
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
[NiIII(OMe)]-mediated reductive activation of CO2 affording a Ni(κ1-OCO) complex†
Tzung-Wen
Chiou
*a,
Yen-Ming
Tseng
a,
Tsai-Te
Lu
b,
Tsu-Chien
Weng
c,
Dimosthenes
Sokaras
c,
Wei-Chieh
Ho
a,
Ting-Shen
Kuo
d,
Ling-Yun
Jang
e,
Jyh-Fu
Lee
e and
Wen-Feng
Liaw
*a
aDepartment of Chemistry, National Tsing Hua University, Hsinchu, 30013, Taiwan. E-mail: d9623817@oz.nthu.edu.tw; wfliaw@mx.nthu.edu.tw
bDepartment of Chemistry, Chung Yuan Christian University, Taoyuan, 32023, Taiwan
cSLAC National Accelerator Laboratory, Menlo Park, CA 94025, USA
dDepartment of Chemistry, National Taiwan Normal University, Taipei, 10610, Taiwan
eNational Synchrotron Radiation Research Center, Hsinchu, 30013, Taiwan
First published on 24th February 2016
Abstract
Carbon dioxide is expected to be employed as an inexpensive and potential feedstock of C1 sources for the mass production of valuable chemicals and fuel. Versatile chemical transformations of CO2, i.e. insertion of CO2 producing bicarbonate/acetate/formate, cleavage of CO2 yielding μ-CO/μ-oxo transition-metal complexes, and electrocatalytic reduction of CO2 affording CO/HCOOH/CH3OH/CH4/C2H4/oxalate were well documented. Herein, we report a novel pathway for the reductive activation of CO2 by the [NiIII(OMe)(P(C6H3-3-SiMe3-2-S)3)]− complex, yielding the [NiIII(κ1-OCO˙−)(P(C6H3-3-SiMe3-2-S)3)]− complex. The formation of this unusual NiIII(κ1-OCO˙−) complex was characterized by single-crystal X-ray diffraction, EPR, IR, SQUID, Ni/S K-edge X-ray absorption spectroscopy, and Ni valence-to-core X-ray emission spectroscopy. The inertness of the analogous complexes [NiIII(SPh)], [NiII(CO)], and [NiII(N2H4)] toward CO2, in contrast, demonstrates that the ionic [NiIII(OMe)] core attracts the binding of weak σ-donor CO2 and triggers the subsequent reduction of CO2 by the nucleophilic [OMe]− in the immediate vicinity. This metal–ligand cooperative activation of CO2 may open a novel pathway promoting the subsequent incorporation of CO2 in the buildup of functionalized products.
Introduction
Carbon dioxide, the waste from human activity embodying the nature of high thermodynamic stability and chemical inertness, is expected to be employed as an inexpensive and potential feedstock of C1 sources for the regeneration of valuable chemicals and fuel.1,2 Nature developed carbon monoxide dehydrogenase (CODH) to harbor a Ni–Fe cluster for the reversible interconversion between CO2 and CO.3–5 To gain insight