
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrong Brønsted acid promoted asymmetric hydrogenation of isoquinolines and quinolines catalyzed by a Rh–thiourea chiral phosphine complex via anion binding†
Jialin
Wen
a,
Renchang
Tan
a,
Shaodong
Liu
a,
Qingyang
Zhao
*b and
Xumu
Zhang
*a
aDepartment of Chemistry & Chemical Biology, Rutgers, The State University of New Jersey, 610 Taylor Road, Piscataway, New Jersey 08854, USA. E-mail: xumu@rci.rutgers.edu
bCollege of Chemistry & Materials Science, Northwest University, 1 Xuefu Road, Xi'an, 710127, China. E-mail: zhaoqy@nwu.edu.cn
First published on 26th January 2016
Abstract
Rhodium catalyzed asymmetric hydrogenation of both isoquinolines and quinolines provides a new method to synthesize chiral tetrahydroisoquinolines and tetrahydroquinolines. By introducing strong Brønsted acid HCl, anion binding between the substrate and the ligand was established to achieve high reactivity and high enantioselectivity (up to 99% conversion and 99% ee). An NMR study suggests an anion binding between the catalyst and the substrate. Deuterium labeling experiments reveal a plausible reaction pathway.
Introduction
As one of the important non-covalent interactions in nature, especially in biological systems, anion binding has been demonstrated as a powerful tool in organocatalysis.1 Triggered by the pioneering work done by Jacobsen and other groups, many successful asymmetric catalytic systems involving anion binding have been developed in recent years.2 On the other hand, utilizing the secondary interaction has been one of the fundamental ideas in catalyst design, which combines more catalytic centers (transition metal catalysis, organocatalysis and enzyme catalysis) in single molecules to provide an efficient chiral environment. Inspired by this strategy, we recently developed a novel ferrocene–thiourea chiral bisphosphine ligand (L1). Through a covalent linker, cooperation of transition metal catalysis and organocatalysis could be achieved. This strategy has been successfully applied in rhodium-catalyzed asymmetric hydrogenation of nitroalkenes and unprotected iminiums.3–5 Control experiments implied that the covalent linker was essential for high activities and enantioselectivities.4,5Tetrahydroquinolines (THQs) and tetrahydroisoquinolines (THIQs) are an important family of biologically active molecules, including natural alkaloids and important pharmaceutical products.6 Among various synthetic approaches for synthesizing chiral THQs and THIQs, direct hydrogenation, with a high atom economy, relatively simple procedure and easy work-up, deserves special attention. To the best of our knowledge, however, asymmetric hydrogenation of N-heteroaromatics, especially isoquinolines, remains a challenging task.7 Although quinolines have been successfully hydrogenated in several cases,8 there are only a few examples of isoquinolines. Zhou's group used an iridium–bisphosphine catalyst to obtain high enantioselectivity, but this transformation needs activation by chloroformate or the addition of BCDMH.9 By introducing chiral phosphoric acid, transfer hydrogenation of isoquinoline was achieved by Zhou in 2014 and N-protected 1,2-dihydroisoquinolines were synthesized with moderate enantioselectivities.10 In 2013, Mashima's group synthesized chiral THIQ using a dinuclear iridium(III)–bisphosphine complex, but the substrate scope is still limited to aromatic or bulky substituents (Scheme 1).11 Isoquinolines with less hindered alkyl substituents on the pro-chiral carbons are still challenging. Most of these catalysis cases for N-heteroaromatics are performed with ruthenium, iridium and palladium complexes.6 Rhodium was rarely reported in direct asymmetric hydrogenation of N-heteroaromatics with high turnovers and ee's.7 This is in sharp contrast to the broad application of Rh complexes in asymmetric hydrogenation of olefins, enamides and imines.12
