
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sub-5 nm lanthanide-doped lutetium oxyfluoride nanoprobes for ultrasensitive detection of prostate specific antigen†
Jin
Xu
abc,
Shanyong
Zhou
a,
Datao
Tu
a,
Wei
Zheng
a,
Ping
Huang
a,
Renfu
Li
a,
Zhuo
Chen
b,
Mingdong
Huang
b and
Xueyuan
Chen
 *abc
*abc
aKey Laboratory of Optoelectronic Materials Chemistry and Physics, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China. E-mail: xchen@fjirsm.ac.cn
bState Key Laboratory of Structural Chemistry, Danish-Chinese Centre for Proteases and Cancer, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 12th January 2016
Abstract
It remains challenging to develop ultrasmall (<5 nm) but highly luminescent bioprobes with a large linear detection range for the early diagnosis and monitoring of prostate cancer (PCa). Benefiting from the high molar density of lanthanide ions in an oxyfluoride matrix and the superior dissolution capability of Lu6O5F8 nanoparticles in the enhancer solution, we demonstrated the successful use of novel sub-5 nm Lu6O5F8:Eu3+ nanoprobes for the detection of prostate specific antigen (PSA) in clinical serum samples. The limit of detection for PSA is as low as 0.52 pg mL−1, which is almost a 200-fold improvement relative to that of a commercial dissociation-enhanced lanthanide fluoroimmunoassay (DELFIA) kit. The PSA levels detected in 23 patient serum samples were consistent with those measured independently by the DELFIA kit, showing the assay's reliability with a correlation coefficient of 97%. A linear range of 4 orders of magnitude ranging from 8.5 × 10−4 to 5.6 ng mL−1 for the assay of PSA was achieved, which is highly promising for the early diagnosis of PCa and monitoring of PCa relapse of patients after radical prostatectomy.
Introduction
Prostate cancer (PCa) is the third most common cancer in men worldwide.1 Prostate specific antigen (PSA) has been commonly used as a tumour marker for the early diagnosis and monitoring of PCa.2 For early diagnostic purposes, a PSA level of >4 ng mL−1 is considered as an indicator of suspicion of PCa. For the monitoring of PCa relapse, the PSA level in the serum usually decreases to ∼1 pg mL−1 after radical prostatectomy, and a serial increase in the PSA level indicates the recurrence or metastasis of PCa.3 Thus, ultrasensitive detection of PSA featuring a large linear range from ∼1 pg mL−1 to >4 ng mL−1 is crucial for the theranostics of PCa.Tremendous efforts have been devoted to developing sensitive PSA-detecting approaches with a large linear range. Therein, fluorescence immunoassays are most commonly employed owing to their good compatibility with currently available analytical platforms in the clinic.4 However, commercial fluorescence bioassays have detection limits around 0.1 ng mL−1, which are insufficient for the monitoring of PCa relapse after radical prostatectomy. Therefore, an increased emphasis has been exerted on designing ultrasensitive assays with much better analytical sensitivity than commercial kits.5 Among fluorescence immunoassays previously established for the detection of PSA, time-resolved (TR) photoluminescence (PL) bioassays based on lanthanide-chelate embedded polystyrene or silica nanoparticles (NPs) are capable of achieving improved sensitivity than a commercial kit.6 Nevertheless, such nanoprobes (usually >40 nm) have a tendency to agglomerate and swell in aqueous solution, and potential chelate leakage concerns have also been documented.6,7 Compared with lanthanide chelates, lanthanide-doped inorganic NPs have become a research hotspot in biodetection owing to their relatively high stability and greater flexibility for bioconjugation.8–18 In particular, ultrasmall (<5 nm) nano-bioprobes in the size range of biological molecules are desired due to their minimized interference with the antigen–antibody binding process.19 Hitherto, it is still a challenge to develop ultrasmall lanthanide luminescent nano-bioprobes with both a high detection sensitivity and a large linear detection range. Recently, we have developed the dissolution-enhanced luminescent bioassay (DELBA) technique based on inorganic lanthanide fluoride NPs.20 Upon the addition of NPs to the enhancer solution (i.e. Triton X-100 micelle solution), the trivalent lanthanide (Ln3+) ions in the NPs are extracted into micelles containing chelating ligands to form highly luminescent lanthanide complexes. As such, the TRPL signals are amplified, improving the detection sensitivity. However, there is still great demand for exploring inorganic NPs with more highly concentrated Ln3+ ions and better dissolution performance to further improve the detection sensitivity and linear range of bioprobes. In comparison with fluorides, Ln3+ doped lutetium oxyfluorides, like Lu6O5F8, have a much higher molar density of Ln3+ ions in the matrix (ESI Table S1†). A larger amount of lanthanide luminescent micelles can be transformed from a single oxyfluoride NP. Besides, the Ln–O bond in Lu6O5F8 can be more easily loosened than the Ln–F bond via the protonation reaction with H+,21 which may facilitate Ln3+ ion extraction from the NP surface into micelles. As a result, Ln3+ doped Lu6O5F8 is expected to be more suitable than fluorides as an ultrasmall bioprobe for the detection of PSA.
Herein, monodisperse and ultrasmall Ln3+ (Ln = Eu, Yb/Er) doped lutetium oxyfluoride NPs, which had never been explored before, were synthesized via a modified thermal decomposition route.22 By utilizing the Lu6O5F8:Eu3+ nanoprobes featuring a high molar density of Ln3+ ions and superior dissolution properties in the enhancer solution, we demonstrate the ultrasensitive and accurate detection of PSA in patient serum samples through heterogeneous sandwich bioassays in the TRPL detection mode, as depicted in Fig. 1. Furthermore, we reveal the great potential of ultrasmall Lu6O5F8:Yb3+/Er3+ nanoprobes in upconversion luminescence (UCL) and computed tomography (CT) dual-modal bioimaging.