into the mechanism for the conversion of CO2 to CO in CODH, several Ni–CO2 adducts derived from the reaction of a low-valent Ni complex and CO2 were reported.6–9 The direct electrochemical reduction of CO2 affords oxalate, carbon monoxide, formic acid, methanol, methane, and ethylene.10 To gain insight into the transformation of CO2 at a molecular level, the chemistry of the activation of CO2via nucleophilic attack/interaction on the polarized C center, in addition to the reduction of the coordinated CO2 ligand by low-valence transition metal complexes, has grown explosively over past years.11 The versatile chemical transformations of CO2, i.e. insertion of CO2 producing bicarbonate/acetate/formate,12–18 cleavage of CO2 yielding μ-CO/μ-oxo transition-metal complexes,19–23 reduction of CO2 affording CO/HCOOH/CH3OH/CH4/C2H4/C2H6/methylene, and electrocatalysis of CO2 converting it into oxalate, were well documented.24–26 The direct electrochemical reduction or electrocatalytic transformation of CO2 for the mass production of valuable chemicals and fuel, however, relies on the consumption of sustainable electric potential energy. Here we show a novel pathway for the reductive activation of CO2 by a mononuclear Ni(III) complex [NiIII(OMe)(P(C6H3-3-SiMe3-2-S)3)]−.27 This [NiIII-(OMe)]-mediated reduction of CO2 yields the complex NiIII(κ1-OCO˙−), evidenced by single-crystal X-ray diffraction, EPR, SQUID, Ni/S K-edge X-ray absorption spectroscopy, IR and Ni valence-to-core X-ray emission spectroscopy. The ionic [NiIII(OMe)] core provides a kinetic pathway to induce the binding of CO2 and trigger the subsequent reduction of CO2 by the nucleophilic [OMe]− in the immediate vicinity. The covalent [NiIII(SPh)] core as well as Ni(II) center in complexes [NiII(L)(P(C6H3-3-SiMe3-2-S)3)]− (L = CO or N2H4), in contrast, are inert toward CO2.28Results and discussion
Synthesis and characterization of nickel κ1-OCO complex
When CO2(g) was bubbled into the thermally stable [NiIII(OMe)(PS3)]− (1) (PS3 = P(C6H3-3-SiMe3-2-S)3) in THF,27 a pronounced color change from blue green to yellow green occurred to yield the O-bound κ1-CO2 complex [Ni(κ1-OCO)(PS3)]− (2), instead of complexes [Ni(OC(O)OCH3)-(PS3)]− or [Ni(OC(O)H)(PS3)]−via the classical insertion or β-H migration mechanisms (Scheme 1a).9,12–16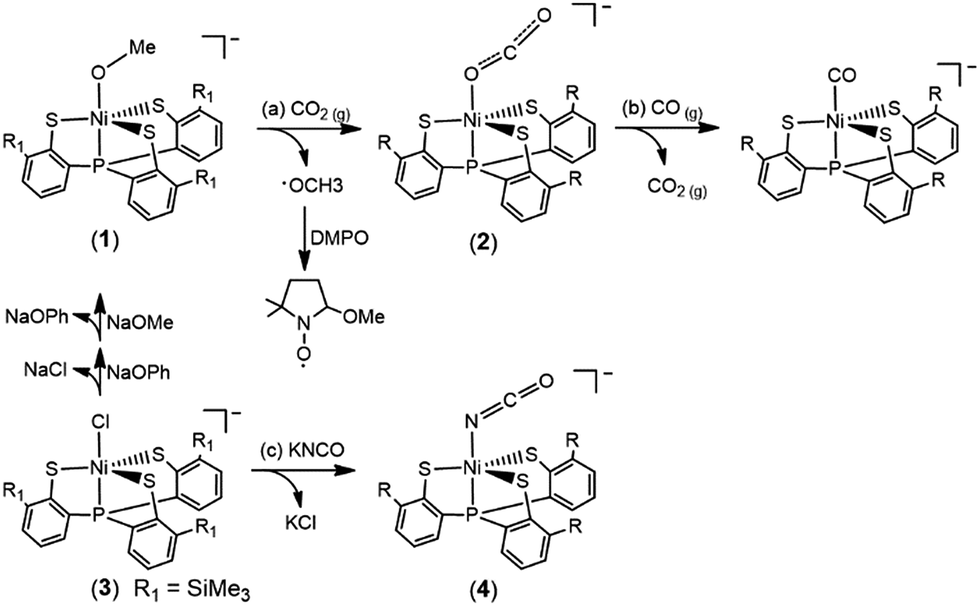 | ||
| Scheme 1 | ||