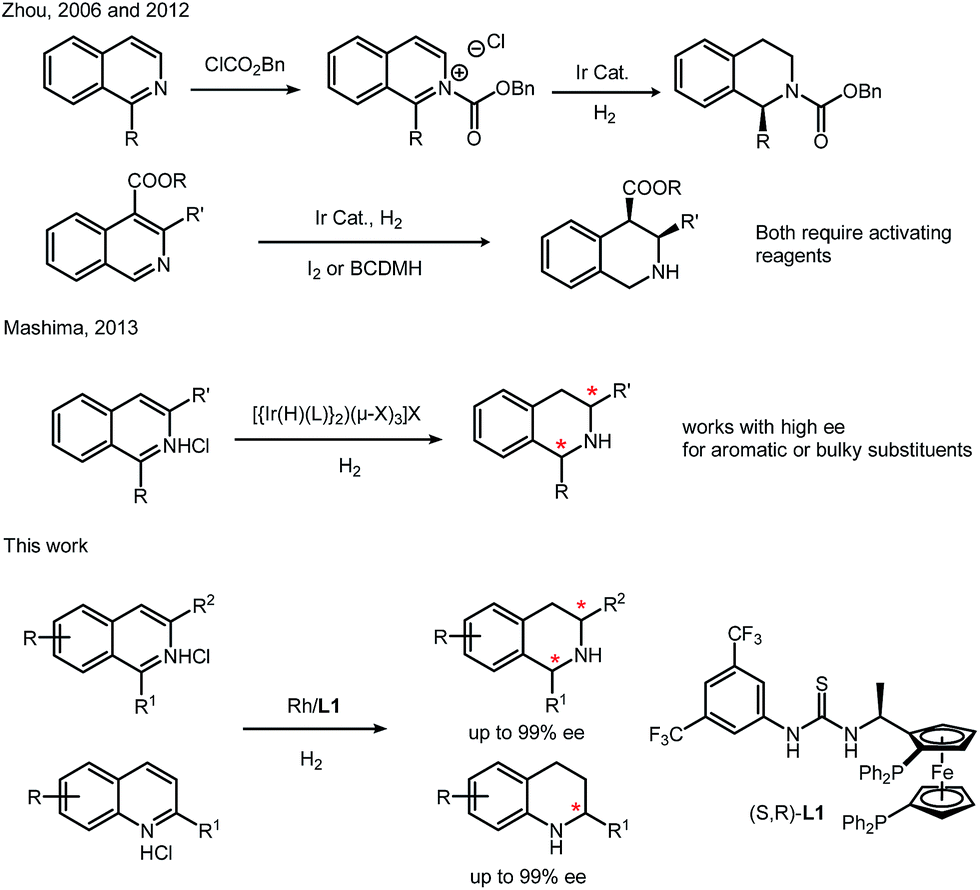 | ||
| Scheme 1 Asymmetric hydrogenation of isoquinolines. | ||
Continuing our research on the ferrocene-based thiourea chiral phosphine system, we herein report a highly reactive and enantioselective example of rhodium/bisphosphine-catalyzed asymmetric hydrogenation of isoquinolines and quinolines. In this catalytic system, a secondary interaction between the catalyst and the substrates was believed to occur via anion binding of ion-pair intermediates. We envision that the introduction of a strong Brønsted acid such as HCl, brings dual benefits by: (1) activating the aromatic ring,13 and (2) establishing a salt bridge between the catalyst and the substrates.
Results and discussion
Our study was initiated with 3-methylisoquinoline hydrochloride as a model substrate. The Rh catalyst was prepared in situ by mixing the metal precursor [Rh(COD)Cl]2 with (S, R)-L1. In accordance with our previous reports,4,5 this catalytic system is solvent-dependent. After screening of alkyl halide, alcohols and ethers, we found that under 40 atm H2 pressure at 40 °C, dichloromethane gives the best result (>99% conversion and 92% ee were observed with 1% catalyst loading). In addition to using single solvents, solvent pairs were also tested. A mixture of dichloromethane and isopropanol (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at 25 °C gives the optimal condition with 99% ee and full conversion. An iridium complex [Ir(COD)Cl]2/L1 also shows high activity with this solvent, but lower enantioselectivity than its Rh counterpart (Table 1, entry 5 vs. entry 8). Halide effect, which causes a considerable difference in some cases,14 was evaluated as well. Using [Rh(COD)I]2 as a metal precursor resulted in small changes in conversion and enantioselectivity (Table 1, entry 6 vs. entry 7).
:1, v/v) at 25 °C gives the optimal condition with 99% ee and full conversion. An iridium complex [Ir(COD)Cl]2/L1 also shows high activity with this solvent, but lower enantioselectivity than its Rh counterpart (Table 1, entry 5 vs. entry 8). Halide effect, which causes a considerable difference in some cases,14 was evaluated as well. Using [Rh(COD)I]2 as a metal precursor resulted in small changes in conversion and enantioselectivity (Table 1, entry 6 vs. entry 7).
|
|
||||
|---|---|---|---|---|
| Entry | Metal | Solvent | Conversionb | eec |
| a Reaction conditions: 1a (0.1 mmol) in 1.0 mL solvent, 1/[Rh(COD)Cl]2/L1 ratio = 100/0.5/1, 40 atm H2, 40 °C, 24 h. b Conversion was determined by 1H NMR analysis, no by product was observed. c ee was determined by GC with a chiral stationary phase. d Performed at 25 °C. | ||||
| 1 | [Rh(COD)Cl]2 | i-PrOH | >99% | 70% |
| 2 | [Rh(COD)Cl]2 | MeOH | 85% | 30% |
| 3 | [Rh(COD)Cl]2 | DCM | >99% | 92% |
| 4 | [Rh(COD)Cl]2 | Dioxane | 95% | 90% |
| 5 | [Rh(COD)Cl]2 | Dioxane/i-PrOH = 2:1 |
>99% | 90% |
| 6d | [Rh(COD)Cl]2 | DCM/i-PrOH = 2:1 |
>99% | 99% |
| 7d | [Rh(COD)I]2 | DCM/i-PrOH = 2:1 |
99% | 93% |
| 8 | [Ir(COD)Cl]2 | Dioxane/i-PrOH = 2:1 |
>99% | 83% |
Counterion effects were also investigated. Bromide or iodide anions do not have a significant influence on the conversion or enantioselectivity, while fluoride anions dramatically inhibit this reaction (for more details, see ESI, section 6†).