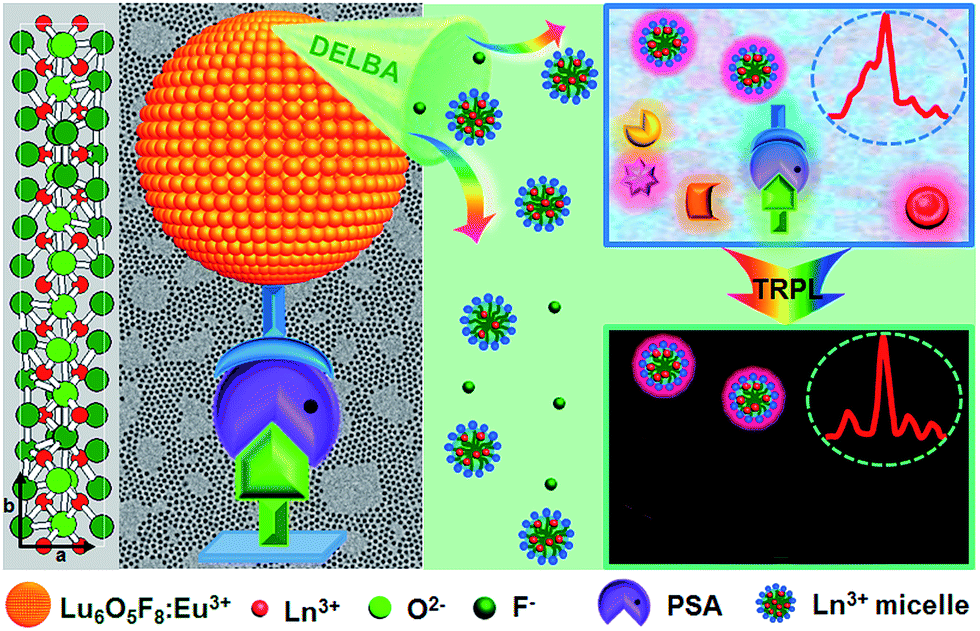 | ||
| Fig. 1 Schematic illustration of PSA detection utilizing sub-5 nm orthorhombic-phase Lu6O5F8:Eu3+ NPs as nanoprobes in TRPL mode. | ||
Results and discussion
Monodisperse and ultrasmall Ln3+ (Ln = Eu, Yb/Er) doped lutetium oxyfluoride NPs were synthesized via a modified thermal decomposition route. The as-prepared Eu3+ (5 mol%) doped Lu6O5F8 NPs are readily dispersed in cyclohexane (Fig. 2a). TEM analysis shows that these NPs are nearly spherical with an average diameter of 4.8 ± 0.5 nm (Fig. 2b), as corroborated by the broad powder X-ray diffraction (XRD) peaks, which are well indexed as the orthorhombic phase of Lu6O5F8 (ESI Fig. S1†). The thermogravimetric analysis of the as-prepared NPs shows a weight loss of 5.17% between 450 and 750 °C (ESI Fig. S2†), which is consistent with the theoretical weight loss (5.19%) resulting from the decomposition of Lu6O5F8 to Lu2O3, thus verifying the chemical composition of the Lu6O5F8 NPs. The high-resolution TEM (HRTEM) image shows very clear lattice fringes with an observed d-spacing of 0.31 nm (Fig. 2c), which is in good agreement with the lattice spacing of the (161) plane of Lu6O5F8. Moreover, the sizes and morphologies of NPs with different Ln3+ doping levels remained essentially unchanged (ESI Fig. S3–S5†). Compositional analyses by energy-dispersive X-ray (EDX) spectroscopy and inductively coupled plasma-atomic emission spectroscopy (ICP-AES) revealed the existence of Lu, O, F, and the doping Ln3+ in the Lu6O5F8 NPs (ESI Fig. S6, Table S2†). Fig. 2d shows the characteristic Eu3+ downshifting (DS) emissions assigned to the 5D0 → 7F0–4 transitions in Lu6O5F8:Eu3+ (5 mol%) NPs upon excitation at 394 nm (7F0 → 5L6). The electric-dipole 5D0 → 7F2 transitions at ∼620 nm are much stronger than the magnetic-dipole 5D0 → 7F1 transitions at ∼590 nm, indicating that the doped Eu3+ ions occupy low-symmetry sites.23 The appearance of the 5D0 → 7F0 transitions infers that the site symmetry of Eu3+ in Lu6O5F8 should be restricted to noncentrosymmetric Cs, Cn, or Cnv (n = 1, 2, 3, 4, 6) groups,23 which is in accordance with the structural analysis that the doped Ln3+ ions may occupy several sites with symmetries of C1, Cs or C2 in Lu6O5F8.24 Meanwhile, in the corresponding excitation spectrum of the NPs (ESI Fig. S1b†), a broad O2−–Eu3+ charge transfer (CT) absorption in the 230–300 nm region was observed, which is typical of Eu3+ doped oxyfluoride hosts.24 Besides the intense DS luminescence, the colloidal cyclohexane solution of Lu6O5F8:Yb/Er (10/2 