
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic structure and reactivity of nickel(I) pincer complexes: their aerobic transformation to peroxo species and site selective C–H oxygenation†
Christoph A.
Rettenmeier
,
Hubert
Wadepohl
and
Lutz H.
Gade
*
Anorganisch-Chemisches Institut, University of Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany. E-mail: lutz.gade@uni-hd.de
First published on 11th February 2016
Abstract
The study is aimed at a deeper understanding of the electronic structure of the T-shaped nickel(I) complex [LigiPr(iso)Ni] (1b), bearing the iso-PyrrMeBox (bis(oxazolinylmethylidene)pyrrolidinido) pincer ligand, and its CO adduct [LigiPr(iso)Ni(CO)] (2b) as well as to provide insight into the mechanism of autoxidation of the different nickel peroxo species of this ligand type. CO was found to react reversibly with complex 1b resulting in the corresponding CO adduct 2b. The EPR data as well as the results of DFT modeling revealed significant differences in the electronic structure of 1b and 2b. Reaction of [LigPh(iso)Ni] and [LigiPr(iso)Ni] (1a and b) with dioxygen yielded the 1,2-μ-peroxo complexes [Lig(iso)NiO]23a and b which reacted with hydrogen peroxide to give the hydroperoxo complexes [Lig(iso)NiOOH] 5a and b. Thermal aerobic decomposition of the peroxo species 3a and 5a in the presence of O2 led to a C–H activation of the ligand at the benzylic position of the oxazoline ring forming diastereomeric cyclic peroxo complexes 6 and 6′. For the 1,2-μ-peroxo complex 3b the autoxidation of the pincer in the absence of O2 occurred at the tertiary C–H bond of the iPr-group and led to a selective formation of the terminal hydroxo complex [LigiPr(iso)NiOH] 7b and the cyclic alkoxy complex 8 in equimolar quantities, while the corresponding cyclic peroxo species 9 was formed along with 7b in the presence of oxygen. Whether or not O–O bond cleavage occurred in the generation of 9 was established upon performing labeling experiments which indicate that the transformation does not involve an initial O–O bond cleaving step. Based on these observations and a series of stoichiometric transformations a tentative proposal for the processes involved in the anaerobic and aerobic decomposition of 3b has been put forward. Finally, the nickel(II) methyl complex [LigPh(iso)NiMe] 14 reacted with O2 to give the methylperoxo complex [LigPh(iso)NiOOMe] 15 which slowly converted to a mixture of near equal amounts of the formato and the hydroxo complexes, [LigPh(iso)NiOOCH] 16 and [LigPh(iso)NiOH] 7a, along with half an equivalent of methanol. The formato complex 16 itself decomposed at elevated temperatures to CO2, dihydrogen as well as the nickel(I) species 1a.
Introduction
Redox active nickel containing enzymes such as [Ni/Fe] hydrogenase,1,2 acetyl CoA synthase (ACS),3,4 CO dehydrogenase and methyl coenzyme M reductase (MCR)5,6 are mainly found in biological systems existing under anaerobic conditions where these nickel enzymes play a crucial role in methanogenesis.7,8 In contrast to the diverse redox chemistry of nickel enzymes under anaerobic conditions, the importance of nickel in enzymatic, aerobic redox processes is limited to those of nickel containing superoxide dismutase (SOD).9,10 Whether nickel oxygen intermediates are involved in the catalytic cycle is currently under debate.11,12Reactive nickel oxo, peroxo and superoxo intermediates have been synthesized in recent years via the direct reaction of nickel(I) complexes with oxygen.13–21 Such nickel oxygen species were found to be capable to intra-22–28 and intermolecularly13,29–33 activate C–H bonds.
The monoanionic iso-PyrrMeBox ligands developed in our group34,35 bear key structural features found in the hydrocorphin system of cofactor F430 of methyl coenzyme M reductase (Fig. 1). Both ligand systems contain an almost identical delocalized 10-electron-π-system involving three nitrogen donor atoms and unsaturated carbon-linkers which interacts with the central metal ion upon its coordination.36
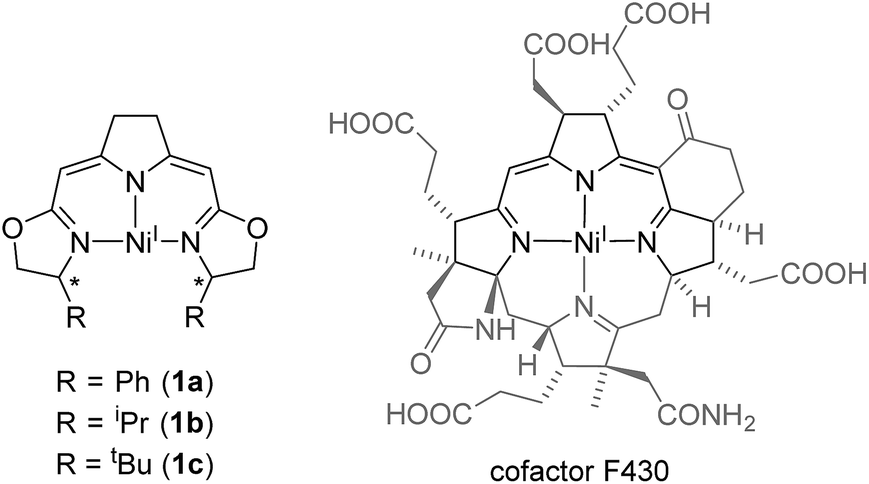 | ||
| Fig. 1 Comparison of nickel(I) complexes bearing the monoanionic iso-PyrrMeBox pincer ligands (left) and the hydrocorphin system found in cofactor F430 (right).6 | ||
While both the F430-hydrocorphin and the iso-PyrrMeBox ligands are capable of stabilizing nickel(I) complexes (Fig. 1), the absence of the fourth donor function of the iso-PyrrMeBox ligand leads to complexes with an available coordination site in the ligand plane in close proximity to the chiral centers of the oxazoline rings. This combination of electronic and structural features makes the iso-PyrrMeBox ligand an ideal candidate for the systematic investigation of its reactivity in catalysis and at the same time opens up the possibility to isolate reactive intermediates.37–39
Recently, we reported the synthesis of the first nickel hydroperoxo complex characterized to date as well as rare nickel-1,2-μ-peroxo complexes using this iso-PyrrMeBox ligand.37 The current work is aimed at a deeper understanding of the electronic structure of the T-shaped nickel(I) pincer complexes, how it is modified by the coordination at the fourth in-plane coordination site and to provide insight into the mechanism of autoxidation of the different nickel peroxo species of this ligand type.
Results and discussion
Coordination at the free coordination site of the T-shaped nickel(I) complexes and its impact on the electronic structure
The electron rich nickel(I) complex 1 showed little tendency to coordinate small molecules such as N2 or CO2 or classical donor ligands such as THF or N-heterocycles at the available fourth coordination site, and no such derivatives were found to be isolable or even detectible. This observation raised the question whether the occupation of the fourth coordination site by the pyrroline donor in the cofactor F430 is enforced by the macrocyclic hydrocorphin (Fig. 1), thus giving rise to only a minor additional metal ligand stabilization.On the other hand, the strong π-acceptor CO was found to react reversibly with the T-shaped nickel(I) complex 1b resulting in a CO pressure dependent equilibrium between the threefold-coordinated nickel(I) species 1b and the corresponding CO adduct 2b (Scheme 1). Exposure of solutions of 2b to an evaporated headspace led to the complete loss of CO, reforming the nickel(I) complex 1b. We note that nickel(I) carbonyl species are believed to be involved in the acetyl-CoA synthesis by ACS as part of the Wood–Ljungdahl pathway in methanogenesis40,41 and it has been shown recently that PNP pincer complexes were able to mimic a key step, namely the methyl transfer onto the carbonyl species to form the corresponding acetyl complex.42
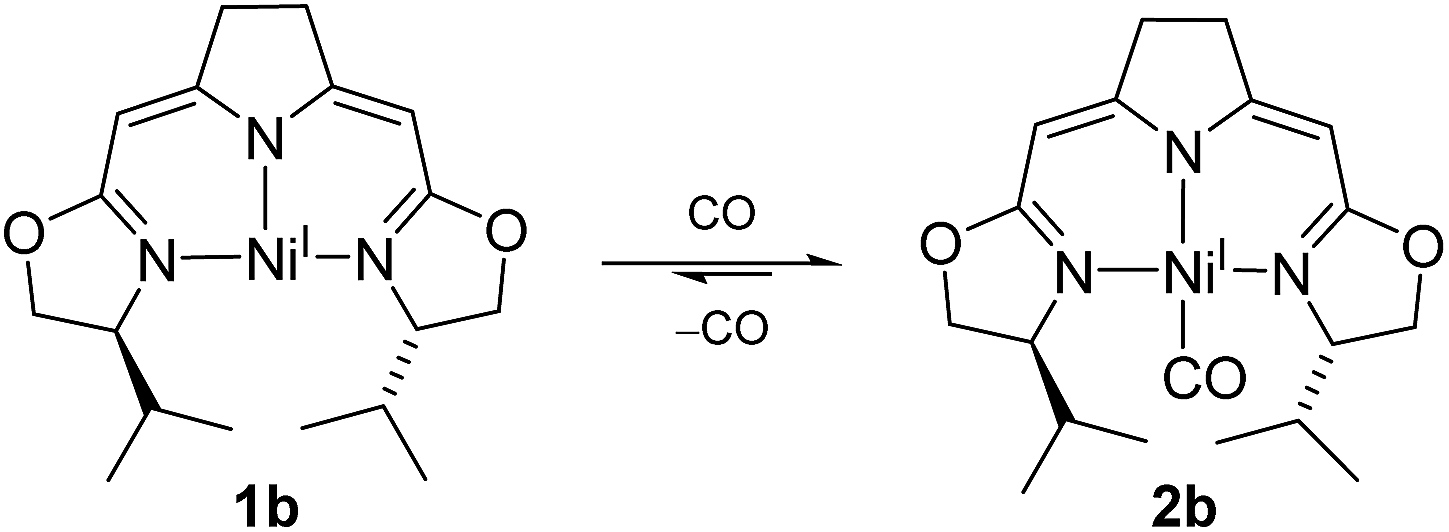 | ||
| Scheme 1 CO dependent equilibrium between nickel(I) complexes 1b and 2b. | ||
The coordination of CO to the nickel(I) center is accompanied by a change in color from red to green so that the reversible reaction can be monitored by UV/Vis and IR spectroscopy (ESI†). The characteristic band of the CO vibration was found at 1955 cm−1 in the IR spectrum (KBr) of the precipitate obtained from the equilibrium mixture at 10 bar CO at −78 °C indicating moderate back-bonding from the nickel(I) center to the CO ligand.