The accompanied formation of [˙OMe] in the reaction described above was corroborated using the spin-trapping reagent DMPO (ESI Fig. S1†).29 The IR νOCO stretching peak at 2177 cm−1 (KBr) (νOCO: 2226 cm−1 in THF) exhibited by complex 2 supports the formation of [Ni(κ1-OCO)(PS3)]−, which is consistent with the isotopic shift of the IR νOCO stretching peak to 2117 cm−1 (KBr) observed in the 13CO2 labeling experiment (ESI Fig. S2†). The conversion of complex 1 to complex 2 under a CO2 atmosphere was also monitored by UV-vis spectrometry; the intense bands at 419 and 605 nm disappeared with the simultaneous formation of absorption bands at 425 and 610 nm (THF) (ESI Fig. S3†). The green needle crystals of complex 2 were isolated when complex 2 was recrystallized from THF–diethyl ether at room temperature. As shown in Scheme 1b, treatment of complex 2 with CO(g) led to the formation of the reported complex [NiII(CO)(PS3)]− accompanied by the release of CO2(g) characterized by IR and GC (Fig. 1).28
 | ||
| Fig. 1 (A) IR spectra for the transformation of complex 2 to [NiII(CO)(PS3)]− in THF. The decrease in the intensity of the IR νCO2 peak at 2226 cm−1 exhibited by complex 2 with the simultaneous formation of an IR νCO peak at 2027 cm−1 indicated the formation of complex [NiII(CO)(PS3)]−. (B) GC chromatogram, derived from the sample collected from the headspace of the tube containing the reaction solution of complex 2 and CO(g), indicating the release of CO2(g) during the transformation of complex 2 to [NiII(CO)(PS3)]−. | ||
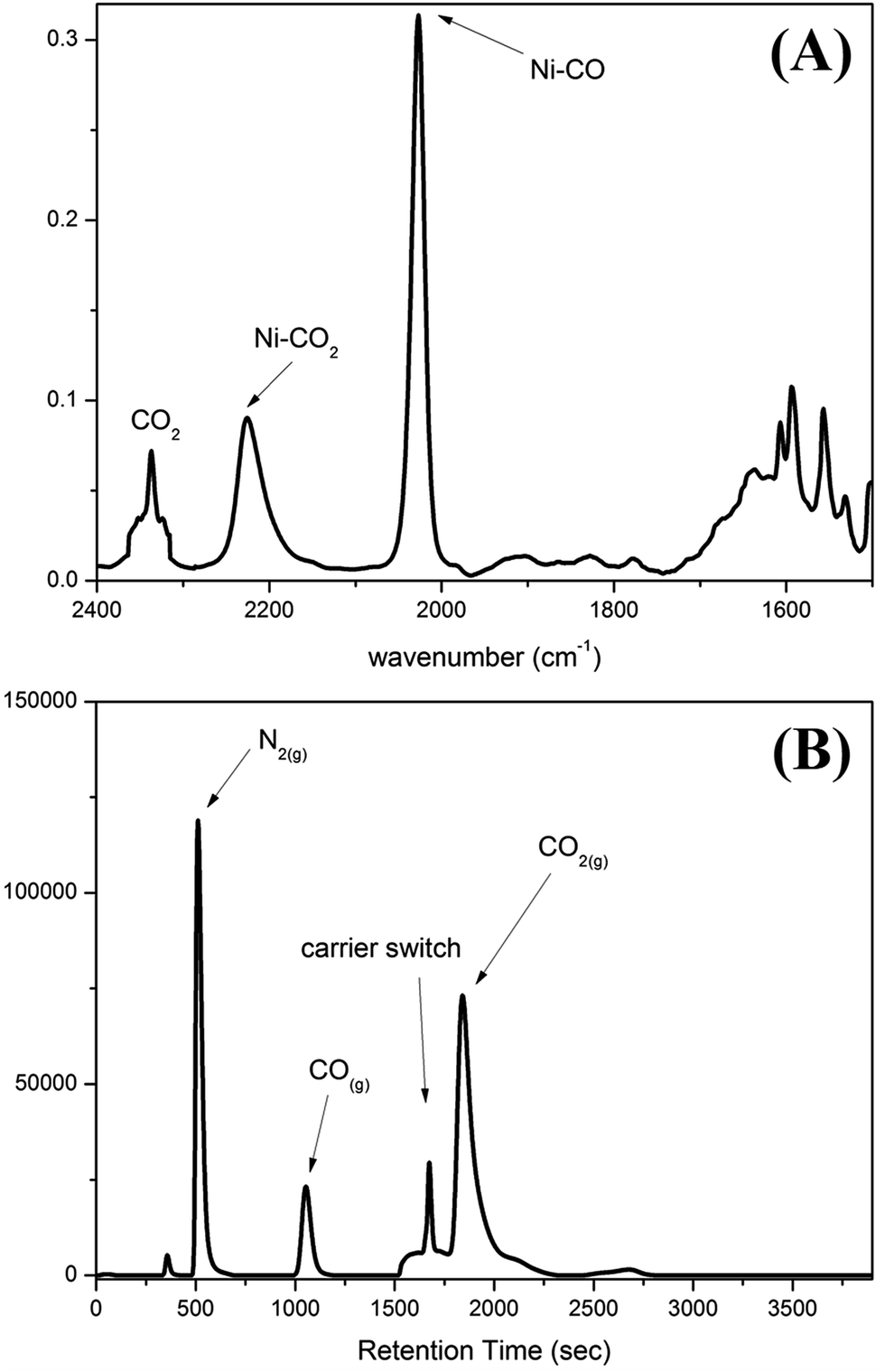
To contrast complex 2 containing a [NiIII:CO2˙−] or [NiII:CO2] center, complex [NiIII(NCO)(PS3)]− (4) was synthesized via the reaction of [Ni(Cl)(PS3)]− (3) and [K][NCO] to mimic the isolobal [NiIII:CO2] (Scheme 1c). Fig. 2 displays ORTEP plots of complexes 2 and 4, with the selected bond distances and angles given in the caption. The strain effect of the chelating ligand ([PS3]3−) in the coordination sphere of complexes 2 and 4 explains that the Ni is in a distorted trigonal bipyramidal geometry with three thiolates locating equatorial positions and the phosphorus is occupying an axial position trans to the [OCO] and [NCO] ligands. In contrast to the linear N–C–O (177.2(3)°) bond observed in complex 4, complex 2 displays a bent O–C–O bond with a bond angle of 171.7(7)°. Compared to the similar O–C and N–C bond distances of 1.200(3) and 1.181(3) Å in complex 4, the dramatic difference (∼0.1 Å) in O–C bond lengths, 1.132(6) vs. 1.240(7) Å, found in complex 2 moreover indicates the polarization of CO2via reductive activation affording a [NiIII:CO2˙−] species.22,30,31 A similar polarization of CO2 was reported in the O-bound κ1-CO2 coordinated complex [((AdArO)3tacn)-UIV(CO2˙−)] ((AdArOH)3tacn = 1,4,7-tris(3-adamantyl-5-tert-butyl-2-hydroxybenzyl)-1,4,7-triazacyclononane), O–C = 1.122(4) and 1.277(7) Å, with the linear U–O–C and O–C–O bonds stabilized by the sterically encumbering ligand framework.30,31 Besides, complex 2 displays a significantly longer Ni–O bond distance (2.028(3) Å) than those observed in [NiII(L)(pyN2Me2)]1− complexes (1.857(5) Å for L = HCO2−; 1.817(4) Å for L = HCO3−).32
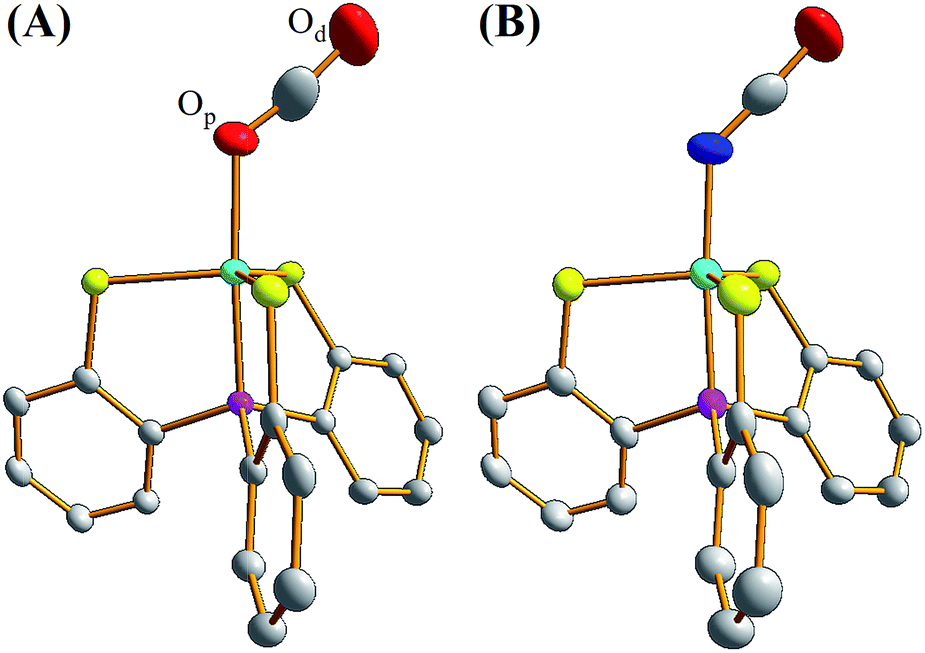 | ||
| Fig. 2 ORTEP drawing schemes of (A) complex 2 and (B) complex 4 with thermal ellipsoids drawn at a 50% probability level. The Ni, P, S, O, N, and C atoms are shown as light blue, purple, yellow, red, blue, and white ellipsoids. The H atom and TMS group are omitted for clarity. Selected bond distances (Å) and angles (°) for complex 2: Ni–Op, 2.028(3); Ni–P, 2.122(1); Ni–S, 2.221(1), 2.287(1), and 2.285(1); Op–C, 1.132(6); Od–C, 1.240(7); O–Ni–P, 176.2(1); Ni–O–C, 127.0(4); Op–C–Od, 171.7(7). Selected bond distances (Å) and angles (°) for complex 4: Ni–N, 1.933(2); Ni–P, 2.120(1); Ni–S, 2.226(1), 2.286(1), and 2.291(1); O–C, 1.200(3); N–C, 1.181(3); N–Ni–P, 175.0(1); Ni–N–C, 135.1(2); O–C–N, 177.2(3). | ||
X-ray absorption/emission spectrum
A Ni and S K-edge X-ray absorption spectroscopic (XAS) study of complex 2 was further attempted to investigate its electronic structure using complexes 1 and 4 as reference complexes. As shown in Fig. 3A, the Ni K-edge XAS of complex 2 (8333.1 eV) together with analogous complexes 1 (8332.9 eV) and 4 (8332.7 eV) shows a similar Ni1s-to-Ni3d transition energy. Accordingly, the formal oxidation state of Ni in complex 2 is similar to those of complexes 1 and 4, which are generally known as a L ligand bound to a d7 Ni(III) center in [NiIII(L) (PS3)]−.28Fig. 3B and ESI Fig. S4† depict the S K-edge XAS spectra of complexes 1, 2, and 4, whereas ESI Fig. S5† shows the calculated S K-edge XAS spectra and spectral deconvolution. As shown in Table 1, the intensity-weighted average energy of the S1s-to-Ni3d transitions in combination with the S1s-to-S*C–S transition energy demonstrate that the Ni3d manifold orbital energy of complex 2 is 0.3 eV higher than those of complexes 1 and 4.33,34 With further regard to the Ni1s-to-Ni3d pre-edge transition energy observed in the Ni K-edge XAS, Ni Zeff of complexes 1, 2, and 4 are all comparable. That is, the