mol%) NPs displayed intense red UCL (inset of Fig. 2e). The UCL spectrum shows that the UCNPs exhibited strong red emission at ∼650 nm and relatively weak green emission at ∼540 nm with a red-to-green (R/G) ratio of ∼13, which are assigned to the 4F9/2 → 4I15/2 and 2H11/2/4S3/2 → 4I15/2 transitions of Er3+, respectively.25–27 Meanwhile, we observed a much shorter UCL lifetime of 4S3/2 (2.3 μs) than that of 4F9/2 (15.2 μs) of Er3+ (ESI Fig. S7†). The enhanced red UC emission may be attributed to the efficient energy back transfer (EBT) process from Er3+ to Yb3+ in the Lu6O5F8:Yb/Er lattice (ESI Fig. S8†).28,29 Such a high R/G ratio in UC phosphors is highly desired for in vivo bioimaging because the red UCL exhibits deeper tissue penetration than green light.29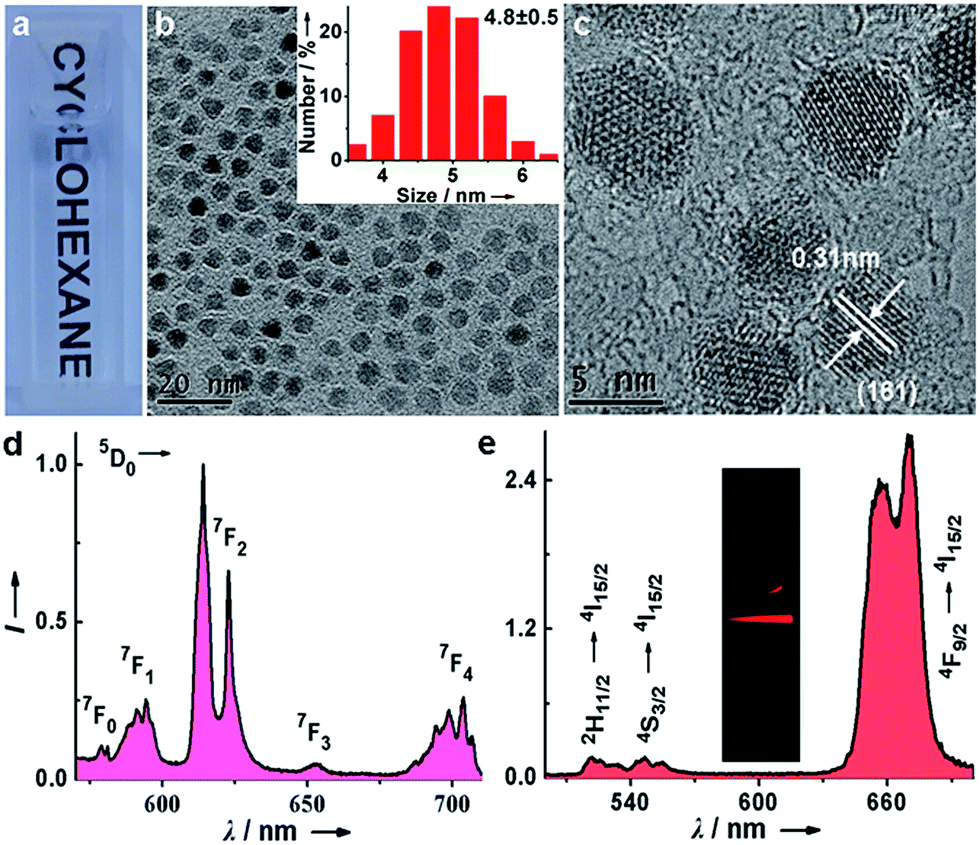 | ||
| Fig. 2 (a) Photograph showing the transparency of the as-prepared Lu6O5F8:Eu3+ (5 mol%) NPs dispersed in cyclohexane. (b) TEM and (c) HRTEM images of the as-prepared NPs. The inset shows a histogram of the size distribution. (d) PL emission spectrum for the Lu6O5F8:Eu3+ NPs upon excitation at 394 nm. (e) UCL spectrum of the as-prepared Lu6O5F8:Yb3+/Er3+ (10/2 mol%) NPs under 980 nm laser irradiation at a power density of ∼50 W cm−2. The inset shows the UCL photograph of the NPs dispersed in cyclohexane. | ||
To evaluate the dissolution performance of the Lu6O5F8:Eu3+ NPs, we firstly removed the oleate acid (OA) ligands from their surface by an acid-washing treatment to render the NPs hydrophilic.30 The successful removal of the surface ligands was confirmed by the Fourier transform infrared (FTIR) spectra (ESI Fig. S9†). Then, we added the solution of ligand-free Lu6O5F8:Eu3+ (40 mol%) NPs (20 μg mL−1) to the enhancer solution (i.e. the solution of Triton X-100 micelle containing 2-naphthoyltrifluoroacetone (β-NTA) and tri-n-octylphosphine oxide (TOPO) chelating ligands in the inner cavity, pH = 2.76). The appearance of a myriad of tiny NPs (<3 nm) in the TEM images indicates the occurrence of the dissolution reaction of the NPs, which was evidenced by the strong deep pink emission under ultraviolet (UV) lamp illumination upon addition of the NPs to the enhancer solution (Fig. 3a–c). To elucidate the origin of the dissolution-enhanced PL, we compared the steady-state PL excitation/emission spectra and the PL decays of the NPs before and after dissolution. The results show unambiguously that the enhanced Eu3+ PL of the NPs dissociated in the enhancer solution originated from the lanthanide complex, instead of the NPs (ESI Fig. S10†).