Interestingly, a very similar scenario of a reversible CO coordination to a T-shaped nickel(I) species with analogous electronic changes had been described by Caulton and coworkers for a PNP pincer system bearing two Si atoms in the backbone of the ligand.43 In their case and other related pincer complexes, structural data by X-ray analysis of the CO adducts were obtained.42,43 The main geometric features of this as well as Lee's more recent example are analogous to those of the DFT optimized structure of 2b (Fig. 2 and ESI†).
 | ||
Fig. 2 Top: EPR spectra of nickel(I) complexes 1b and 2b (963![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 284 GHz, toluene, 30 K) and the corresponding anisotropic g values and coupling constants a. Bottom: DFT calculated (ROB3LYP/6-311G(d,p))45–57 SOMOs (isovalue: 0.04) of nickel(I) complexes 1b and 2b and the corresponding atomic contributions (threshold 0.01). 284 GHz, toluene, 30 K) and the corresponding anisotropic g values and coupling constants a. Bottom: DFT calculated (ROB3LYP/6-311G(d,p))45–57 SOMOs (isovalue: 0.04) of nickel(I) complexes 1b and 2b and the corresponding atomic contributions (threshold 0.01). | ||
In order to obtain insight into the degree to which this occupation of the fourth coordination site by the carbonyl ligand modified the electronic structure of the complex, ESR spectra of 2b were recorded under a pressure of 10 bar CO. The EPR spectrum of 2b recorded at 30 K displayed one dominant signal with rhombic symmetry (gx = 2.022, gy = 2.111, gz = 2.171; cofactor F430:44gII = 2.224, g⊥ = 2.061) which differed from the resonance of the T-shaped nickel(I) complex 1b (gx = 2.041, gy = 2.123, gz = 2.311, Fig. 2). In comparison to the EPR signal of the latter a significant shift of the gz value to higher field was observed when the fourth coordination site was occupied by the CO ligand. Furthermore, significant differences in the superhyperfine coupling to the N atoms were observed. While a triplet superhyperfine splitting of the x component of the signal caused by the central pyrrolidine N atom was observed in the EPR spectrum of complex 1b,39 the coupling of all three N donor atoms with the unpaired electron is resolved in the case of the CO adduct 2b leading to a more complicated coupling pattern (Fig. 2). Assuming C2-symmetry for 2b (which is slightly broken by the out-of-plane-coordination mode of the CO ligand), coupling to the central N-atom (N(central)) as well as to two (near)-equivalent oxazoline N-atoms (N(oxazoline)) accounts for a maximum multiplicity of 15 (triplet of quintet). Due to the different superhyperfine coupling constants at hand, however, superpositioning leads to the pattern that is actually observed experimentally which could be successfully simulated (values are given in Fig. 2, the simulated spectrum is depicted in the ESI†).
The superhyperfine splitting in the EPR spectra of 1b and 2b are well reflected in the results of restricted open shell DFT calculations. The isotropic Fermi contact couplings indicate a significantly higher spin density at the two oxazoline N cores in the CO adduct than in the T-shaped nickel(I) complex [(1b) aN(oxazoline) 2.4 G and 2.4 G, aN(central) 10.2 G (2b); aN(oxazoline) = 9.5 G and 8.7 G, aN(central) = 6.7 G]. Both complexes are best described as nickel(I) species (calc. Mulliken spin densities: (1b) Ni 0.849; (2b) Ni 0.821) in which the unpaired electron resides in molecular orbitals with dominant dx2−y2 character. The SOMOs of complexes 1b and 2b are depicted in Fig. 2 along with the major atomic contributions to each orbital.
The arrangement of the iPr-substituent found in the modulated minimum structure of 1b causes one of the methyl C–H moieties to be located in close proximity to the nickel(I) center (DFT: d(C–H⋯Ni) = 2.232 Å, d(C–H⋯Ni) = 3.097 Å, d(C–H) = 1.097 Å, X-ray:58d(C–H⋯Ni) = 2.38(3) Å (refined H-atom))59 In this set-up the C–H group appears to be interacting with the nickel(I) center with a considerable amount of spin density located at the C–H bond (Fig. 2). Indeed, the corresponding C–H bond is slightly elongated compared to the residual average over both iPr-substituents (d(C–H)av = 1.094 Å with σ = 0.001 (DFT)). However, from an energetic perspective the orientation of the iPr-substituent is mainly a result of minimizing steric repulsion between vicinal groups. Therefore, a staggered conformation for all dihedral angles of the substituent is preferred (ESI†). In contrast to 2b and all other fourfold coordinated complexes of this type analyzed so far, the available coordination site in 1b allows an iPr-substituent to adopt a conformation in which one of the methyl groups is located in the open coordination site at the nickel center. The same orientation is observed in the minimum structure of the metal-free iPr-oxazoline itself with only small differences in the rotational barriers (RB3LYP/6-311(d,p), ESI†). A possible effect on the H NMR shifts was not observed and is expected to be very small due to the rapid exchange of all 12 (6 + 6) methyl proton signals of the iPr-group even at low temperature. Furthermore, no superhyperfine coupling to the proton is resolved in the EPR spectrum of 1b (Fig. 2, Fermi contact coupling C 5.0 G, H 1.4 G).
Occupation of the fourth coordination site in the plane of 2b by the π-acceptor CO thus leads to a significant change in the electronic properties of the complexes. While the g tensor of 1b is highly anisotropic the occupation of the fourth coordination site reduces the anisotropy significantly towards an axial symmetry as observed for the cofactor F430.44 A main feature of the T-shaped nickel(I) complex 1b is the high amount of unpaired electron density at the empty coordination site of the nickel center, which makes the system an attractive candidate for the activation of small molecules/C–X bonds and/in catalysis. This also renders such three-coordinate Ni complexes interesting objects of study for the interaction with O2 and resulting oxidation and autoxidation processes.