Ni and S K-edge XAS study supports the [NiIII:CO2˙−] electronic structure in complex 2. As shown in Fig. S6,† the cyclic voltammogram of a 2 mM solution of complex 4 in CH3CN indicates a reversible interconversion between NiIII/NiII at E1/2 = −0.58 V and an irreversible oxidation at Epa = −0.21 V, whereas complex 2 exhibits a reversible interconversion between NiIII/NiII at E1/2 = −0.70 V and an irreversible oxidation at Epa = −0.29 V (vs. Fc/Fc+).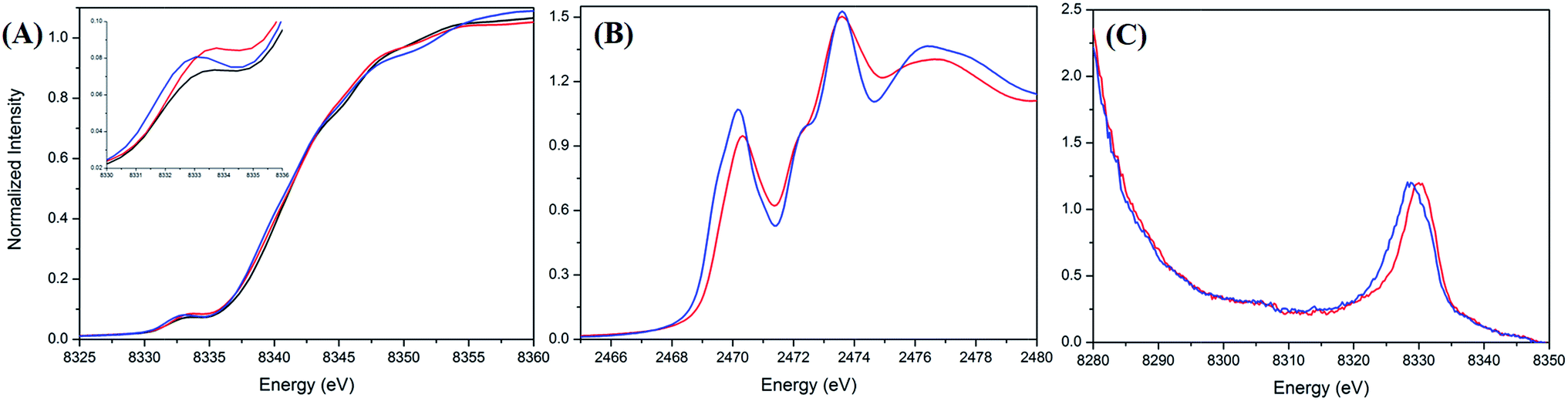 | ||
| Fig. 3 (A) Ni K-edge X-ray absorption spectra of complexes 1 (black), 2 (red), and 4 (blue). (B) S K-edge X-ray absorption and (C) Ni valence-to-core X-ray emission spectra of complexes 2 (red) and 4 (blue). | ||
| Complexes | Ni1s → Ni3d energya (eV) | S1s → Ni3d energyb (eV) | S1s → Ni3d intensityb | S1s → S*C–S energyb (eV) | Relative d-manifold energy shiftd (eV) | |||
|---|---|---|---|---|---|---|---|---|
| 1st peak | 2nd peak | Avgc | 1st peak | 2nd peak | ||||
| a The peak energy is determined by the minimum of the second derivative. b The peak energy and intensity is determined based on the spectral deconvolution. c The intensity-weighted average energy is given here. d Calculated from the difference of the thiolate peak energy and the intensity-weighted pre-edge peak energy. | ||||||||
| 1 | 8332.9 | 2469.7 | 2470.4 | 2470.0 | 0.29 | 1.34 | 2472.1 | 0 |
| 2 | 8333.1 | 2469.9 | 2470.5 | 2470.3 | 0.47 | 1.15 | 2472.1 | 0.3 |
| 4 | 8332.7 | 2469.5 | 2470.2 | 2470.1 | 0.33 | 1.77 | 2472.2 | 0 |
| [Ni(SPh) (PS3)]− | 8333.0 | 2469.8 | 2470.4 | 2470.3 | 0.52 | 1.91 | 2472.3 | 0.1 |
With regard to complex 4 as an isolobal equivalent to [NiIII:CO2], complex 2 is a NiIII complex bearing a 17-valence-electron [CO2]˙− ligand. The significantly lower intensity of the second S1s-to-Ni3d transition peak observed in the S K-edge XAS spectrum of complex 2, compared to that of complex 4, discloses that the one extra electron shared by the axial Ni3d orbital and 2π*u orbital of CO2 leads to a strengthening of the NiIII–CO2˙− bond and stabilizes the coordination of κ1-[CO2]˙− toward the NiIII center (Fig. 3B and Table 1). As observed in complex [NiIII(L)(PS3)]− (L = OMe, SEt, SPh), complex 4 displays an EPR silence at 300 K, an axial signal at g = 2.27 and 2.04 at 77 K, and an effective magnetic moment of 1.74 μB at 300 K (Fig. 4C and S7A†).27,28,35 The stabilization of the [CO2]˙− radical through coordination to the NiIII center in complex 2 was further evidenced by EPR spectroscopy.