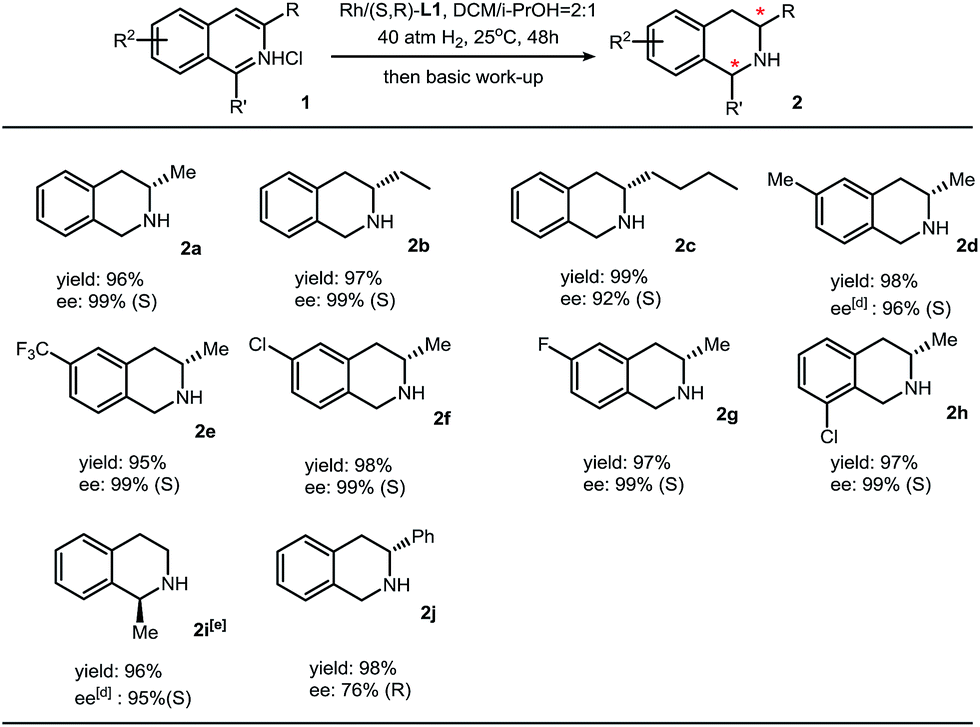
We then applied this method to synthesize various chiral THIQs. Different alkyl substituents at the 3-position or benzo ring do not show obvious changes in the yields and enantioselectivities. Aryl substituents, such as phenyl groups, on the 3-position lead to full conversion and moderate enantioselectivity (76% ee). In addition to 3-alkyl isoquinolines, 1-methylisouinoline was also hydrogenated with high enantioselectivity (Table 2).
|
a Reaction conditions: 1 (0.2 mmol) in 1.2 mL solvent, 1/[Rh(COD)Cl]2/L1 ratio = 100/0.5/1; yield was determined with isolated THIQ products; ee was determined by GC or HPLC with a chiral stationary phase; eperformed in dioxane/i-PrOH = 2:1 (v/v) at 60 °C under 60 atm H2 with 2% catalyst.
|
|---|
|
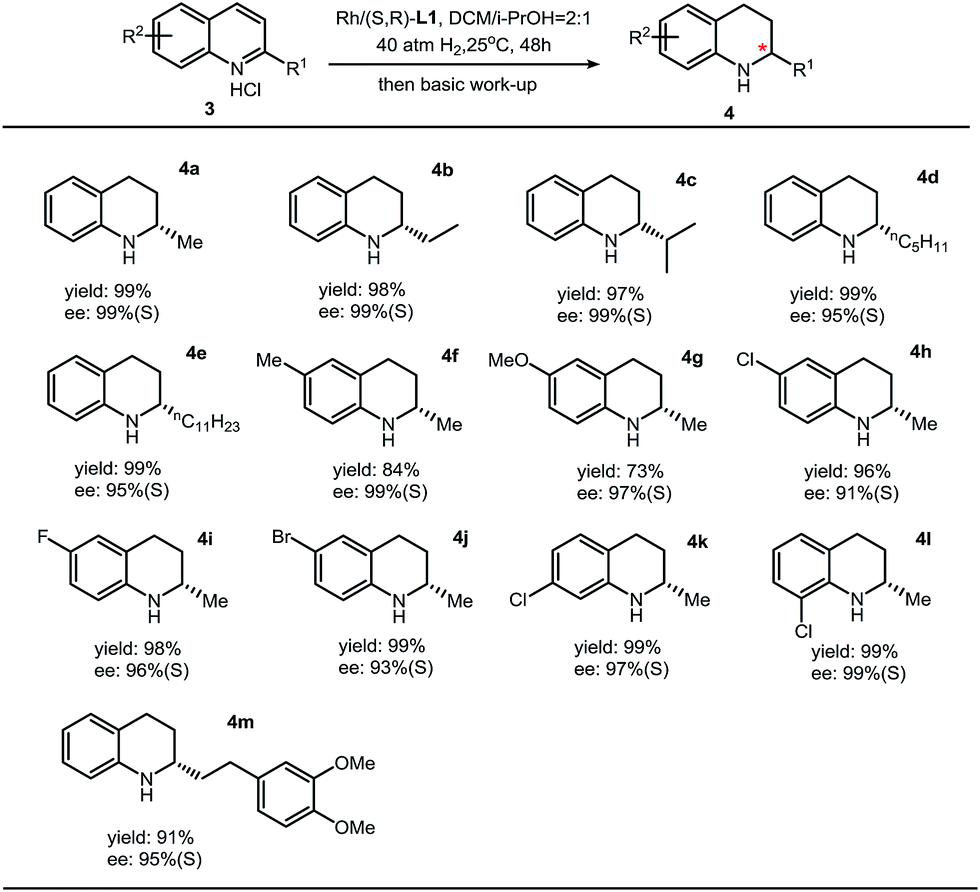
As far as we know, few catalytic systems can be applied for asymmetric hydrogenation of both isoquinolines and quinolines with both high turn-overs and excellent enantioselectivities.7,8 Then a question came to us: can this synthetic protocol be used to synthesize chiral THQs with high ee? By employing the optimized condition (DCM/iPrOH = 2:1, v/v) in the case of asymmetric hydrogenation of isoquinolines, we found that the enantioselectivity was dramatically increased to 99% ee.5 This observation, in return, suggests that this thiourea–Rh–bisphosphine catalytic system is highly solvent-dependent. The substrate scope of asymmetric hydrogenation of quinolines is broad. High enantioselectivities have been obtained with various substituents (Table 3).
| a Reaction conditions: 3 (0.2 mmol) in 1.2 mL solvent, 3/[Rh(COD)Cl]2/L1 ratio = 100/0.5/1; yield was determined with isolated THQ products; ee was determined by GC or HPLC with a chiral stationary phase. |
|---|
|
The potential application of this synthetic protocol was estimated: we have scaled up this asymmetric hydrogenation reaction into gram scale. No significant signs of changes in conversion or enantioselectivity were observed with 0.5% catalyst loading.15 In addition, the role of strong Brønsted acid HCl is essential in this chemical transformation. Without HCl, no hydrogenation product was observed.16