Synthesis of peroxo intermediates by the reaction of nickel(I) with oxygen and their thermal decomposition
We previously reported the reaction of nickel(I) complexes 1a and b with oxygen at low temperature which leads to 1,2-μ-peroxo complexes 3a and b (Scheme 2).37 These bulky dinuclear species were found to exist in an oxygen pressure dependent equilibrium with the corresponding mononuclear paramagnetic superoxo complexes 4a and b.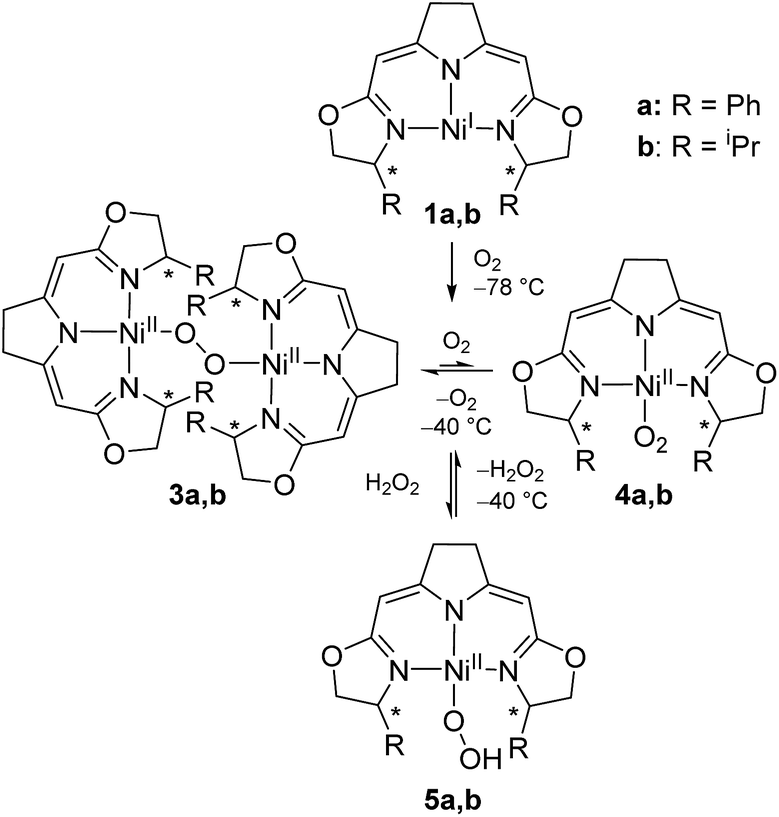 | ||
| Scheme 2 Aerobic formation of the different nickel(II) peroxo complexes 3a and b, superoxo complexes 4a and b and hydroperoxo complexes 5a and b at low temperatures. | ||
The addition of hydrogen peroxide to a solution of 1,2-μ-peroxo complexes 3a and b gave the hydroperoxo complexes 5a and b, of which complex 5a, bearing the phenyl-substituted pincer ligand, could be isolated and structurally characterized by X-ray diffraction.37 However, we subsequently observed that the hydroperoxo complex bearing the iPr-substituted pincer ligand 5b was unstable in the absence of hydrogen peroxide even at low temperature (−20 °C) and upon attempted isolation its transformation back to the 1,2-μ-peroxo complex 3b occurred.
Thermal decomposition of the hydroperoxo complex 5a
As reported previously,37 the thermal aerobic decomposition of hydroperoxo complex 5a leads to a C–H activation of the ligand at the benzylic position of the oxazoline ring forming cyclic peroxo complexes 6 and 6′ (Scheme 3). The reaction involves the chiral carbon center, and a partial racemization of its configuration takes place during the transformation leading to two diastereomeric species. The loss of stereoinformation is attributed to the intermediate formation of a configurationally labile benzylic radical which then further reacts with O2 in a diastereoselective manner to the products 6 and 6′ in a ratio of 1 to 2.3. Furthermore, slow formation of the mixture of the same diastereomers 6 and 6′ occurred when a solution of the hydroxo complex 7a was stirred under an atmosphere of oxygen.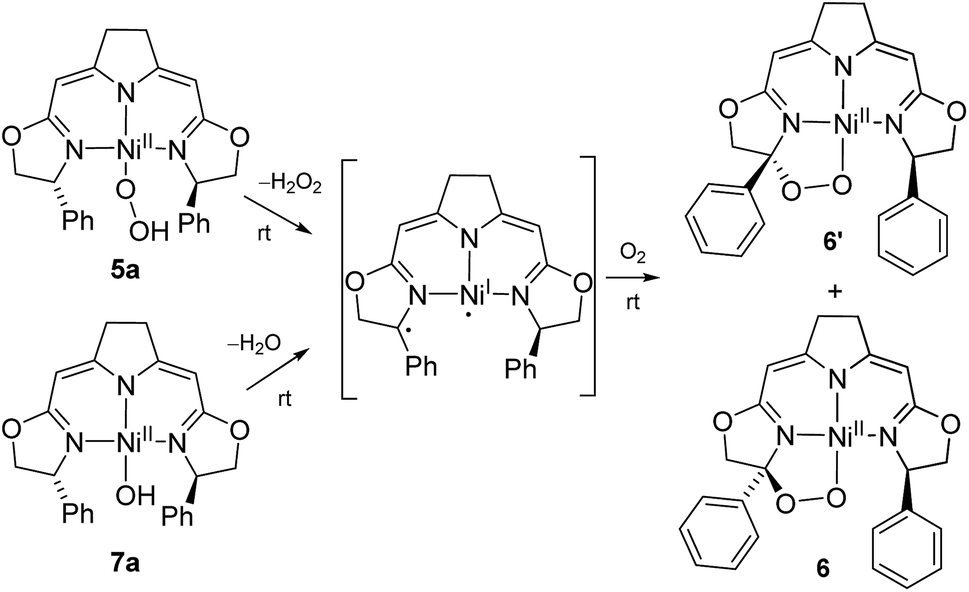 | ||
| Scheme 3 Mechanistic proposal for the formation of the cyclic peroxo complexes 6/6′ in the aerobic degradation of the nickel(II) peroxo and hydroxo complexes 5a and 7a at room temperature. | ||
Since these earlier observations were pertinent to the reactions studied in this work we aimed to substantiate our proposal of a formal O2 insertion into a diradical species.37 To this end we carried out labeling experiments which showed that, as proposed previously, in both cases the oxygen atoms of the cyclic peroxo species 6 and 6′ originate from the molecular oxygen present in the reaction mixture (Scheme 4). The thermal decomposition of the 18O-labeled hydroperoxo complex 5a[18O] in the presence of 16O2 led exclusively to the homo-isotopologous species bearing two 16O atoms. Similarly, when a solution of the hydroxo complex 7a was held under an atmosphere of 16O2/18O2 the homo-isotopologous peroxo species 6 and 6′ were exclusively obtained. Possible scrambling of the O2 fragment indicating a homolytic O–O bond dissociation in the course of the transformation was not observed.
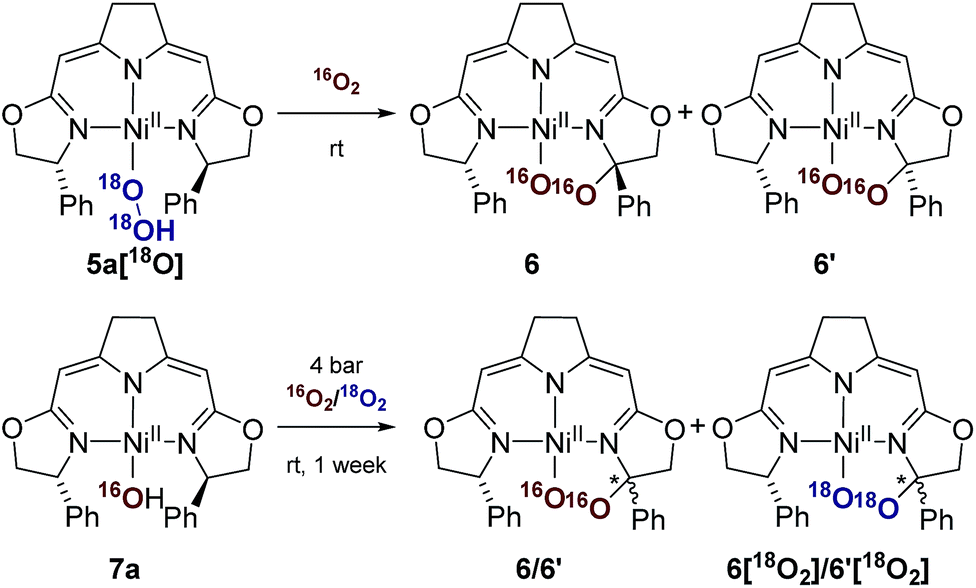 | ||
| Scheme 4 Labeling experiments in the aerobic thermal decomposition of the hydroperoxo and the hydroxo complexes 5a and 7a at room temperature. | ||
Thermal decomposition of the 1,2-μ-peroxo complexes
In contrast to the hydroperoxo complex 5a containing the Ph-substituted iso-PyrrMeBox pincer, which could be isolated and fully characterized, the iPr-substituted hydroperoxo complex 5b was found to convert to the corresponding 1,2-μ-peroxo complex 3b even at low temperature. This led us to investigate the thermal autoxidative transformation of this 1,2-μ-peroxo complex 3b (Scheme 5).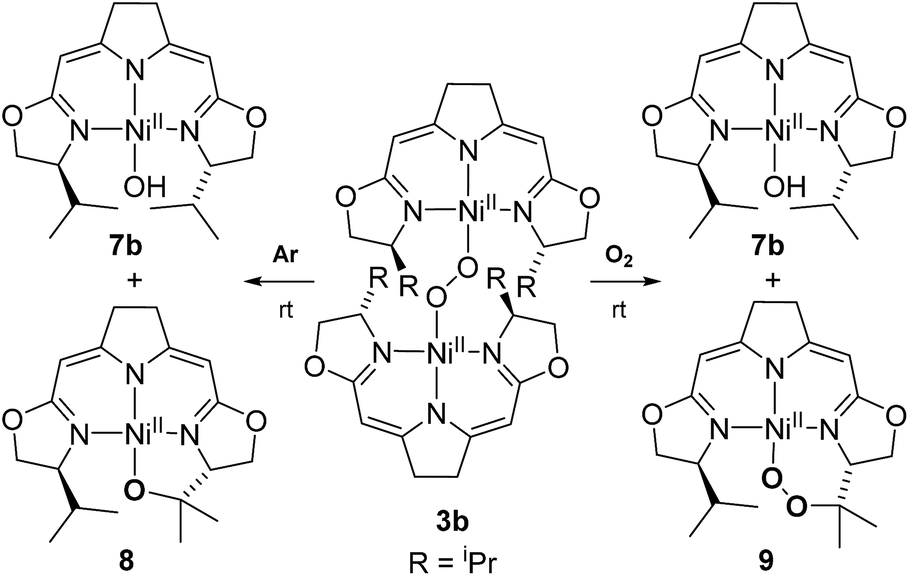 | ||
| Scheme 5 Thermal decomposition of 1,2-μ-peroxo complex 3b in the absence and presence of oxygen. | ||
For the 1,2-μ-peroxo complex 3b the autoxidation of the pincer occurred at the tertiary C–H bond of the iPr-group instead of the C–H group adjacent to the nitrogen donor of the oxazoline ring. Under anaerobic conditions decomposition of 3b led to a selective formation of the terminal hydroxo complex 7b and the cyclic alkoxy complex 8 in equimolar quantities. However, in the presence of O2 the corresponding cyclic peroxo species 9 was formed together with the hydroxo complex 7b, again in a 1:1 ratio. We note that a similar reactive behaviour had been observed for the autoxidation of the bis(μ-oxo)dinickel(III) complex bearing the Me3-tpa ligand (tpa = tris(6-methyl-2-pyridylmethyl)amine).24,25,27
Both metallacycles were isolated and structurally characterized by X-ray diffraction (Fig. 3). In both complexes a distorted square-planar coordination sphere of the nickel center is found. The prominent structural motif of complex 8 is an oxazametallacycle, which adopts a distorted envelope conformation. Due to the geometric constraints of the five-membered ring the N3–Ni–O3 angle [84,34(9)°] is narrowed compared to the ideal square-planar geometry resulting in an in-plane-distortion of the whole coordination sphere around the nickel center.
