS; (d) J. Casas, M. Engqvist, I. Ibrahem, B. Kaynak and A. Cordova, Angew. Chem., Int. Ed., 2005, 44, 1343 CrossRef CAS PubMed; (e) R. Thayumanavan, F. Tanaka and C. F. Barbas III, Org. Lett., 2004, 6, 3541 CrossRef CAS PubMed; (f) M. Markert, U. Scheffler and R. Mahrwald, J. Am. Chem. Soc., 2009, 131, 16642 CrossRef CAS PubMed; (g) Y. Hayashi, Y. Yasui, T. Kawamura, M. Kojima and H. Ishikawa, Angew. Chem., Int. Ed., 2011, 50, 2804 CrossRef CAS PubMed; (h) T. Kano, H. Sugimoto and K. Maruoka, J. Am. Chem. Soc., 2011, 133, 18130 CrossRef CAS PubMed; (i) U. Scheffler and R. Mahrwald, J. Org. Chem., 2012, 77, 2310 CrossRef CAS PubMed; (j) K. Rohr and R. Mahrwald, Org. Lett., 2012, 14, 2180 CrossRef CAS PubMed.
- In the cases employing α-branched aldehyde, intermolecular cross-aldol reactions without employing excess amount and slow addition of one aldehyde were developed by Mahrwald and Maruoka. See, ref. 3f and h–j.
- Only the examples of regio- and diastereoselective intramolecular cross-aldol reaction of the 1,6- and 1,7-dial bearing α-branched formyl group were reported. See, (a) R. Breslow, J. Desper and Y. Huang, Tetrahedron Lett., 1996, 37, 2541 CrossRef CAS; (b) I. Kumar and C. V. Rode, Tetrahedron: Asymmetry, 2010, 21, 2703 CrossRef CAS; (c) G. Coulthard, W. Erb and V. K. Aggarwal, Nature, 2012, 489, 278 CrossRef CAS PubMed; (d) S. Prévost, K. Thai, N. Schützenmeister, G. Coulthard, W. Erb and V. K. Aggarwal, Org. Lett., 2015, 17, 504 CrossRef PubMed.
- The utility of a catalyst with mild reactivity was also suggested in ref. 3h.
- (a) T. Furuta, J. Yamamoto, Y. Kitamura, A. Hashimoto, H. Masu, I. Azumaya, T. Kan and T. Kawabata, J. Org. Chem., 2010, 75, 7010 CrossRef CAS PubMed; (b) T. Furuta, M. Nikaido, J. Yamamoto, T. Kuribayashi and T. Kawabata, Synthesis, 2013, 45, 1312 CrossRef CAS.
- The preparation of axially chiral anilines (R)-11, 12a, 12b, 12c and 13 is described in the ESI†. Aniline 12a was reported, see, (a) F. Taran, C. Gauchet, B. Mohar, S. Meunier, A. Valleix, P. Y. Renard, C. Créminon, J. Grassi, A. Wagner and C. Mioskowski, Angew. Chem., Int. Ed., 2002, 41, 124–127 CrossRef CAS. Aniline (S)-14 has been reported, see, (b) A. Sakakura, K. Suzuki and K. Ishihara, Adv. Synth. Catal., 2006, 348, 2457 CrossRef CAS; (c) T. Chen, J. Gao and M. Shi, Tetrahedron, 2006, 62, 6289 CrossRef CAS.
- For a review on iminium catalysis, see, (a) A. Erkkil, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416 CrossRef PubMed. For examples of aniline-type catalysts for iminium activation, see, (b) A. Sakakura, K. Suzuki, K. Nakano and K. Ishihara, Org. Lett., 2006, 8, 2229 CrossRef CAS PubMed; (c) A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581 CrossRef CAS PubMed; (d) A. Erkkilä, P. M. Pihko and M.-R. Clarke, Adv. Synth. Catal., 2007, 349, 802 CrossRef; (e) T. Kano, Y. Tanaka, K. Osawa, T. Yurino and K. Maruoka, Chem. Commun., 2009, 1956 RSC; (f) P. Galzerano, G. Bencivenni, F. Pesciaioli, A. Mazzanti, B. Giannichi, L. Sambri, G. Bartoli and P. Melchiorre, Chem.–Eur. J., 2009, 15, 7846 CrossRef CAS PubMed; (g) P. Crisalli and E. T. Kool, Org. Lett., 2013, 15, 1646 CrossRef CAS PubMed.
- Few examples have proposed aniline derived enamine intermediate, see (a) S. Brandes, B. Niess, M. Bella, A. Prieto, J. Overgaard and K. A. Jørgensen, Chem.–Eur. J., 2006, 12, 6039 CrossRef CAS PubMed; (b) W. Schrader, P. P. Handayani, J. Zhou and B. List, Angew. Chem., Int. Ed., 2009, 48, 1463 CrossRef CAS PubMed; (c) L. Ren, T. Lei and L.-Z. Gong, Chem. Commun., 2011, 47, 11683 RSC; (d) Y. Deng, L. Liu, R. G. Sarkisian, K. Wheeler, H. Wang and Z. Xu, Angew. Chem., Int. Ed., 2013, 52, 3663 CrossRef CAS PubMed.
- Only an example of moderate stereoselective enolexo-intramolecular aldol reaction of 15 catalyzed by L-proline was reported. Highly stereoselective reactions of the 1,7-heptanedials were achieved in the same paper. See, (a) C. Pidathala, L. Hoang, N. Vignola and B. List, Angew. Chem., Int. Ed., 2003, 42, 2785 CrossRef CAS PubMed. The desymmetrization of the meso-1,6-hexanedials via an enolexo-intramolecular aldol condensations in the presence of proline-related organocatalysts were reported; however, only moderate enantioselectivities were observed. See, (b) V. B. Kurteva and C. A. M. Afonso, Tetrahedron, 2005, 61, 267 CrossRef CAS.
- Cat. 11 did not show catalytic activity in the self-aldol reaction of dodecanal. This result suggested the intrinsic low reactivity of aniline-type catalyst. See, ESI.†.
- These results are consistent with the previous report by List. See, ref. 11a.
- (a) K. Clinch, G. B. Evans, G. W. J. Fleet, R. H. Furneaux, S. W. Johnson, D. H. Lenz, S. P. H. Mee, P. R. Rands, V. L. Schramm, E. A. Taylor Ringia and P. C. Tyler, Org. Biomol. Chem., 2006, 4, 1131 RSC; (b) V. P. Kamath, J. J. Juarez-Brambila, C. B. Morris, C. D. Winslow and P. E. Morris Jr, Org. Process Res. Dev., 2009, 13, 928 CrossRef CAS.
- H. J. M. Gijsen, M. J. A. De Cleyn, C. J. Love, M. Surkyn, S. F. A. Van Brandt, M. G. C. Verdonck, L. Moens, J. Cuypers and J.-P. R. M. A. Bosmans, Tetrahedron, 2008, 64, 2456 CrossRef CAS.
- Due to the long reaction time, H/D exchange between the sulfonamide moiety of (R)-13 and substrate 1a-D might take place to lead to the deuterated catalyst. In this situation, the possibility that the KIE value was derived from the other steps including C–C bond formation cannot be ruled out.
- The higher reactivity of the enamine resulting from iminium intermediate 29 than that from intermediate 30 would be alternative explanation of the regioselectivity, although it seems unlikely because the C–C bond formation might not be involved as rate-determining step.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and DFT studies. See DOI: 10.1039/c5sc04594k |
| This journal is © The Royal Society of Chemistry 2016 |