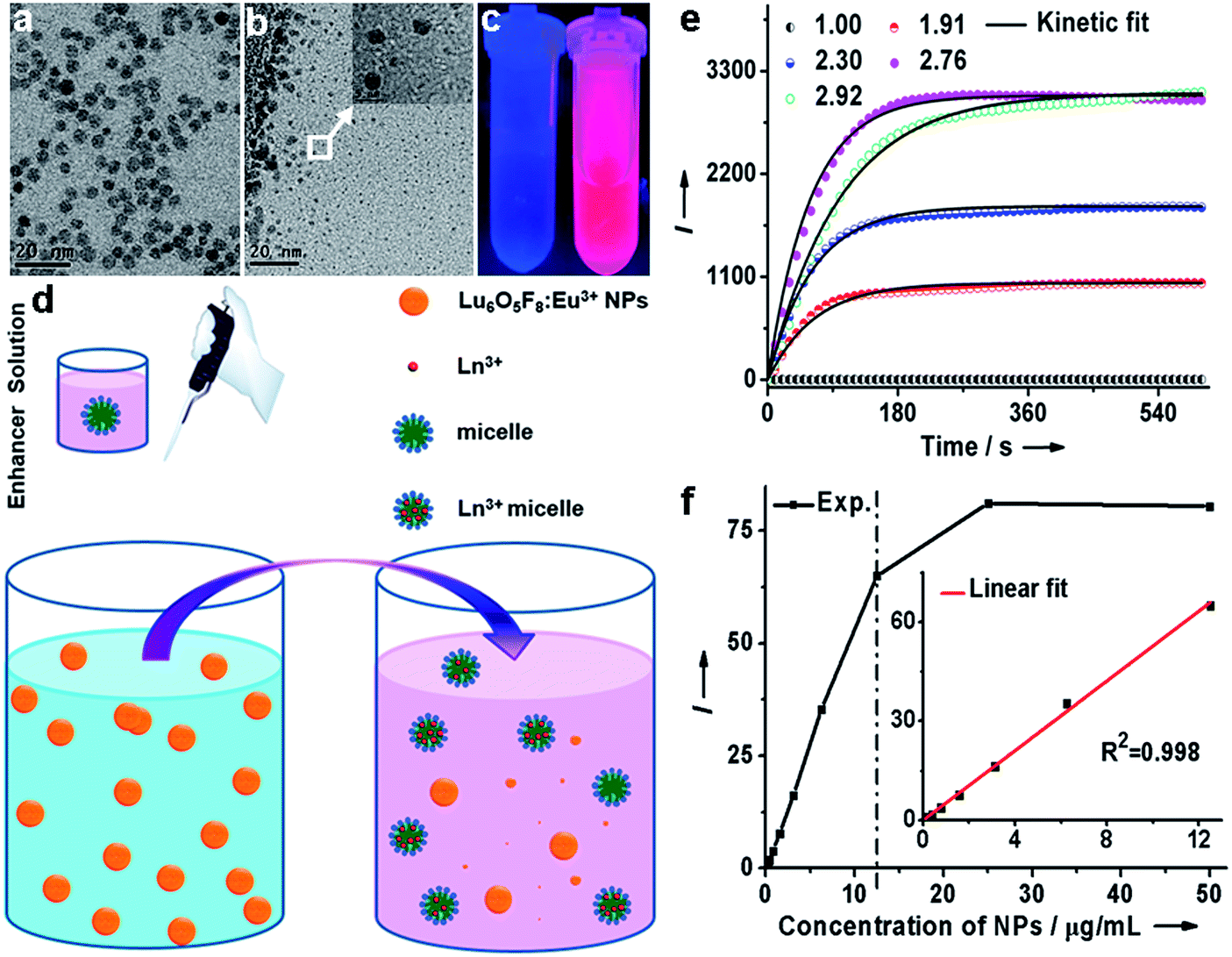 | ||
| Fig. 3 (a) Ligand-free Lu6O5F8:Eu3+ (40 mol%) NPs and (b) ligand-free NPs added to the enhancer solution. The inset shows an enlarged HRTEM image of the NPs, indicated by the white square. (c) Photograph showing the PL of ligand-free Lu6O5F8:Eu3+ NPs in a buffer solution (left) and the enhancer solution (right) under UV lamp illumination at 300 nm. (d) A schematic diagram for the mechanism of Lu6O5F8:Eu3+ NP dissolution. Upon addition of the enhancer solution to the solution of NPs, the massive Ln3+ ions accommodated in the NPs are extracted by the chelating ligands into micelles through spontaneous ligand–metal coordination reactions to form highly luminescent β-NTA–Ln3+–TOPO ternary complexes in the NP-micelle collisions. (e) Kinetic fit for the dissolution-enhanced PL signal of the ligand-free Lu6O5F8:Eu3+ (40 mol%) NPs (50 μg mL−1) dissolved in the enhancer solution at pH 1.00, 1.91, 2.30, 2.76 and 2.92. (f) Concentration-dependent dissolution-enhanced PL signal of ligand-free Lu6O5F8:Eu3+ NPs dissolved in the enhancer solution. Inset: the linear range of the PL signal versus the NP concentration (0–12.5 μg mL−1). (e–f) Each data point represents the mean of triplicate experiments. | ||
Since the lanthanide complexes are formed through the coordination reaction of the chelating ligands with the Ln3+ ions in the NPs (Fig. 3d), the dissolution-enhanced PL intensity of the Lu6O5F8:Eu3+ NPs depends critically on the molar concentration of Eu3+ in the NPs. Specifically, we found that Lu6O5F8 NPs doped with 40 mol% Eu3+ proved the most satisfactory (ESI Fig. S11†), due to the fact that the inert Lu3+ ion complex may decrease the concentration quenching of Eu3+ and enhance the luminescence of Eu3+ complex through a co-fluorescence effect.31 Similarly, the pH value of the enhancer solution has a complicated effect on the dissolution process of the NPs and the PL intensity of the Eu3+ complex (Fig. 3e).31 To gain deep insight into the dissolution dynamics of the NPs and to screen out optimal pH conditions for NP dissolution, we derived a kinetic equation (equation (S7) in ESI†) to simulate the dissolution process of the NPs. As shown in Fig. 3e and ESI Fig. S12,† the PL signal was well fitted by the kinetic equation. The kinetic parameters derived from the fitting are listed in ESI Table S3.† The results show that the equilibrium constant Keq is the highest at pH 2.76. The higher value of Keq indicates that a larger amount of NPs is dissolved when reaction equilibrium is reached. Consistently, the dissociation/association rate constants (Kd/Ka) are relatively high at pH 2.76, which means only a short time is needed to reach equilibrium in the dissolution reaction. As such, 2.76 was chosen as the optimal pH value for the dissolution of Lu6O5F8. Under these optimized conditions, we measured the linear dynamic range of dissolution-enhanced PL intensity versus the NP concentration. The PL signal increased linearly with the NP concentration in the large range of 0–12.5 μg mL−1 (i.e. 0–61.1 nmol Ln3+ per mL) (Fig. 3f). As shown in Fig. 3e, the dissolution-enhanced PL of NPs with a concentration even as high as 50 μg mL−1 was effectively stable within 5 minutes under optimum conditions. These results reveal the excellent dissolution performance of ultrasmall Lu6O5F8:Eu3+ nanoprobes for bioassays.