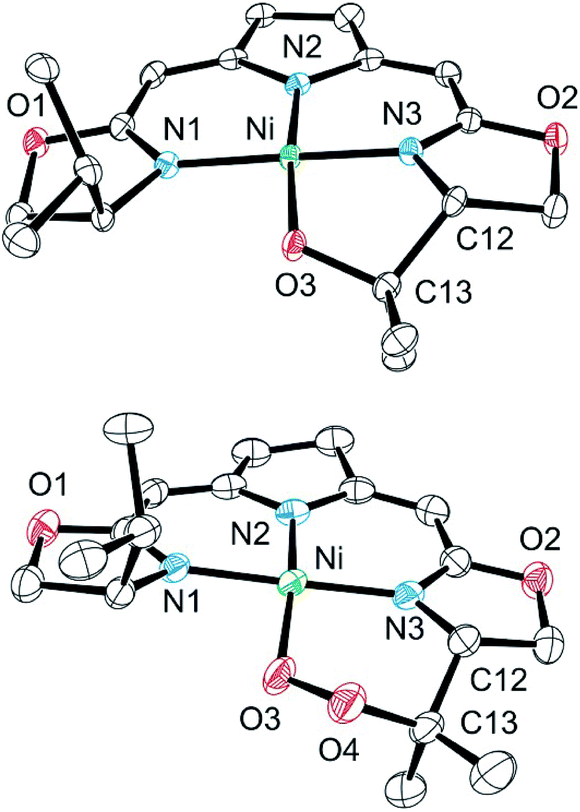 | ||
| Fig. 3 Molecular structure of 8 (top) and 9 (bottom). Hydrogen atoms were omitted for clarity. Selected bond lengths [Å] and angles [°]: 8: Ni–N(3) 1.811(3), Ni–O(3) 1.857(2), Ni–N(1) 1.868(3), Ni–N(2) 1.903(2), N(3)–Ni–O(3) 84.34(10), N(3)–Ni–N(1) 174.40(11), O(3)–Ni–N(1) 90.90(10), N(3)–Ni–N(2) 90.79(11), O(3)–Ni–N(2) 173.21(10), N(1)–Ni–N(2) 94.19(11); 9: Ni–O(3) 1.842(3), Ni–N(1) 1.890(3), Ni–N(2) 1.920(3), Ni–N(3) 1.868(3), O(3)–O(4) 1.443(4), O(3)–Ni–N(1) 84.41(12), O(3)–Ni–N(2) 166.68(14), O(3)–Ni–N(3) 92.10(12), N(1)–Ni–N(2) 92.06(13), N(1)–Ni–N(3) 173.17(13), N(3)–Ni–N(2) 92.63(13). | ||
However, the additional oxygen atom in the cyclic peroxo complex 9 leads to a larger six membered dioxazametallacyle. Thus, a wider N3–Ni–O3 angle [92,10(12)°] is observed resulting in-plane-distortion of the square planar coordination geometry in the opposite sense [sum of the N2–Ni–N3 and N3–Ni–O3 angle 184,73°], as well as a slight out–of plane displacement of the oxygen atom bound to the nickel center. The O–O bond length was found to be 1.443(4) Å.
The formation of 9 from the aerobic degradation of the dinuclear peroxo complex 3b mirrors the generation of the cyclic peroxide 6/6′ from hydroperoxo species 5a. It was therefore of interest to probe for analogous reactivity of the corresponding peroxo species 3a. Indeed, this dinuclear complex, containing the Ph-substituted pincer cleanly converted to a 1:1 mixture of the cyclic alkyl peroxide 6/6′ and hydroxo complex 7a (Scheme 6).
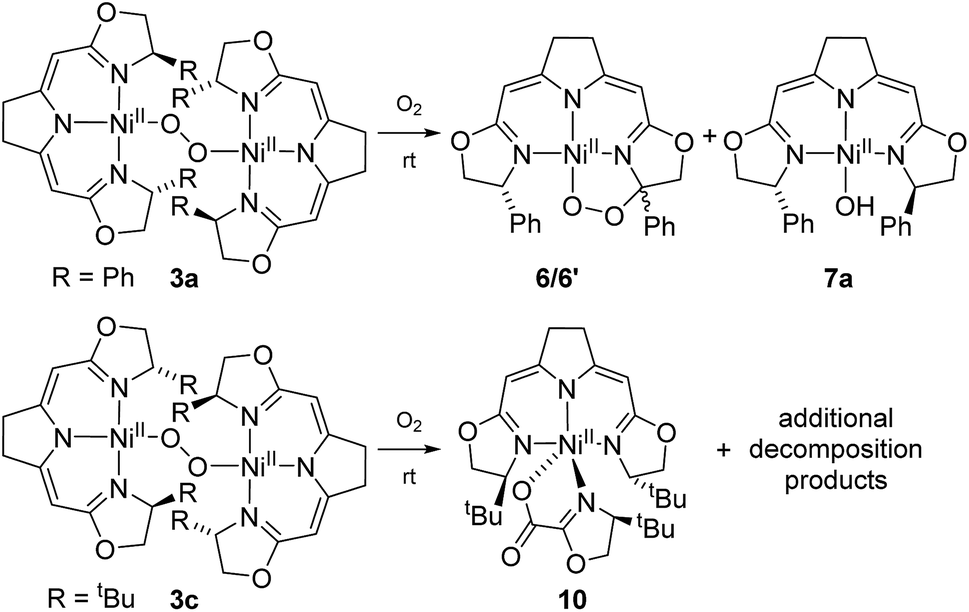 | ||
| Scheme 6 Aerobic autoxidation of 1,2-μ-peroxo complexes 3a and 3c at room temperature. | ||
The two nickel 1,2-μ-peroxo complexes 3a and b contain fairly reactive C–H bonds in close proximity to the Ni–O–O–Ni moiety which undergo bond dissociation in the course of their thermal decay. In the absence of such reactive C–H bonds, as in the corresponding nickel peroxo complex 3c bearing the tBu-substituted iso-PyrrMeBox pincer ligand, non-specific thermal oxidative degradation was observed leading to a mixture of compounds, of which one major product 10 could be isolated and structurally characterized by X-ray diffraction (ESI†). In this complex the bidentate coordination of an oxazolinylcarboxylate to the nickel pincer fragment is observed which is thought to result from the partial oxidative cleavage of the C–C double bond in the backbone of the pincer ligand.