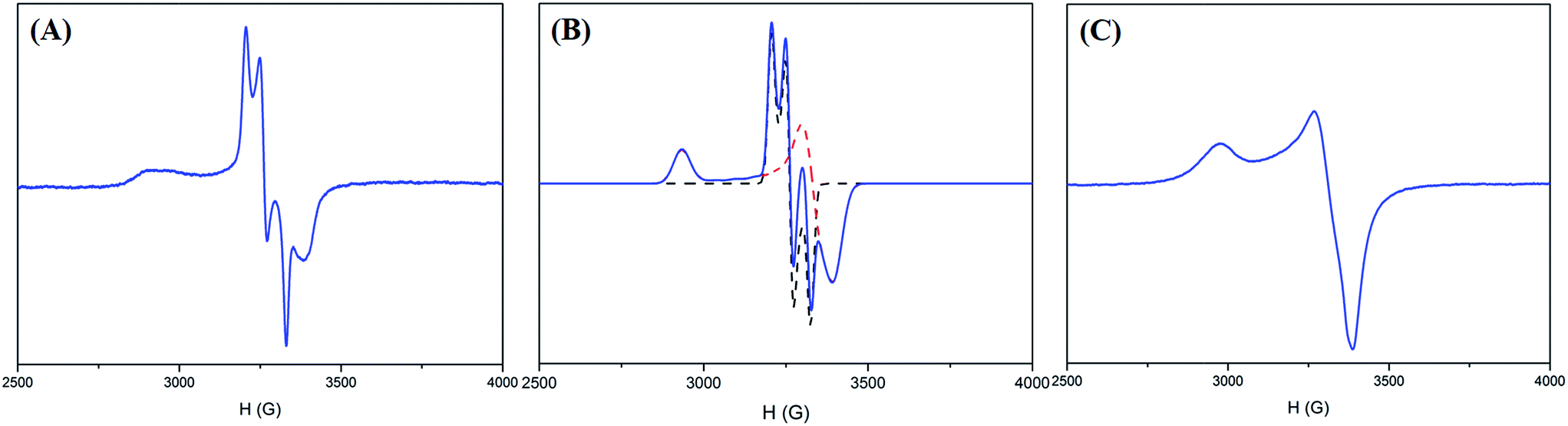 | ||
| Fig. 4 (A) EPR spectrum of complex 2 at 77 K, (B) simulated EPR spectrum (blue) of complex 2 combining [NiIII(L)(PS3)]− (dashed red line) and the [CO2]˙− radical (dashed black line), and (C) EPR spectrum of complex 4 at 77 K. | ||
As shown in Fig. 4A and B, the EPR spectrum of complex 2 at 77 K apparently resembles a combination of the typical EPR signal of [NiIII(L) (PS3)]− (g = 2.31, 2.03, and 2.00) and the [CO2]˙− radical with a contribution of Ni3d leading to the observed g anisotropy (Fig. 4A and B).36 The spin quantitation of complex 2, using complex 4 as a reference, demonstrates that the electronic structure of complex 2 is best described as a resonance hybrid between [NiIII:CO2˙−] and [NiII:CO2], which is supported by the effective magnetic moment of 1.59 μB exhibited by complex 2 at 300 K (ESI Fig. S7B and S7C†).
The experimental valence-to-core X-ray emission (V2C XES) spectra of complexes 2 and 4 are presented in Fig. 3C. In comparison with complex 4, the broad V2C transition peak of complex 2 at 8330.0 eV shifts from 8328.8 eV upon replacement of the [NCO]− by the [CO2]˙− ligand. DFT calculation was further pursued to verify the nature of the V2C transition(s). As shown in ESI Fig. S8A and S8B,† the DFT calculated V2C XES spectra resembles the experimental V2C features and, in particular, the trend of the energy shift comparing complexes 2 and 4. The contribution of the 4σg, 3σu, and 1πg orbitals of [NCO]− and Ni3d–S3p orbitals results in the V2C features of complex 4.37 For complex 2, the absence of transitions from the 3σu and 1πg orbitals and an additional transition from the occupied 2πu orbital of [CO2]˙−, in addition to the upward shift of the Ni3d–S3p orbitals in complex 2, rationalizes the higher V2C transition energy of complex 2 in comparison with complex 4.