Prior to the luminescent bioassay, the OA-capped Lu6O5F8:Eu3+ NPs were surface-modified via ligand exchange with citrate, and then coupled with avidin following the EDC/NHS protocol.32,33 The successful conjugation of avidin to the surface of the NPs was corroborated by the appearance of amide bands in the FTIR spectrum, the increase in dynamic light scattering (DLS) size and the decrease of the zeta-potential for the NPs after surface modification (ESI Fig. S13 and S14†). By utilizing the bicinchoninic acid (BCA) protein assay kit, the number of avidin molecules conjugated to each NP was estimated to be ∼1.8 (ESI Fig. S15 and S16†).
By virtue of the specific recognition of the anti-PSA antibody with PSA, we employed avidin-conjugated Lu6O5F8:Eu3+ nanoprobes in a sandwich-type bioassay for the detection of PSA. As illustrated in Fig. 4a and ESI Movie S1,† the capture antibody was first bound to the microplate well, and the biotinylated detection antibody (which was bound to PSA) was coupled to the nanoprobe through biotin–avidin interactions. PSA was quantified by measuring the dissolution-enhanced TRPL signal of the oxyfluoride nanoprobes upon addition of the enhancer solution on a microplate reader. For comparison, control experiments by replacing PSA with bovine serum albumin (BSA) under otherwise identical conditions, were also conducted. These control experiments showed negligible TRPL signal (Fig. 4b), verifying the high specificity. The limit of detection (LOD), defined as the concentration that corresponds to three times the standard deviation above the signal measured in the control experiment, was determined to be 0.52 pg mL−1 (15.2 fM). The derived LOD exhibits an almost 200-fold improvement relative to that of a commercial dissociation-enhanced lanthanide fluoroimmunoassay (DELFIA) kit (0.1 ng mL−1),34 and is the lowest among lanthanide nanoprobes ever reported for the luminescent bioassay of PSA (ESI Table S4†). Such a low LOD is attributed to the high molar density of Eu3+ ions in a single oxyfluoride NP and the high specificity of nanoprobes in the bioassays.
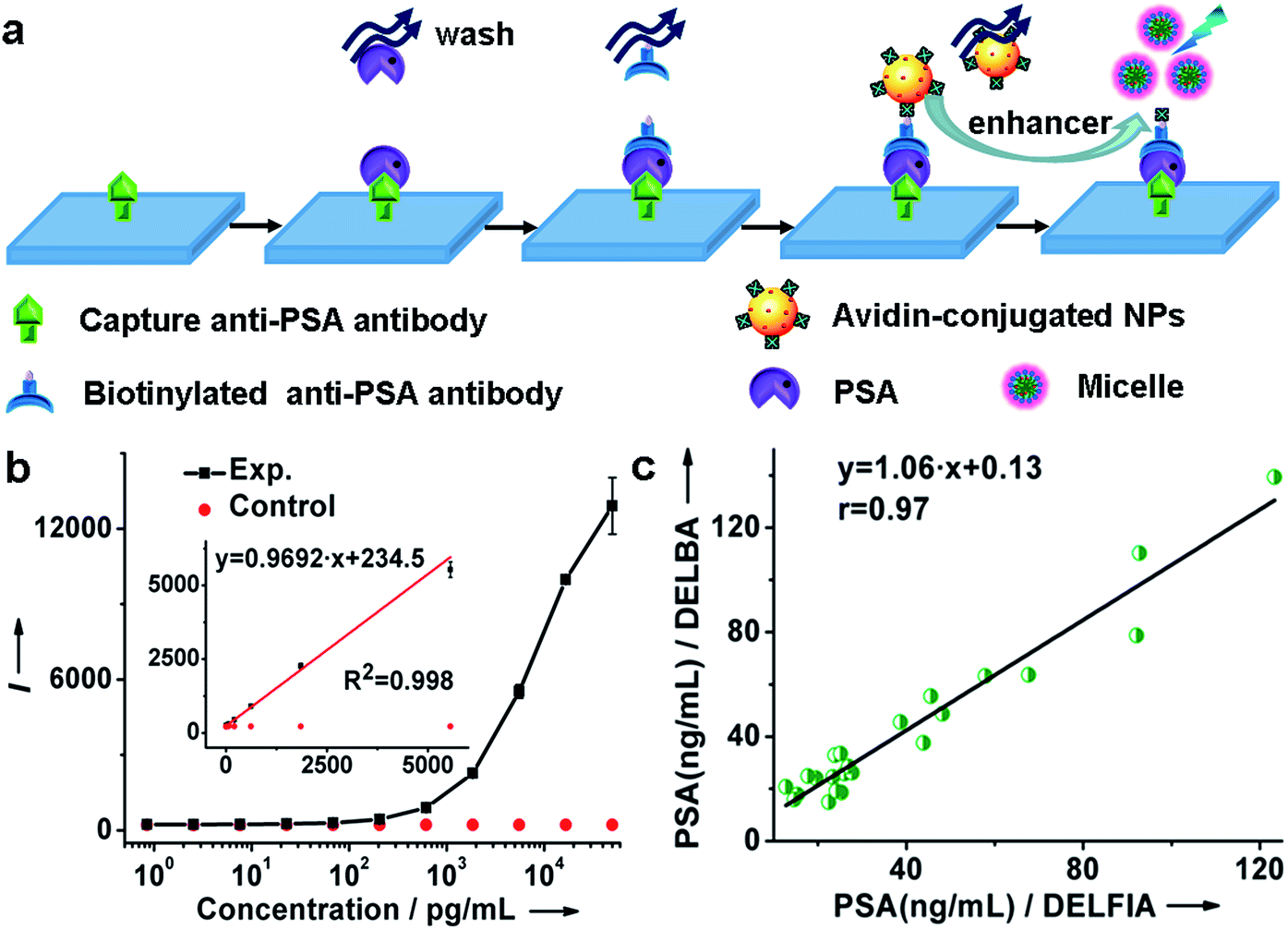 | ||
| Fig. 4 (a) The process and principle of a heterogeneous assay for the detection of PSA. (b) Calibration curve for the PSA assay based on Lu6O5F8:Eu3+ (40 mol%) NPs. Inset: the linear range (8.5 × 10−4 to 5.6 ng mL−1) of the calibration curve with the regression equation of y = 0.9692x + 234.5 (R2 = 0.998). (c) Correlation between the NP-based assay and commercial DELFIA kit for the detection of PSA in 23 patient serum samples. (b and c) Each data point represents the mean (±standard deviation) of triplicate experiments. | ||