Mechanistic aspects of the autoxidation of complex 3b
Whereas the transformation of 3b to 7b and 8 under anaerobic conditions is balanced, the mass balance of the reaction under oxygen atmosphere leading to 7b and 9 is not readily accounted for. The generation of the cyclic peroxide species 9 raises the question of whether O–O bond cleavage occurred during this transformation or not. In the former case isotope scrambling would be expected upon performing the reaction using the homoisotopically labeled peroxo complexes 3b[16O2]/3b[18O2] under an atmosphere of a mixture of homoisotopic 16O2 and 18O2 (Scheme 7).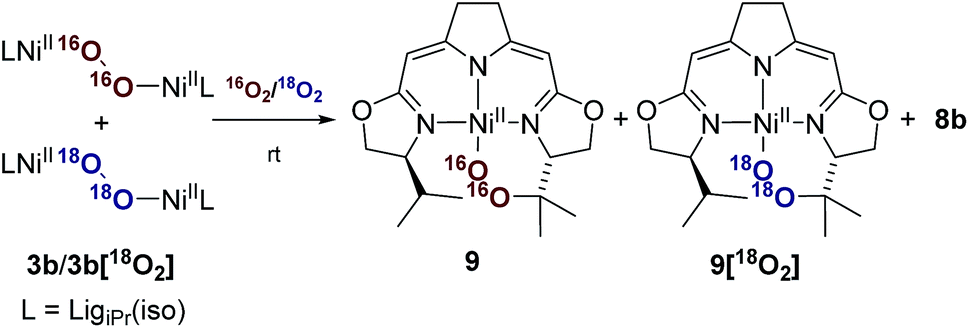 | ||
| Scheme 7 Labeling experiment for the investigation of the oxygen insertion in the course of the thermal decomposition of 3b applying a mixed atmosphere of 16O2/18O2. | ||
The outcome of this reaction clearly indicated that such scrambling did not occur, i.e. that the generation of 9 under O2 does not involve an initial O–O bond cleaving step. It was therefore of interest to probe to which extent the presence or absence of external O2 influenced the rates of the anaerobic and aerobic transformation of 3b to the two sets of reaction products depicted in Scheme 5.
The transformation of 3b in solution at room temperature in the presence of an internal standard (1,4-dimethoxybenzene) was monitored by 1H NMR spectroscopy, first under an atmosphere of argon and then at 5 bar oxygen pressure. In both cases the data obtained neatly followed the kinetic law of a first order decay. Significantly, oxygen had no effect on the rate of the decomposition (Fig. 4). In the course of the reaction the only species observed in the 1H NMR spectra were the starting μ-1,2-peroxo-complex 3b and the reaction products, while no intermediate could be detected. The identical decay rates under anaerobic and aerobic conditions indicated that 3b decomposes via an initial rate determining step which does not involve an external attack and/or insertion of oxygen.
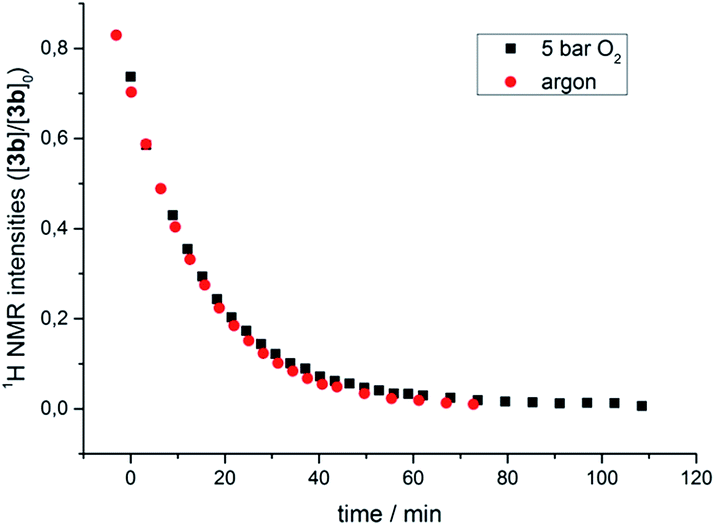 | ||
| Fig. 4 Course of the thermal decomposition of iPr-substituted μ-1,2-peroxo complex 3b under argon and in the presence of oxygen (5 bar) in THF monitored by 1H NMR spectroscopy. | ||
This observation, along with the labeling study described above, implies a similar early (rate determining) step in the reaction sequence for both cases and contradicts a reaction model for the mass balanced anaerobic conversion of 3b to 7b and 8 which would involve an initial O–O bond cleavage, hydrogen abstraction from one iso-propyl group in a pincer ligand (with concomitant formation of the hydroxo complex) and final cyclization of the diradical species generated to give the metallacyclic complex 8. It also contradicts a decomposition pathway via the superoxo species 4 which are in oxygen dependent equilibrium with complexes 3 (Scheme 2).
Possible alternative mechanistic models could involve the cyclic alkyl peroxo species 9 as key intermediate for both transformations. To probe for this possibility several stoichiometric test reactions were carried out (Scheme 8). Reaction of the cyclic alkyl peroxo complex 9 with an equimolar quantity of nickel hydrido compound 11b, generated in situ by pressurizing a solution of 3b with H2 gave a 1:1 mixture of compounds 7b and 8 as observed in the anaerobic degradation of 3b. These findings demonstrate that 9 could be a precursor in the formation of 8, provided that a nickel hydrido species was generated in an earlier step. On the other hand, the hydroperoxo complex 5a reacted with the Ni–H complex 11a to give the hydroxo species 7a.
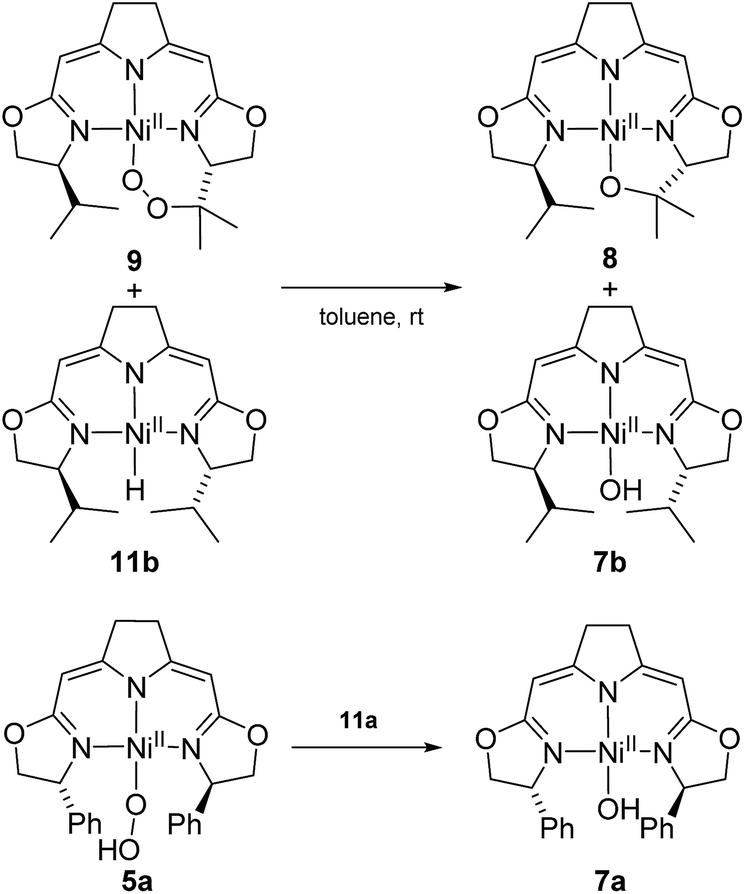 | ||
| Scheme 8 Stoichiometric conversions of cyclic peroxido complex 9 to the alkoxido compound 8 as well as the oxidation of the hydrido complex 11a to the hydroperoxido complex 5a and its reduction to 7a. | ||
These observations may imply that the formation of 8 or 9 as represented in Scheme 5 is dependent on the presence or absence of a hydrido species 11b generated in the early stages of the autoxidation and which is consumed in the presence of O2, a transformation previously reported for palladium and platinum hydrido complexes,60–62 thus leaving the cyclic alkyl peroxide 9 as isolated from the reaction. The formation of such a hydrido species in an autoxidation process appears counterintuitive, and in fact, could not be proved directly in case at hand. However, we were able to detect such a nickel hydride directly in the reaction of nickel(I) compound 1b with the oxidant N2O at −78 °C indicating that the hydride formation under oxidative conditions may occur (Scheme 9).
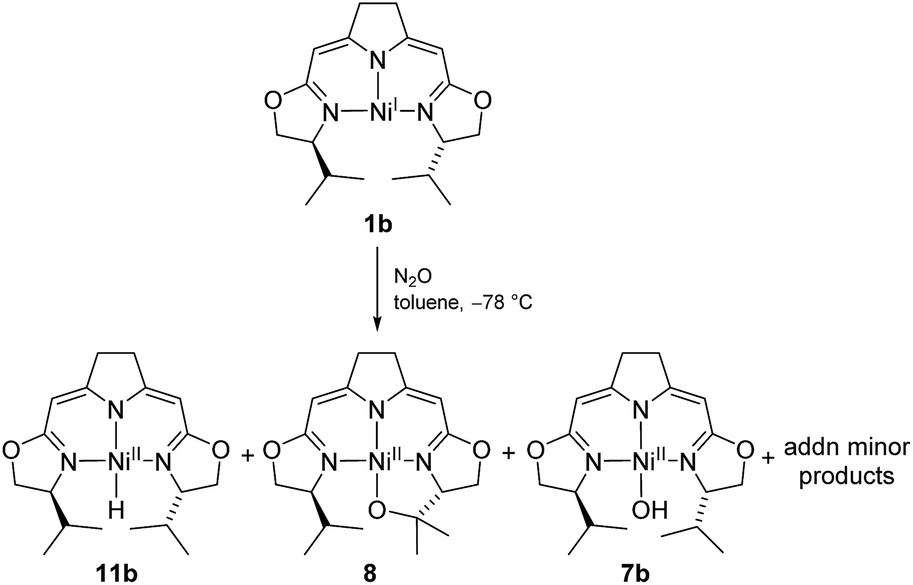 | ||
| Scheme 9 Formation of the hydrido complex 11b in the reaction of the nickel(I) complex with N2O at low temperatures. | ||
Whilst the experimental pieces of evidence gained for this highly reactive and sensitive system do not allow for a complete mechanistic picture, the tentative proposal for the processes involved in the anaerobic and aerobic decomposition of 3b as represented in Scheme 5 is given above (Scheme 10). The reaction scheme accounts for all of the observations described above.