Complex [Ni(L)(P(C6H3-3-SiMe3-2-S)3)]−, embedded in a distorted trigonal bipyramidal geometry, features a wealth of chemical reactivity tailored by the oxidation state of Ni and coordinating ligand L (L = OPh, SPh, SePh and Cl for NiIII; L = CO, N2H4 for NiII).27,28,35 To dissect the unique reactivity of [NiIII(OMe)(PS3)]− (1) toward CO2 activation, the addition of CO2 into a THF solution of the representative NiIII-chalcogenate complex [Ni(SPh)(PS3)]− was investigated. In contrast to the reaction of complex 1 and CO2 yielding complex 2, complex [NiIII(SPh)(PS3)]− is inert toward CO2. In addition, despite the potential reduction power of the NiII center in combination with the labile nature of the CO or N2H4 ligand, neither complex [NiII(CO)(PS3)]− nor complex [NiII(N2H4)(PS3)]− showed a reactivity toward CO2 when the THF solution of these Ni complexes was treated with CO2, respectively, at ambient temperature for 3 days. As shown in ESI Fig. S4† and Table 1, the covalent character of the [NiIII(SPh)] core, compared to the [NiIII(OMe)] core, derived from the σ-/π-electron-donating nature of the coordinated phenylthiolate ligand, rationalizes the inertness of [NiIII(SPh)(PS3)]− toward CO2.33,34 Despite the labile nature of CO and N2H4, the inert reactivity of the NiII center toward CO2 demonstrates that the lowered Ni3d manifold orbitals in NiIII complex 1 attracts the binding of weak σ-donor CO2 and triggers the subsequent reduction of CO2 by the nucleophilic [OMe]− in the immediate vicinity. The reactivity of complex 1 toward CO2, affording an O-bound [NiIII:CO2˙−] species, uncovers a novel strategy for the immobilization and reductive activation of CO2, contrary to the typical interaction of unoccupied CO2 2π*u orbitals with filled high-lying metal d orbitals in low-valence metal complexes.38,39 Theoretically, lowering the energy of the 2π*u (6a1) (LUMO) orbital on CO2 for interaction with nickel orbitals binding by way of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– unit may be responsible for the coordinated CO2 reduction and the nonlinearity of the triatomic CO2 molecule which contains 17 valence electrons, as reported by McGlynn and co-workers.37 These results illustrate aspects of how a coordinated ligand and the electronic state of the nickel center work in concert to trigger coordination and activation of CO2.
C– unit may be responsible for the coordinated CO2 reduction and the nonlinearity of the triatomic CO2 molecule which contains 17 valence electrons, as reported by McGlynn and co-workers.37 These results illustrate aspects of how a coordinated ligand and the electronic state of the nickel center work in concert to trigger coordination and activation of CO2.
Conclusions
Complex 1, with the inherent combination of an electrophilic [NiIII(PS3)] core and a properly positioned [OMe]− nucleophile, was employed to provide an optimum electronic condition to trap and activate CO2 to afford complex 2, containing the O-coordinated [κ1-CO2]˙− ligand. The NiIII-mediated reduction of CO2 by an adjacent [OMe]− ligand immobilizes CO2 in the form of [NiIII:CO2˙−] and may open a novel CO2 activation pathway promoting the subsequent incorporation of CO2 in the buildup of functionalized products.Acknowledgements
We gratefully acknowledge the support on the hardware and software from staff at BL-16A and BL-17C of NSRRC, and BL 6-2 of SLAC. Authors thank Ms Pei-Lin Chen for single-crystal X-ray structural determinations, Mr Yu-Huan Lu and Prof Hsin-Tsung Chen for the help on theoretical calculation. We also thank the Ministry of Science and Technology (Taiwan) for the financial support.Notes and references
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388 CrossRef CAS PubMed.
- J.-H. Jeoung and H. Dobbek, Science, 2007, 318, 1461 CrossRef CAS PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149 CrossRef CAS PubMed.
- J. Fesseler, J.-H. Jeoung and H. Dobbek, Angew. Chem., 2015, 54, 8560 CrossRef CAS PubMed.
- M. Aresta, C. F. Nobile, V. G. Albano, E. Forni and M. Manassero, J. Chem. Soc., Chem. Commun., 1975, 636 RSC.
- J. S. Anderson, V. M. Iluc and G. L. Hillhouse, Inorg. Chem., 2010, 49, 10203 CrossRef CAS PubMed.
- Y.-E. Kim, J. Kim and Y. Lee, Chem. Commun., 2014, 50, 11458 RSC.