It is worth emphasizing that the luminescent bioassay based on ultrasmall oxyfluoride nanoprobes demonstrates an excellent linear dependence of the PL intensity on PSA concentration over a rather wide range, namely, 8.5 × 10−4 to 5.6 ng mL−1 (R2 = 0.998) (Fig. 4b and ESI Fig. S17†). According to Diamandis et al., the PSA levels in ∼60% of patients suffering from PCa decreased to <5 pg mL−1 after radical prostatectomy,35 and a postoperative increase of serum PSA indicates the recurrence or metastasis of cancer.3 Benefiting from the ultrahigh sensitivity of our nanoprobe, such a large linear dynamic range spanning four orders of magnitude for a PSA bioassay might fulfil the critical requirement for tracing the serum PSA level for the early diagnosis of PCa and for monitoring PCa relapse after prostatectomy.
To further verify the applicability of the Lu6O5F8:Eu3+ nanoprobes in practical bioassays for PSA, we firstly studied the response of the oxyfluoride probes towards potential interfering proteins coexisting in serum samples. PSA (300 pM) and noncognate proteins (10 nM), including human serum albumin (HSA), carcinoembryonic antigen (CEA), alpha fetoprotein (AFP), and beta-human chorionic gonadotropin (β-HCG), were added to prepared serum samples from a healthy male with an original PSA level of 0.54 ng mL−1 (15.88 pM) determined by a commercial DELFIA kit, respectively. In the blank control, no targets were added to the samples. As shown in ESI Fig. S18,† the PL signals of all the samples with noncognate proteins added were indistinguishable from that of the blank control. By contrast, when the standard solution of PSA was added, even with its concentration more than 30-fold lower than that of the noncognate protein, a greater than 7 times stronger PL signal was observed. These results indicate that nonspecific binding is negligible, thus confirming the great specificity of our proposed method for the detection of PSA in complex human serum samples. Then, we carried out in vitro detection of PSA in 23 serum samples of patients with PSA levels from 12.00 to 140.00 ng mL−1. The PSA levels determined were compared with those independently detected using a commercial DELFIA kit. As shown in Fig. 4c and Table 1, the PSA levels determined from the DELBA assay based on Lu6O5F8:Eu3+ NPs are consistent with those from the DELFIA. The correlation coefficient between both kinds of assays was determined to be 0.97, indicating that the NPs-based assay results are as reliable as that of the commercial DELFIA. The coefficients of variation (CV) of the Lu6O5F8:Eu3+-based assay ranged from 0.2% to 8.5% (Table 1), showing the assay's good reproducibility. Moreover, we evaluated the analytical accuracy and precision of the Lu6O5F8:Eu3+ bioprobe through the determination of PSA levels, CV, and the recovery of two serum samples from healthy males upon the addition of PSA standard solutions with different concentrations. The CVs of all the assays are below 8% and the analytical recoveries are in the range of 92–108% (Table 2), both of which are within the acceptance criteria (CVs ≤ 15%; recoveries in the range of 90–110%) set for bioanalytical method validation.20 These results verify that the Lu6O5F8:Eu3+ bioprobe has high reliability and practicability for PSA detection.