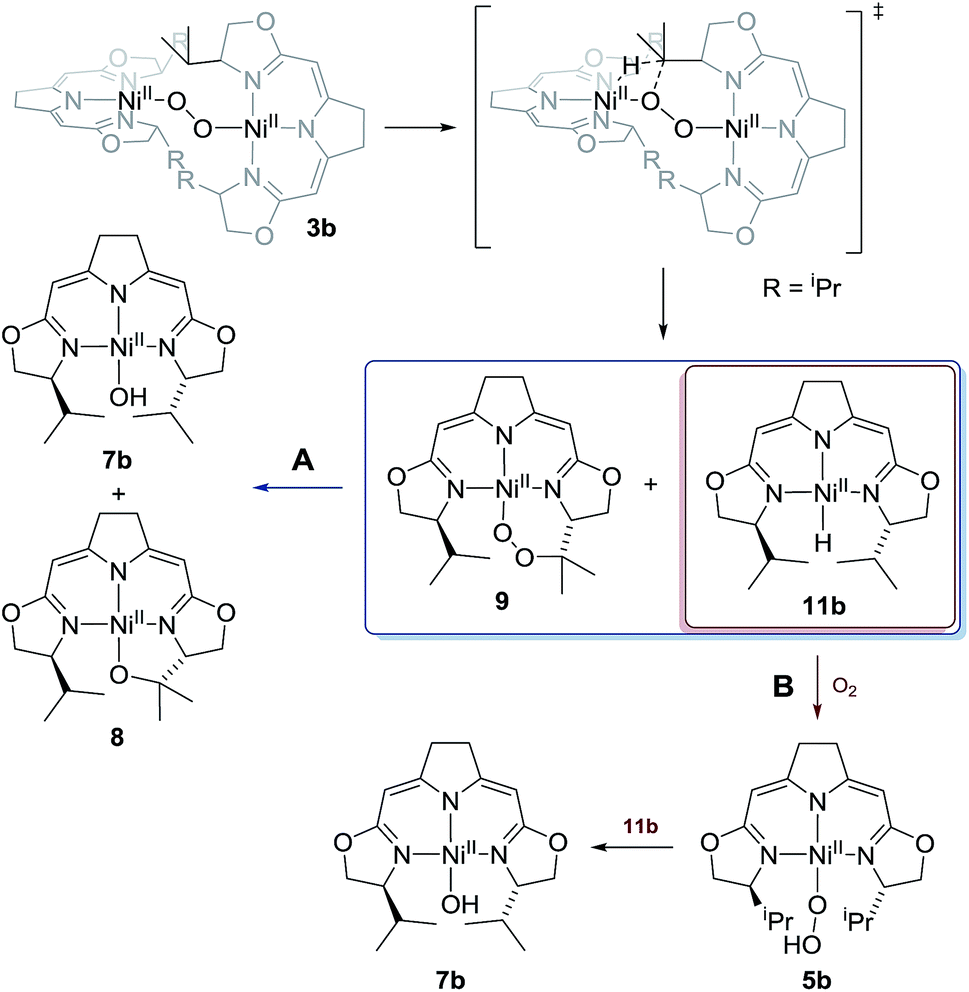 | ||
| Scheme 10 Proposed mechanism for the thermal decomposition in case of the 1,2-μ-peroxo complex 3b in absence (reaction path A) and presence of oxygen (reaction path B). | ||
Formation and thermal decomposition of a nickel methylperoxo complex
The autoxidation of the pincer ligand in the nickel complexes described above raised the question about the reactivity of their alkyl derivatives towards oxygen. Exposure of a solution of the nickel ethyl complex 12 to an atmosphere of oxygen at −78 °C led to a rapid elimination of ethylene to form the hydroperoxo complex 5b (Scheme 11), which was subsequently converted to the dinuclear 1,2-μ-peroxo complex 3b as described above. In order to probe for a possible alkyl radical intermediate in the reaction with oxygen, the hexenyl complex 13 was synthesized and subjected to oxygen at low temperature. However, no rearrangement occurred under these conditions and 1,5-hexadien was the only alkene formed during the reaction indicating that no long-lived radical species were involved.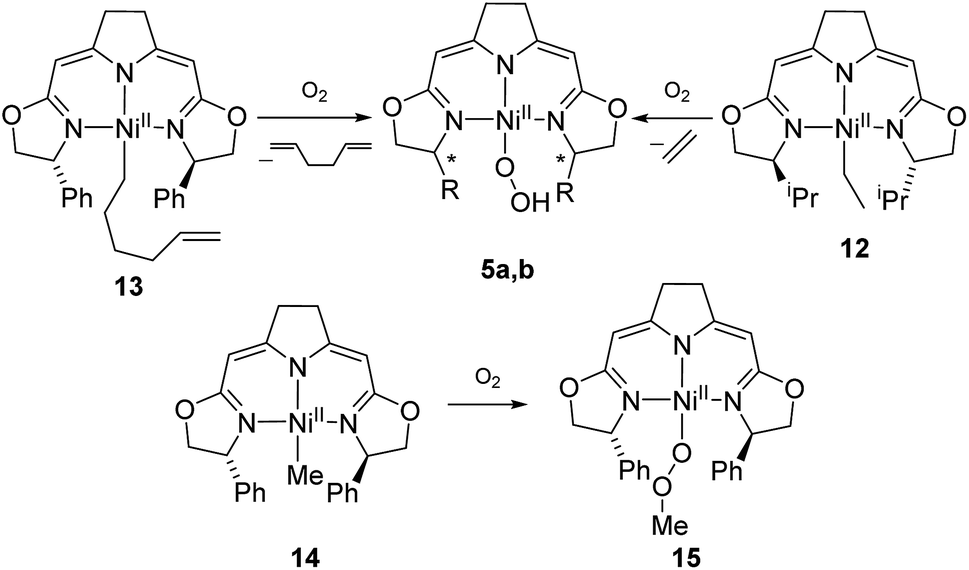 | ||
| Scheme 11 Reaction of alkylnickel complexes with oxygen. | ||
The methyl complex 14 slowly reacted with oxygen to give the methylperoxo complex 15. In an analogous manner, oxygen has been shown previously to insert into the Zn–R and Pt–R bonds leading to the corresponding alkylperoxo complexes.63,64 The methylperoxo complex 15 was isolated and characterized. Similar to the hydroperoxo complex 5a the apparent weakness of the O–O vibrational Raman band hampered the identification of this mode. However, suitable single crystals for X-ray diffraction were obtained (Fig. 5).
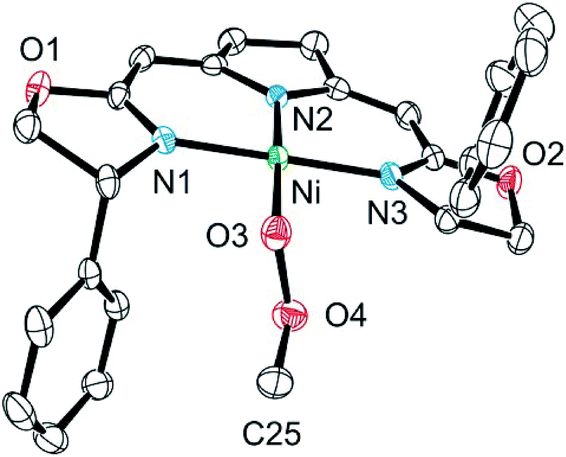 | ||
| Fig. 5 Molecular structure of the methylperoxo complex 15. Hydrogen atoms were omitted for clarity. Selected bond lengths [Å] and angles [°]: Ni–O(3) 1.8497(16), Ni–N(1) 1.8927(19), Ni–N(2) 1.9246(19), Ni–N(3) 1.8948(19), O(3)–O(4) 1.513(2), O(4)–C(25) 1.398(3), O(3)–Ni–N(1) 87.57(8), O(3)–Ni–N(2) 173.91(8), O(3)–Ni–N(3) 87.96(8), N(1)–Ni–N(2) 92.43(8), N(1)–Ni–N(3) 174.55(8), N(3)–Ni–N(2) 92.35(8), O(4)–O(3)–Ni 102.51(11), C(25)–O(4)–O(3) 104.97(17), Ni–O(3)–O(4)–C(25) −170.27(15). | ||
The molecular structure of 15 resembles to a large extent that of the corresponding hydroperoxo complex 5a.37 Both complexes possess a square-planar coordination geometry with almost identical Ni–N and Ni–O bond lengths [Ni–O(3) 1.8497(16) (15), 1.8456(16) (5a); Ni–N(1) 1.8927(19) (15), 1.8962(18) (5a); Ni–N(2) 1.9246(19) (15), 1.9264(18) (5a); Ni–N(3) 1.8948(19) (15), 1.8889(19) (5a)]. However, the torsion angle Ni–O(3)–O(4)–C(25) of −170.27(15) of the peroxo ligand in 15 reflects the repulsive nature of the interaction of the methylperoxo ligand with the Ph-substituent of the oxazoline ring. The O–O bond length in 15 is elongated compared to 5a [15: O(3)–O(4) 1.513(2), 5a: 1.492(2)]. Both interatomic distances are rather large compared to Akita's tBu-peroxo species65 and other related complexes found in the literature.66–69
The methylperoxo complex 15 was found to be fairly stable at room temperature but slowly converted to a mixture of near equal amounts of the formato and the hydroxo complexes 16 and 7a along with half an equivalent of methanol (Scheme 12).