- Y.-E. Kim, S. Oh, S. Kim, O. Kim, J. Kim, S. W. Han and Y. Lee, J. Am. Chem. Soc., 2015, 137, 4280 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J. M. Saveant, Chem. Soc. Rev., 2013, 42, 2423 RSC.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975 RSC.
- A. Looney, R. Han, K. Mcneill and G. Parkin, J. Am. Chem. Soc., 1993, 115, 4690 CrossRef CAS.
- O. R. Allen, S. J. Dalgarno, L. D. Field, P. Jensen, A. J. Turnbull and A. C. Willis, Organometallics, 2008, 27, 2092 CrossRef CAS.
- A. Jana, D. Ghoshal, H. W. Roesky, I. Objartel, G. Schwab and D. Stalke, J. Am. Chem. Soc., 2009, 131, 1288 CrossRef CAS PubMed.
- S. F. Yin, J. Maruyama, T. Yamashita and S. Shimada, Angew. Chem., 2008, 47, 6590 CrossRef CAS PubMed.
- B. Kersting, Angew. Chem., 2001, 40, 3987 CrossRef CAS.
- M. Vogt, A. Nerush, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Chem. Sci., 2014, 5, 2043 RSC.
- G. A. Filonenko, M. P. Conley, C. Copéret, M. Lutz, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2013, 3, 2522 CrossRef CAS.
- C. C. Lu, C. T. Saouma, M. W. Day and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 4 CrossRef CAS PubMed.
- M. T. Whited and R. H. Grubbs, J. Am. Chem. Soc., 2008, 130, 5874 CrossRef CAS PubMed.
- B. C. Fullmer, H. J. Fan, M. Pink and K. G. Caulton, Inorg. Chem., 2008, 47, 1865 CrossRef CAS PubMed.
- I. Castro-Rodriguez and K. Meyer, J. Am. Chem. Soc., 2005, 127, 11242 CrossRef CAS PubMed.
- S. C. Bart, C. Anthon, F. W. Heinemann, E. Bill, N. M. Edelstein and K. Meyer, J. Am. Chem. Soc., 2008, 130, 12536 CrossRef CAS PubMed.
- J. G. Rebelein, Y. Hu and M. W. Ribbe, Angew. Chem., 2014, 53, 11543 CrossRef CAS PubMed.
- R. Angamuthu, P. Byers, M. Lutz, A. L. Spek and E. Bouwman, Science, 2010, 327, 313 CrossRef CAS PubMed.
- G. H. Jin, C. G. Werncke, Y. Escudiee, S. Sabo-Etienne and S. Bontemps, J. Am. Chem. Soc., 2015, 137, 9563 CrossRef CAS PubMed.
- T.-W. Chiou and W.-F. Liaw, Inorg. Chem., 2008, 47, 7908 CrossRef CAS PubMed.
- C.-M. Lee, Y.-L. Chuang, C.-Y. Chiang, G.-H. Lee and W.-F. Liaw, Inorg. Chem., 2006, 45, 10895 CrossRef CAS PubMed.
- N. A. M. Azman, S. Peiro, L. Fajari, L. Julia and M. P. Almajano, J. Agric. Food Chem., 2014, 62, 5743 CrossRef CAS PubMed.
- I. Castro-Rodriguez, H. Nakai, L. N. Zakharov, A. L. Rheingold and K. A Meyer, Science, 2004, 305, 1757 CrossRef CAS PubMed.
- W. J. Evans, C. A. Seibel and J. W. Ziller, Inorg. Chem., 1998, 37, 770 CrossRef CAS.
- D. G. Huang and R. H. Holm, J. Am. Chem. Soc., 2010, 132, 4693 CrossRef CAS PubMed.
- T.-T. Lu, S.-H. Lai, Y.-W. Li, I.-J. Hsu, L.-Y. Jang, J.-F. Lee, I.-C. Chen and W.-F. Liaw, Inorg. Chem., 2011, 50, 5396 CrossRef CAS PubMed.
- E. I. Solomon, B. Hedman, K. O. Hodgson, A. Dey and R. K. Szilagyi, Coord. Chem. Rev., 2005, 249, 97 CrossRef CAS.
- C.-M. Lee, C.-H. Chen, S.-C. Ke, G.-H. Lee and W.-F. Liaw, J. Am. Chem. Soc., 2004, 126, 8406 CrossRef CAS PubMed.
- J. H. Lunsford and J. P. Jayne, J. Phys. Chem., 1965, 69, 2182 CrossRef CAS.
- J. W. Rabalais, J. M. Mcdonald, V. Scherr and S. P. Mcglynn, Chem. Rev., 1971, 71, 73 CrossRef.
- X. L. Yin and J. R. Moss, Coord. Chem. Rev., 1999, 181, 27 CrossRef CAS.
- D. H. Gibson, Chem. Rev., 1996, 96, 2063 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 785531, 1435237 and 1435238. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04652a |
| This journal is © The Royal Society of Chemistry 2016 |