To gain evidence of a secondary interaction of L1 with the chloride anion of the substrates, we mixed L1 and 1a (3 eq.) in CDCl3. The 1HNMR showed similar obvious changes of chemical shifts (Fig. 1). The original thiourea N–H peaks (hidden in the aromatic peaks within 7.3–7.0 ppm) shift downfield to 9.10 and 7.62 (marked with red circles in Fig. 1). In addition, the three protons on the 3,5-bis(trifluoromethyl)phenyl group also shift downfield a little by 0.19 ppm (marked by an arrow in Fig. 1). These changes of chemical shifts, consistent with the case of L1 with Cl− from TBAC,5 suggest the anion binding between the ligand and the substrate.
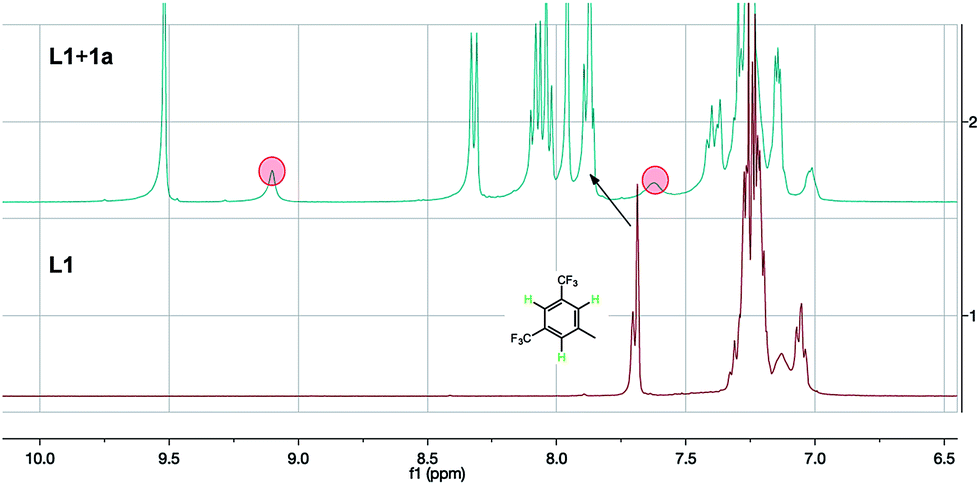 | ||
| Fig. 1 NMR study of the interaction of L1 and 1a. | ||
We synthesized a series of analogues of L1 and conducted control experiments to evaluate the collaboration manner of each unit of L1 (Table 4). Urea bisphosphine ligand L2 only gives trace product, and this sharp contrast suggests the crucial role of the thiourea moiety in L1. Compared to H (L3) and methyl (L4), a more electron-withdrawing trifluoromethyl group at the 3- and 5-position on the phenyl ring increases the enantioselectivity, which is probably due to the stronger acidity of the N–H proton on the thiourea. After N-methylation of the less acidic thiourea N–H proton, enantioselectivity results in a minor decrease. This observation reveals that the more acidic thiourea N–H proton contributes mostly in anion binding with chloride. Furthermore, monophosphine ligand L6 and the mixture of ferrocene–thiourea compound L7 with triphenylphosphine can hardly catalyze the hydrogenation reaction. On the other hand, the mixture of thiourea molecules and bisphosphine–Ugi's amine L8 failed to show catalytic activity. These results (L1vs.L7/PPh3 and L8/thiourea) demonstrate the importance of a covalent incorporation of a bisphosphine moiety and thiourea. The idea of secondary interactions offers an alternative strategy for asymmetric hydrogenation.