| No. | NPs-based assay | DELFIA | No. | NPs-based assay | DELFIA | ||
|---|---|---|---|---|---|---|---|
| Mean ± SD (ng mL−1) | CV% | Mean ± SD (ng mL−1) | Mean ± SD (ng mL−1) | CV% | Mean ± SD (ng mL−1) | ||
| 1 | 25.84 ± 1.12 | 4.3 | 26.03 ± 0.50 | 13 | 37.61 ± 0.83 | 2.2 | 43.88 ± 0.38 |
| 2 | 24.06 ± 0.22 | 0.9 | 19.60 ± 0.43 | 14 | 45.60 ± 0.98 | 2.1 | 38.69 ± 0.60 |
| 3 | 17.75 ± 0.39 | 2.2 | 15.36 ± 0.27 | 15 | 110.32 ± 0.63 | 0.6 | 92.76 ± 0.70 |
| 4 | 63.20 ± 1.77 | 2.8 | 57.86 ± 1.01 | 16 | 55.49 ± 1.31 | 2.4 | 45.56 ± 0.67 |
| 5 | 20.72 ± 0.94 | 4.5 | 12.78 ± 0.65 | 17 | 32.83 ± 1.85 | 5.6 | 23.86 ± 0.44 |
| 6 | 139.39 ± 1.89 | 1.4 | 123.32 ± 0.79 | 18 | 24.86 ± 1.21 | 4.9 | 17.74 ± 0.16 |
| 7 | 63.64 ± 0.68 | 1.1 | 67.65 ± 0.62 | 19 | 15.92 ± 1.35 | 8.5 | 14.66 ± 0.31 |
| 8 | 28.58 ± 0.54 | 1.9 | 26.93 ± 0.61 | 20 | 19.20 ± 0.53 | 2.8 | 24.12 ± 0.29 |
| 9 | 78.67 ± 1.16 | 1.5 | 92.09 ± 0.96 | 21 | 26.22 ± 0.06 | 0.2 | 27.74 ± 0.35 |
| 10 | 18.64 ± 1.48 | 7.9 | 25.26 ± 0.25 | 22 | 33.42 ± 0.08 | 0.2 | 25.15 ± 0.56 |
| 11 | 14.82 ± 0.40 | 2.7 | 22.44 ± 0.48 | 23 | 24.50 ± 1.21 | 4.9 | 23.40 ± 0.32 |
| 12 | 48.73 ± 0.54 | 1.1 | 48.20 ± 0.82 | ||||
| Added (ng mL−1) | Found (ng mL−1) | CV (%) n = 4 | Recovery (%) |
|---|---|---|---|
| Sample 1 | 2.99 | 3.57 | — |
| 2.50 | 5.65 | 3.98 | 106.51 |
| 3.00 | 5.78 | 2.13 | 93.24 |
| 12.00 | 15.87 | 1.06 | 107.36 |
| Sample 2 | 1.83 | 5.09 | — |
| 1.00 | 2.81 | 7.36 | 98.17 |
| 7.00 | 9.01 | 2.88 | 102.59 |
| 28.00 | 27.76 | 2.03 | 92.60 |
To demonstrate the multifunctionality of the oxyfluoride nanoprobes, we also employed ultrasmall Lu6O5F8:Yb/Er NPs in proof-of-concept UCL and CT bioimaging, in view of the high R/G UC emissions and the high mass density of the Lu6O5F8:Yb/Er NPs (∼9 g cm−3). After incubation with human lung cancer (H1299) cells, strong red UC emissions of Er3+ were clearly visualized in the cells upon 980 nm irradiation, in sharp contrast to the weak green luminescence observed in the dark (ESI Fig. S19a†), which agrees well with the corresponding nearly single-band UC spectrum (Fig. 2e). In addition, as a proof-of-concept experiment, we recorded in vitro color-mapped CT images and measured the CT values using an aqueous solution of Lu6O5F8:Yb/Er NPs. The CT value of NPs at 12.5 mg mL−1 was 319 Hounsfield units (HU), which is much higher than that of LiLuF4 NPs (181 HU) and commercial iopromide (186 HU) at the same concentration (ESI Fig. S19b and c†).36 To further evaluate the cytotoxicity of the citrate-capped Lu6O5F8:Yb/Er NPs for potential bioimaging applications, the dark/photo toxicity of the NPs were measured against HELF cells using a standard methylthiazolyltetrazolium (MTT) assay. There is no significant cytotoxicity of the NPs (ESI Fig. S20†), even at a high concentration of 1 mg mL−1, either in the dark or upon 980 nm irradiation for 2 min. These results demonstrate the potential of the UC NPs as bioprobes for UCL/CT dual-modal bioimaging.
Conclusions
In summary, we have developed sub-5 nm Lu6O5F8:Eu3+ NPs as the first inorganic oxyfluoride nanoprobe for the detection of PSA. Because of the high molar density of Ln3+ ions in the Lu6O5F8 NPs, with superior dissolution capability in the enhancer solution and the minimized non-specific binding of the NPs in TRPL bioassays, we have achieved an exceptionally large linear dynamic range spanning from 0.85 pg mL−1 to 5.6 ng mL−1, along with a LOD as low as to 0.52 pg mL−1, which is more than two orders of magnitude lower than that of a commercial DELFIA kit. More importantly, we have successfully employed the ultrasmall nanoprobes for the detection of PSA in 23 clinical serum samples of patients suffering from PCa, and the assay results were highly consistent with those measured independently by DELFIA kit, showing the assay's reliability with a correlation coefficient of 0.97. These findings demonstrate the great potential of such a nano-bioprobe in the practical in vitro detection of tumor markers, and particularly, for monitoring PCa relapse of patients after radical prostatectomy, which may eventually open up a new route to the exploitation of oxyfluoride nanoprobes in versatile biomedical applications.Acknowledgements
This work is supported by the 973 program of MOST (No. 2014CB845605), the NSFC (Nos. U1305244, U1405229, 21325104, 11304314 and 21501180), the CAS/SAFEA International Partnership Program for Creative Research Teams, Strategic Priority Research Program of the CAS (No. XDA09030307), and Fujian Provincial Natural Science Foundation for Young Scientists (No. 2015J05116).Notes and references
- H. W. Yang, C. W. Lin, M. Y. Hua, S. S. Liao, Y. T. Chen, H. C. Chen, W. H. Weng, C. K. Chuang, S. T. Pang and C. C. M. Ma, Adv. Mater., 2014, 26, 3662–3666 CrossRef CAS PubMed.
- T. J. Polascik, J. E. Oesterling and A. W. Partin, J. Urol., 1999, 162, 293–306 CrossRef CAS PubMed.
- H. Yu, E. P. Diamandis, A. F. Prestigiacomo and T. A. Stamey, Clin. Chem., 1995, 41, 430–434 CAS.
- D. B. Liu, X. L. Huang, Z. T. Wang, A. Jin, X. L. Sun, L. Zhu, F. Wang, Y. Ma, G. Niu, A. R. H. Walker and X. Y. Chen, ACS Nano, 2013, 7, 5568–5576 CrossRef CAS PubMed.
- B. Acevedo, Y. Perera, M. Ruiz, G. Rojas, J. Benítez, M. Ayala and J. Gavilondo, Clin. Chim. Acta, 2002, 317, 55–63 CrossRef CAS.