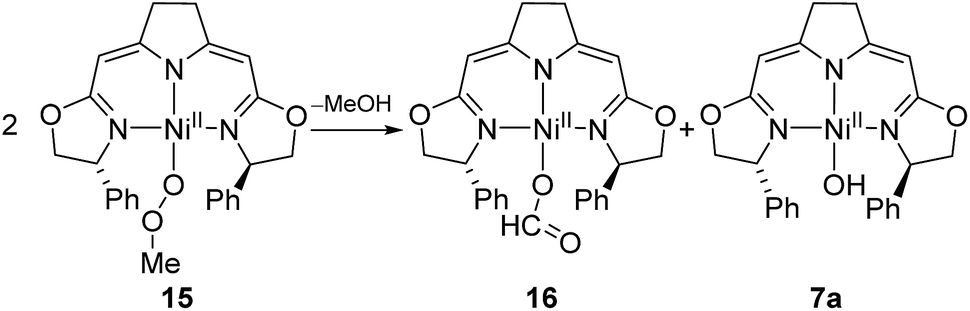 | ||
| Scheme 12 Thermal decomposition of the methylperoxo complex 9. | ||
The formato complex 16 was also separately synthesized by the reaction of the hydroxo complex 7a with formic acid, isolated and characterized by 1H, 13C NMR as well as IR spectroscopy. The molecular structure was established by X-ray diffraction and is depicted in Fig. 6. The C–O bond lengths of the formato ligand were found to be O(3)–C(25A) 1.326(5) Å and O(4)–C(25A) 1.228(5).
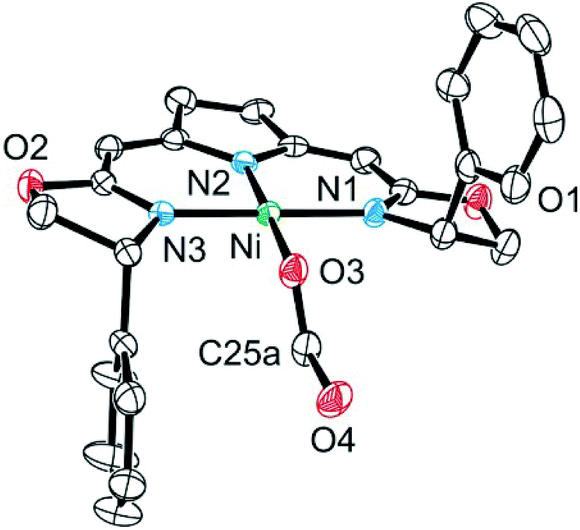 | ||
| Fig. 6 Molecular structure of formato complex 16. Only the major of two disordered sites of the formato ligand is depicted. Hydrogen atoms were omitted for clarity. Selected bond lengths [Å] and angles [°]: Ni–O(3) 1.8850(15), Ni–N(1) 1.8934(17), Ni–N(2) 1.9001(17), Ni–N(3) 1.8894(17), O(3)–C(25A) 1.326(5), O(4)–C(25A) 1.228(5), O(3)–Ni–N(1) 87.35(7), O(3)–Ni–N(2) 178.36(7), O(3)–Ni–N(3) 87.33(7), N(1)–Ni–N(2) 93.00(7), N(3)–Ni–N(1) 173.73(8), N(3)–Ni–N(2) 92.41(8). | ||
The formato complex 16 itself decomposed at elevated temperatures to CO2, dihydrogen as well as the nickel(I) species 1a (Scheme 13). The formation of the latter is thought to occur via the hydrido complex 11a which was detected in trace amounts (1H NMR) after the reaction was completed. Attempts to establish reaction conditions for the reverse reaction, the hydrogenation of CO2 in the presence of the nickel(I) complex, were unsuccessful.
 | ||
| Scheme 13 Thermal decomposition of formato complex 10. | ||
Conclusions
This study has shed further light onto the electronic structure and reactivity of the three- and four-coordinate nickel(I) complexes 1b and 2b bearing the iso-PyrrMeBox pincer ligand which partially models the structure of the hydrocorphin macrocycle in cofactor F430. However, this analogy is limited to the almost identical delocalized 10-electron-π-system involving three nitrogen donor atoms while the coordination of a fourth ligand was found occur only with the strong π-acceptor CO. This additional coordination significantly influences the electronic structure and is reflected in the way the g tensor of 1b, which is highly anisotropic, is modified significantly towards an axial symmetry upon the occupation of the fourth coordination site in the CO-adduct 2b, a spectroscopic characteristic reminiscent of the cofactor F430.These low-valent T-shaped nickel complexes readily form peroxo species in the presence of dioxygen. While their formation and degradation may model aspects of the aerobic deactivation of methyl coenzyme M reductase (MCR), the dominance of dinuclear peroxo-species in the reaction pathways observed in this, limits the relevance of the model system 1b for such processes involving the cofactor F430. However, the reaction patterns and structurally characterized intermediate and final oxidation products established in this study are expected to shed new light on the mechanisms operating in oxidations catalyzed by Ni complexes.
The most notable general principles appear to be the propensity of the Ni–X (X = O, C, H) bonds to be cleaved homolytically and the facile insertion of O2 into Ni–C bonds. The homolytic dissociation of Ni–O bonds in nickel peroxo species is believed to generate highly reactive radical intermediates which initiate the autoxidation process(es) observed. These are complex multistep reaction sequences which are only partially elucidated and define the challenges for future work in this field.
Acknowledgements
We thank Prof. M. Enders and Dr Marion Kerscher for advice and experimental support. We acknowledge funding by the Deutsche Forschungsgemeinschaft (Ga 488/9-1). The computational studies were supported in part by bwGRiD, member of the German D-Grid initiative, funded by the Ministry for Education and Research and the Ministry for Science, Research and Arts Baden-Württemberg and in part by the bwHPC initiative and the bwHPC-C5 project provided through associated compute services of the JUSTUS HPC facility at the University of Ulm. bwHPC and bwHPC-C5 (http://www.bwhpc-c5.de) are funded by the Ministry of Science, Research and the Arts Baden-Württemberg (MWK) and the Germany Research Foundation (DFG).Notes and references
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- R. K. Thauer, A.-K. Kaster, M. Goenrich, M. Schick, T. Hiromoto and S. Shima, Annu. Rev. Biochem., 2010, 79, 507–536 CrossRef CAS PubMed.
- E. L. Hegg, Acc. Chem. Res., 2004, 37, 775–783 CrossRef CAS PubMed.
- S. W. Ragsdale and M. Kumar, Chem. Rev., 1996, 96, 2515–2540 CrossRef CAS PubMed.
- S. Ragsdale, in The Metal-Driven Biogeochemistry of Gaseous Compounds in the Environment, ed. P. M. H. Kroneck and M. E. S. Torres, Springer, Netherlands, 2014, vol. 14, ch. 6, pp. 125–145 Search PubMed.
- B. Jaun and R. K. Thauer, in Nickel and Its Surprising Impact in Nature, John Wiley & Sons, Ltd, 2007, pp. 323–356, DOI:10.1002/9780470028131.ch8.
- S. Scheller, M. Goenrich, R. Boecher, R. K. Thauer and B. Jaun, Nature, 2010, 465, 606–608 CrossRef CAS PubMed.
- R. K. Thauer, A.-K. Kaster, H. Seedorf, W. Buckel and R. Hedderich, Nat. Rev. Microbiol., 2008, 6, 579–591 CrossRef CAS PubMed.
- I. A. Abreu and D. E. Cabelli, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 263–274 CrossRef CAS PubMed.
- M. J. Clarke, Less Common Metals in Proteins and Nucleic Acid Probes, Springer, Berlin, Heidelberg, 1998 Search PubMed.
- S. K. Chatterjee, R. C. Maji, S. K. Barman, M. M. Olmstead and A. K. Patra, Angew. Chem., Int. Ed., 2014, 53, 10184–10189 CrossRef CAS PubMed.