| a Reaction conditions: 1 (0.1 mmol) in 0.6 mL solvent, 1/[Rh(COD)Cl]2/ligand ratio = 100/0.5/1; conversion was determined by 1H NMR analysis; ee was determined by GC with a chiral stationary phase. |
|---|
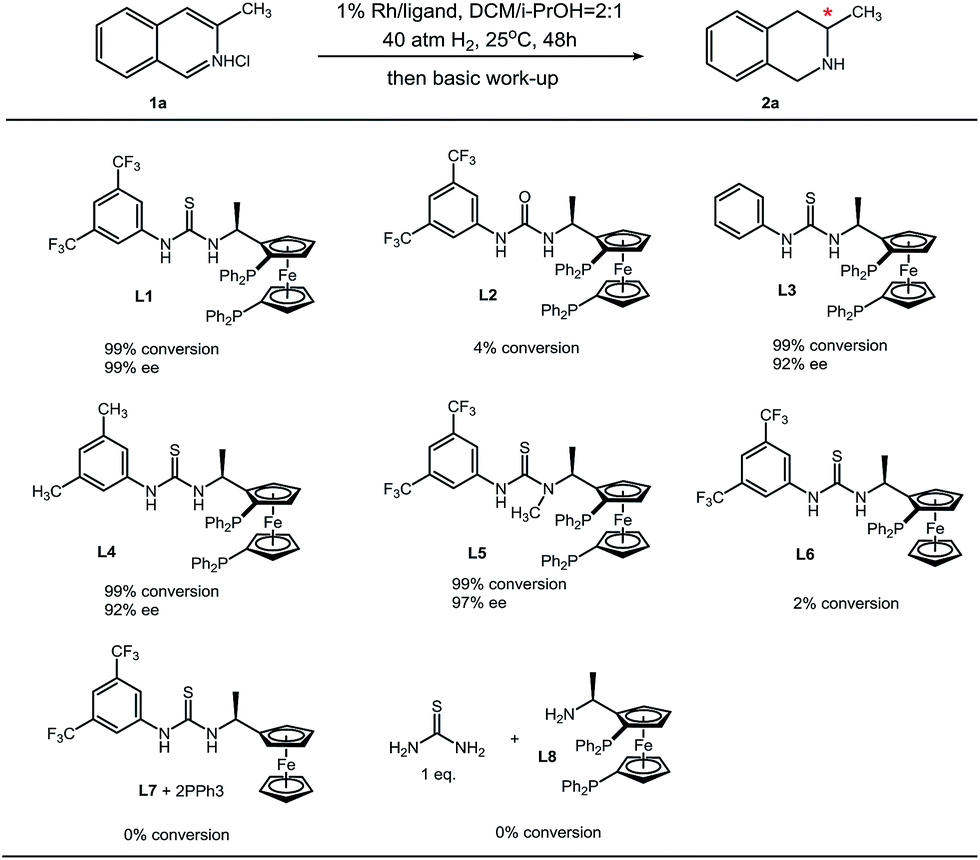
|
In order to obtain insight of this transformation, deuterium labeling experiments were conducted. First, 3-methylisoquinoline hydrochloride was reduced by deuterium gas in methanol (Scheme 2, eqn (1)). D atoms are added only at the 1- and 3-positions. This indicates that the hydrogen atom at the 4-position of the product comes from the methanol solvent. Second, 3-methylisoquinoline deuterochloride was reduced with hydrogen gas in deuterated methanol (Scheme 2, eqn (2)). Hydrogen atoms are added at the 1- and 3-positions. The original H at the 4-position, however, was exchanged with D, resulting in two D atoms at the 4-position. This indicates that the D atoms at the 4-position come from the methanol-d4 solvent. A tautomerization is probably responsible for this H–D exchange. Interestingly, H–D exchange also occurs on the methyl group (53% original H was replaced by D). This observation, in return, proves the existence of tautomerization. Based on these results, we proposed a possible path for this transformation (Scheme 2). After addition of hydrogen at the 1-position, the substrate is partially reduced, leading to a conjugate enamine, which could not be further reduced in this rhodium–thiourea–bisphosphine catalytic system. After tautomerization follows, giving an iminium intermediate, which could be easily hydrogenated.5 Another tautomer, a terminal enamine is less preferred than its conjugate counterpart, because the latter is more stable. This explains why H atoms on the methyl group were partially replaced by D (but H at the 4-position is always replaced by D) (for details, please see ESI†).