- J. Vuojola and T. Soukka, Methods Appl. Fluoresc., 2014, 2, 012001 CrossRef.
- M. Q. Tan, Z. Q. Ye, G. L. Wang and J. L. Yuan, Chem. Mater., 2004, 16, 2494–2498 CrossRef CAS.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- G. F. Wang, Q. Peng and Y. D. Li, Acc. Chem. Res., 2011, 44, 322–332 CrossRef CAS PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Y. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- W. Zheng, P. Huang, D. T. Tu, E. Ma, H. M. Zhu and X. Y. Chen, Chem. Soc. Rev., 2015, 44, 1379–1415 RSC.
- Y. Y. Liu, Y. Liu, W. B. Bu, C. Cheng, C. J. Zuo, Q. F. Xiao, Y. Sun, D. L. Ni, C. Zhang, J. A. Liu and J. L. Shi, Angew. Chem., Int. Ed., 2015, 54, 8105–8109 CrossRef CAS PubMed.
- H. Dong, L. D. Sun and C. H. Yan, Chem. Soc. Rev., 2015, 44, 1608–1634 RSC.
- J. N. Liu, J. W. Bu, W. B. Bu, S. J. Zhang, L. M. Pan, W. P. Fan, F. Chen, L. P. Zhou, W. J. Peng, K. L. Zhao, J. L. Du and J. L. Shi, Angew. Chem., Int. Ed., 2014, 53, 4551–4555 CrossRef CAS PubMed.
- J. N. Liu, Y. Liu, W. B. Bu, J. W. Bu, Y. Sun, J. L. Du and J. L. Shi, J. Am. Chem. Soc., 2014, 136, 9701–9709 CrossRef CAS PubMed.
- L. Zhou, R. Wang, C. Yao, X. M. Li, C. L. Wang, X. Y. Zhang, C. J. Xu, A. J. Zeng, D. Y. Zhao and F. Zhang, Nat. Commun., 2015, 6, 6938 CrossRef CAS PubMed.
- J. C. G. Bünzli and S. V. Eliseeva, Chem. Sci., 2013, 4, 1939–1949 RSC.
- W. P. Fan, B. Shen, W. B. Bu, X. P. Zheng, Q. J. He, Z. W. Cui, K. L. Zhao, S. J. Zhang and J. L. Shi, Chem. Sci., 2015, 6, 1747–1753 RSC.
- G. Rainò, T. Stöferle, C. Park, H. C. Kim, T. Topuria, P. M. Rice, I. J. Chin, R. D. Miller and R. F. Mahrt, ACS Nano, 2011, 5, 3536–3541 CrossRef PubMed.
- S. Y. Zhou, W. Zheng, Z. Chen, D. T. Tu, Y. S. Liu, E. Ma, R. F. Li, H. M. Zhu, M. D. Huang and X. Y. Chen, Angew. Chem., Int. Ed., 2014, 53, 12498–12502 CAS.
- D. Panias, M. Taxiarchou, I. Paspaliaris and A. Kontopoulos, Hydrometallurgy, 1996, 42, 257–265 CrossRef CAS.
- X. Sun, Y. W. Zhang, Y. P. Du, Z. G. Yan, R. Si, L. P. You and C. H. Yan, Chem.–Eur. J., 2007, 13, 2320–2332 CrossRef CAS PubMed.
- P. A. Tanner, Chem. Soc. Rev., 2013, 42, 5090–5101 RSC.
- T. Passuello, F. Piccinelli, M. Trevisani, M. Giarola, G. Mariotto, L. Marciniak, D. Hreniak, M. Guzik, M. Fasoli, A. Vedda, V. Jary, M. Nikl, V. Causin, M. Bettinelli and A. Speghini, J. Mater. Chem., 2012, 22, 10639–10649 RSC.
- H. Schäfer, P. Ptacek, K. Kömpe and M. Haase, Chem. Mater., 2007, 19, 1396–1400 CrossRef.
- H. Schäfer, P. Ptacek, O. Zerzouf and M. Haase, Adv. Funct. Mater., 2008, 18, 2913–2918 CrossRef.
- H. Schäfer, P. Ptacek, H. Eickmeier and M. Haase, Adv. Funct. Mater., 2009, 19, 3091–3097 CrossRef.
- F. Wang and X. G. Liu, J. Am. Chem. Soc., 2008, 130, 5642 CrossRef CAS PubMed.
- R. C. Lv, P. P. Yang, F. He, S. L. Gai, C. X. Li, Y. L. Dai, G. X. Yang and J. Lin, ACS Nano, 2015, 9, 1630–1647 CrossRef CAS PubMed.
- N. Bogdan, F. Vetrone, G. A. Ozin and J. A. Capobianco, Nano Lett., 2011, 11, 835–840 CrossRef CAS PubMed.
- J. C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- M. L. Deng and L. Y. Wang, Nano Res., 2014, 7, 782–793 CrossRef CAS.
- S. Fang, C. Wang, J. Xiang, L. Cheng, X. J. Song, L. G. Xu, R. Peng and Z. Liu, Nano Res., 2014, 7, 1327–1336 CrossRef CAS.
- K. Järås, B. Adler, A. Tojo, J. Malm, G. Marko-Varga, H. Lilja and T. Laurell, Clin. Chim. Acta, 2012, 414, 76–84 CrossRef PubMed.
- E. P. Diamandis, H. Yu and D. N. Melegos, Clin. Chem., 1996, 42, 853–857 CAS.
- P. Huang, W. Zheng, S. Y. Zhou, D. T. Tu, Z. Chen, H. M. Zhu, R. F. Li, E. Ma, M. D. Huang and X. Y. Chen, Angew. Chem., Int. Ed., 2014, 53, 1252–1257 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI, Tables S1–S4, Fig. S1–S20 and Movie S1. See DOI: 10.1039/c5sc04599a |
| This journal is © The Royal Society of Chemistry 2016 |