- K. C. Ryan, O. E. Johnson, D. E. Cabelli, T. C. Brunold and M. J. Maroney, JBIC, J. Biol. Inorg. Chem., 2010, 15, 795–807 CrossRef CAS PubMed.
- J. Cho, H. Y. Kang, L. V. Liu, R. Sarangi, E. I. Solomon and W. Nam, Chem. Sci., 2013, 4, 1502–1508 RSC; T. Corona, F. F. Pfaff, F. Acuña-Parés, A. Draksharapu, C. J. Whiteoak, V. Martin-Diaconescu, J. Lloret-Fillol, W. R. Browne, K. Ray and A. Company, Chem. Eur. J., 21, 15029–15038 CrossRef CAS PubMed.
- S. Yao and M. Driess, Acc. Chem. Res., 2011, 45, 276–287 CrossRef PubMed.
- J. Cho, R. Sarangi, J. Annaraj, S. Y. Kim, M. Kubo, T. Ogura, E. I. Solomon and W. Nam, Nat. Chem., 2009, 1, 568–572 CrossRef CAS PubMed.
- S. Yao, E. Bill, C. Milsmann, K. Wieghardt and M. Driess, Angew. Chem., Int. Ed., 2008, 47, 7110–7113 CrossRef CAS PubMed.
- M. T. Kieber-Emmons and C. G. Riordan, Acc. Chem. Res., 2007, 40, 618–625 CrossRef CAS PubMed.
- M. T. Kieber-Emmons, J. Annaraj, M. S. Seo, K. M. Van Heuvelen, T. Tosha, T. Kitagawa, T. C. Brunold, W. Nam and C. G. Riordan, J. Am. Chem. Soc., 2006, 128, 14230–14231 CrossRef CAS PubMed.
- M. T. Kieber-Emmons, R. Schenker, G. P. A. Yap, T. C. Brunold and C. G. Riordan, Angew. Chem., 2004, 116, 6884–6886 CrossRef.
- K. Fujita, R. Schenker, W. Gu, T. C. Brunold, S. P. Cramer and C. G. Riordan, Inorg. Chem., 2004, 43, 3324–3326 CrossRef CAS PubMed.
- B. S. Mandimutsira, J. L. Yamarik, T. C. Brunold, W. Gu, S. P. Cramer and C. G. Riordan, J. Am. Chem. Soc., 2001, 123, 9194–9195 CrossRef CAS PubMed.
- T. Tano, Y. Doi, M. Inosako, A. Kunishita, M. Kubo, H. Ishimaru, T. Ogura, H. Sugimoto and S. Itoh, Bull. Chem. Soc. Jpn., 2010, 83, 530–538 CrossRef CAS.
- A. Kunishita, Y. Doi, M. Kubo, T. Ogura, H. Sugimoto and S. Itoh, Inorg. Chem., 2009, 48, 4997–5004 CrossRef CAS PubMed.
- J. Cho, H. Furutachi, S. Fujinami, T. Tosha, H. Ohtsu, O. Ikeda, A. Suzuki, M. Nomura, T. Uruga, H. Tanida, T. Kawai, K. Tanaka, T. Kitagawa and M. Suzuki, Inorg. Chem., 2006, 45, 2873–2885 CrossRef CAS PubMed.
- J. Cho, H. Furutachi, S. Fujinami and M. Suzuki, Angew. Chem., Int. Ed., 2004, 43, 3300–3303 CrossRef CAS PubMed.
- S. Itoh, H. Bandoh, M. Nakagawa, S. Nagatomo, T. Kitagawa, K. D. Karlin and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 11168–11178 CrossRef CAS PubMed.
- K. Shiren, S. Ogo, S. Fujinami, H. Hayashi, M. Suzuki, A. Uehara, Y. Watanabe and Y. Moro-oka, J. Am. Chem. Soc., 1999, 122, 254–262 CrossRef.
- S. Itoh, H. Bandoh, S. Nagatomo, T. Kitagawa and S. Fukuzumi, J. Am. Chem. Soc., 1999, 121, 8945–8946 CrossRef CAS.
- Y. Morimoto, S. Bunno, N. Fujieda, H. Sugimoto and S. Itoh, J. Am. Chem. Soc., 2015, 137, 5867–5870 CrossRef CAS PubMed.
- J. Nakazawa, S. Terada, M. Yamada and S. Hikichi, J. Am. Chem. Soc., 2013, 135, 6010–6013 CrossRef CAS PubMed.
- R. Latifi, L. Tahsini, D. Kumar, G. N. Sastry, W. Nam and S. P. de Visser, Chem. Commun., 2011, 47, 10674–10676 RSC.
- T. Nagataki, K. Ishii, Y. Tachi and S. Itoh, Dalton Trans., 2007, 1120–1128, 10.1039/b615503k.
- T. Nagataki, Y. Tachi and S. Itoh, Chem. Commun., 2006, 4016–4018, 10.1039/b608311k.
- F. Konrad, J. Lloret Fillol, H. Wadepohl and L. H. Gade, Inorg. Chem., 2009, 48, 8523–8535 CrossRef CAS PubMed.
- C. Mazet and L. H. Gade, Chem.–Eur. J., 2003, 9, 1759–1767 CrossRef CAS PubMed.
- F. Konrad, J. Lloret Fillol, C. Rettenmeier, H. Wadepohl and L. H. Gade, Eur. J. Inorg. Chem., 2009, 4950–4961 CrossRef CAS.
- C. A. Rettenmeier, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2015, 54, 4880–4884 CrossRef CAS PubMed.
- Q.-H. Deng, R. L. Melen and L. H. Gade, Acc. Chem. Res., 2014, 47, 3162–3173 CrossRef CAS PubMed.
- C. Rettenmeier, H. Wadepohl and L. H. Gade, Chem.–Eur. J., 2014, 20, 9657–9665 CrossRef CAS PubMed.
- G. Bender, E. Pierce, J. A. Hill, J. E. Darty and S. W. Ragsdale, Metallomics, 2011, 3, 797–815 RSC.
- G. Bender, T. A. Stich, L. Yan, R. D. Britt, S. P. Cramer and S. W. Ragsdale, Biochemistry, 2010, 49, 7516–7523 CrossRef CAS PubMed.
- C. Yoo, S. Oh, J. Kim and Y. Lee, Chem. Sci., 2014, 5, 3853–3858 RSC.
- M. J. Ingleson, B. C. Fullmer, D. T. Buschhorn, H. Fan, M. Pink, J. C. Huffman and K. G. Caulton, Inorg. Chem., 2008, 47, 407–409 CrossRef CAS PubMed.
- C. Holliger, A. J. Pierik, E. J. Reijerse and W. R. Hagen, J. Am. Chem. Soc., 1993, 115, 5651–5656 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, 2013 Search PubMed.
- J.-P. Blaudeau, M. P. McGrath, L. A. Curtiss and L. Radom, J. Chem. Phys., 1997, 107, 5016–5021 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. P. McGrath and L. Radom, J. Chem. Phys., 1991, 94, 511–516 CrossRef CAS.
- R. C. Binning and L. A. Curtiss, J. Comput. Chem., 1990, 11, 1206–1216 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- K. Raghavachari and G. W. Trucks, J. Chem. Phys., 1989, 91, 1062–1065 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- P. J. Hay, J. Chem. Phys., 1977, 66, 4377–4384 CrossRef CAS.
- A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033–1036 CrossRef CAS.
- J. Wenz, C. A. Rettenmeier, H. Wadepohl and L. H. Gade, Chem. Commun., 2016, 52, 202–205 RSC.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS PubMed.
- M. M. Konnick, N. Decharin, B. V. Popp and S. S. Stahl, Chem. Sci., 2011, 2, 326–330 RSC.
- M. C. Denney, N. A. Smythe, K. L. Cetto, R. A. Kemp and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 2508–2509 CrossRef CAS PubMed.
- D. D. Wick and K. I. Goldberg, J. Am. Chem. Soc., 1999, 121, 11900–11901 CrossRef CAS.
- D. Mukherjee, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2012, 134, 13018–13026 CrossRef CAS PubMed.
- K. A. Grice and K. I. Goldberg, Organometallics, 2009, 28, 953–955 CrossRef CAS.
- S. Hikichi, H. Okuda, Y. Ohzu and M. Akita, Angew. Chem., Int. Ed., 2009, 48, 188–191 CrossRef CAS PubMed.
- T. Miyaji, M. Kujime, S. Hikichi, Y. Moro-oka and M. Akita, Inorg. Chem., 2002, 41, 5286–5295 CrossRef CAS PubMed.
- M. Akita, T. Miyaji and Y. Moro-oka, Chem. Commun., 1998, 1005–1006, 10.1039/a801111g.
- H. Komatsuzaki, N. Sakamoto, M. Satoh, S. Hikichi, M. Akita and Y. Moro-oka, Inorg. Chem., 1998, 37, 6554–6555 CrossRef CAS PubMed.
- N. Kitajima, T. Katayama, K. Fujisawa, Y. Iwata and Y. Morooka, J. Am. Chem. Soc., 1993, 115, 7872–7873 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1439102–1439106. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04644k |
| This journal is © The Royal Society of Chemistry 2016 |