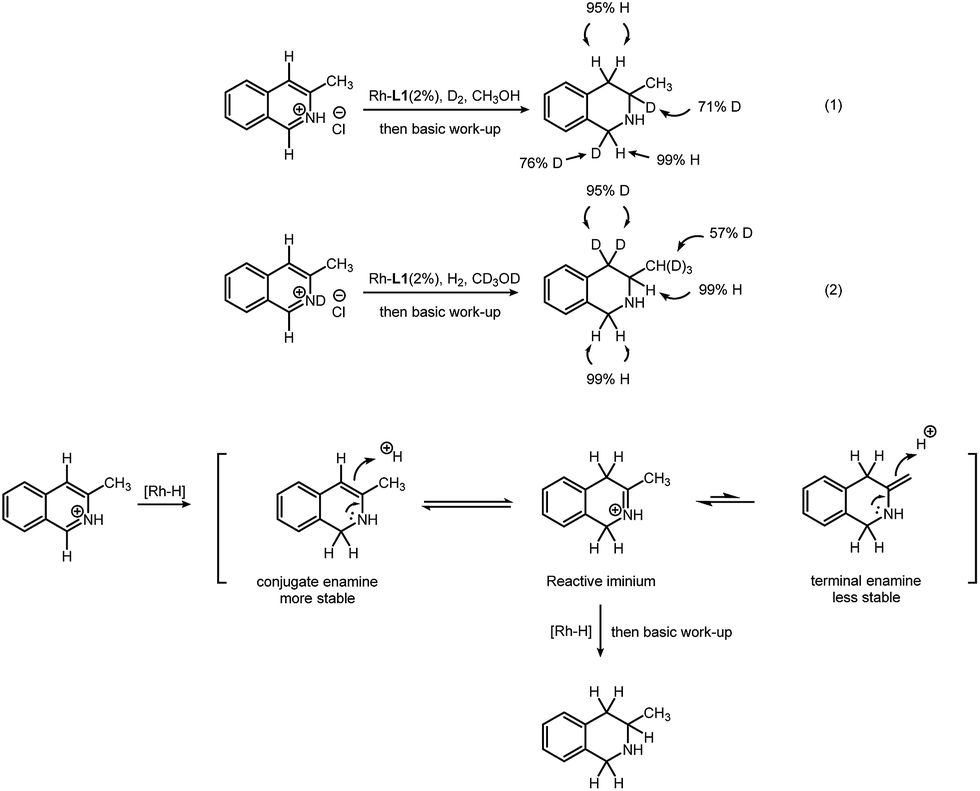 | ||
| Scheme 2 Deuterium labeling experiment and proposed transformation path. | ||
Recent reports revealed an outer-sphere mechanism for bisphosphine–transition metal catalyzed hydrogenation. In 2014, Zhou reported a series of detailed studies on palladium catalyzed asymmetric hydrogenation of protonated indoles. After careful evaluation, hydride transfer from a Pd–H complex to an iminium intermediate was proposed (Scheme 3, eqn (1)).17 In 2015, Mashima's group proposed a mechanism for iridium-catalyzed asymmetric hydrogenation of isoquinoline hydrogen halide salts. Strong experimental evidence was presented to favor an outer-sphere mechanism, in which hydrogen bonding between an Ir–Cl complex and N–H proton was proposed (Scheme 3, eqn (2)).18 Both of these mechanism studies above were focused on hydrogenation of the halide salts of aromatic compounds. Similarly, the nature of protonated (iso)quinolines suggests that it seems not possible for the substrates to coordinate to the rhodium complex in this case, no matter if in a σ-bonding or a π-bonding manner. A hydride transfer mechanism is more likely in this catalytic case, instead of the traditional inner-sphere mechanism that involves the direct coordination of unsaturated bonds to the metal. Based on previous reports and our deuterium labeling experiments, we propose a catalytic cycle to explain a plausible outer-sphere mechanism (Scheme 3). After oxidative addition of H2, a dihydride rhodium(III) complex is formed. The chloride anion of the isoquinolinium salt will bind with the hydrogen of thiourea, forming a Rh dichloro–dihydride anionic complex. The equatorial hydride of the active rhodium complex will be transferred to the isoquinolinium due to the strong trans effect of a phosphine. After this 1,2-addition, a tautomerization step will follow, giving an iminium intermediate. The iminium, instead of an enamine, will undergo the insertion of hydride from a rhodium dihydride complex. The chirality of the product originates in this step. Another molecule of hydrogen will react and form the active rhodium dihydride species, finishing the catalytic cycle. In order to approve the mechanism of this rhodium catalyzed asymmetric hydrogenation of (iso)quinolinium salts, further study is needed in the future.
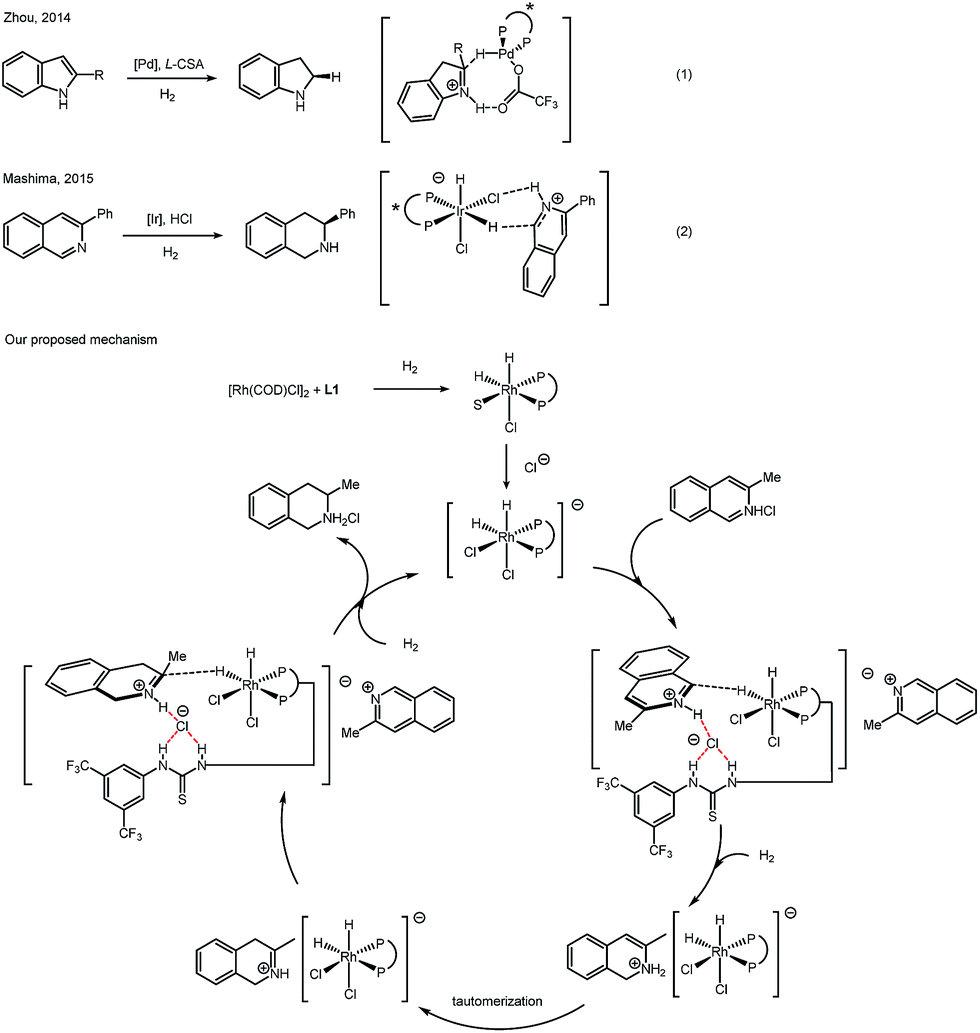 | ||
| Scheme 3 Outer-sphere mechanism in asymmetric hydrogenation of N-heteroaromatics. | ||
Conclusions
In summary, we report a successful example of Rh-catalyzed asymmetric hydrogenation of both isoquinolines and quinolines. Compared with previous catalytic systems, this method has a broader substrate scope. The strong Brønsted acid HCl activates the N-heteroaromatic substrates and introduces anion binding into the catalytic system, which played an important role in this transformation. Deuterium labeling experiments revealed an enamine–iminium tautomerization equilibrium after the first hydride transfer step.Acknowledgements
We are grateful for Mr Weijie Chen's helpful discussion and Mr Yang Gao's technical support.Notes and references
- For reviews, see: (a) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603 CrossRef CAS PubMed; (b) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534 CrossRef CAS PubMed; (c) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518 CrossRef CAS PubMed.
- Recent advances in ion-pair catalysis: (a) S. Lin and E. N. Jacobsen, Nat. Chem., 2012, 4, 817 CrossRef CAS PubMed; (b) M. P. Lalonde, M. A. McGowan, N. S. Rajapaksa and E. N. Jacobsen, J. Am. Chem. Soc., 2013, 135, 1891 CrossRef CAS PubMed; (c) G. Bergonzini, C. S. Schindler, C.-J. Wallentin, E. N. Jacobsen and C. R. Stephenson, Chem. Sci., 2014, 5, 112 RSC; (d) C. Min, N. Mittal, D. X. Sun and D. Seidel, Angew. Chem., Int. Ed., 2013, 52, 14084 CrossRef CAS PubMed; (e) C. Min, C. Lin and D. Seidel, Angew. Chem., Int. Ed., 2015, 54, 6608 CrossRef CAS PubMed; (f) A. G. Schafer, J. M. Wieting, T. J. Fisher and A. E. Mattson, Angew. Chem., Int. Ed., 2013, 52, 11321 CrossRef CAS PubMed.
- X. Dong, Q. Zhao, P. Li, C. Chen and X. Zhang, Org. Chem. Front., 2015, 2, 1424 Search PubMed.
- Q. Zhao, S. Li, K. Huang, R. Wang and X. Zhang, Org. Lett., 2013, 15, 4014 CrossRef CAS PubMed.
- Q. Zhao, J. Wen, R. Tan, K. Huang, P. Metola, R. Wang, E. V. Anslyn and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 8467 CrossRef CAS PubMed.
- (a) V. Sridharan, P. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157–7259 CrossRef CAS PubMed; (b) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef CAS PubMed; (c) K. W. Bentley, Nat. Prod. Rep., 2006, 23, 444 RSC.
- (a) Y. Zhou, Acc. Chem. Res., 2007, 40, 1357 CrossRef CAS PubMed; (b) D. Wang, Q. Chen, S. Lu and Y. Zhou, Chem. Rev., 2012, 112, 2557 CrossRef CAS PubMed.
- Selected successful examples of asymmetric hydrogenation of quinolines: (a) S. Lu, X. Han and Y. Zhou, Adv. Synth. Catal., 2004, 346, 909 CrossRef CAS; (b) K. H. Lam, L. Xu, L. Feng, Q. Fan, F. L. Lam, W. Lo and A. Chan, Adv. Synth. Catal., 2005, 347, 1755 CrossRef CAS; (c) S. M. Lu, Y. Q. Wang, X. W. Han and Y. G. Zhou, Angew. Chem., Int. Ed., 2006, 45, 2260 CrossRef CAS PubMed; (d) H. Zhou, Z. Li, Z. Wang, T. Wang, L. Xu, Y. He, Q. Fan, J. Pan, L. Gu and A. Chan, Angew. Chem., Int. Ed., 2008, 47, 8464 CrossRef CAS PubMed; (e) W. Tang, J. Tan, L. Xu, K. Lam, Q. Fan and A. Chan, Adv. Synth. Catal., 2010, 352, 1055 CrossRef CAS; (f) F. Gou, X. Zhang and Y. Liang, Adv. Synth. Catal., 2010, 352, 244 CrossRef; (g) T. Wang, L. Zhuo, Z. Li, F. Chen, Z. Ding, Y. He, Q. Fan, J. Xiang, Z. Yu and A. Chan, J. Am. Chem. Soc., 2011, 133, 9878 CrossRef CAS PubMed; (h) C. Deport, M. Buchotte, K. Abecassis, H. Tadaoka, T. Ayad, T. Ohshima, J. Genet, K. Mashima and V. Ratovelomanana-Vidal, Synlett, 2007, 17, 2743 Search PubMed.
- (a) S. M. Lu, Y. Q. Wang, X. W. Han and Y. G. Zhou, Angew. Chem., Int. Ed., 2006, 45, 2260 CrossRef CAS PubMed; (b) L. Shi, Z. Ye, L. Cao, R. Guo, Y. Hu and Y. Zhou, Angew. Chem., Int. Ed., 2012, 51, 8286 CrossRef CAS PubMed.
- L. Shi, Y. Ji, W. Huang and Y. Zhou, Acta Chim. Sin., 2014, 72, 820–824 CrossRef CAS.
- A. Iimuro, K. Yamaji, S. Kandula, T. Nagano, Y. Kita and K. Mashima, Angew. Chem., Int. Ed., 2013, 52, 2046 CrossRef CAS PubMed.
- For reviews, see: (a) J. Xie, S. Zhu and Q. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef CAS PubMed; (b) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (c) W. Zhang, Y. Chi and X. Zhang, Acc. Chem. Res., 2007, 40, 1278 CrossRef CAS PubMed.
- Z. Yu, W. Jin and Q. Jiang, Angew. Chem., Int. Ed., 2012, 51, 6060 CrossRef CAS PubMed.
- K. Fagnou and M. Lautens, Angew. Chem., Int. Ed., 2002, 41, 26 CrossRef CAS.
- 99% yield and 98% ee was obtained in asymmetric hydrogenation of 5 mmol 2-methylisoquinolinium chloride with 0.5% catalyst.
- 100% recovery of starting material.
- Y. Duan, L. Li, M. Chen, C. Yu, H. Fan and Y. Zhou, J. Am. Chem. Soc., 2014, 136, 7688 CrossRef CAS PubMed.
- Y. Kita, K. Yamaji, K. Higashida, K. Sathaiah, A. Iimuro and K. Mashima, Chem.–Eur. J., 2015, 21, 1915 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04712a |
| This journal is © The Royal Society of Chemistry 2016 |