
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient blue thermally activated delayed fluorescent OLEDs with record-low driving voltages utilizing high triplet energy hosts with small singlet–triplet splittings†
Dongdong
Zhang
,
Minghan
Cai
,
Zhengyang
Bin
,
Yunge
Zhang
,
Deqiang
Zhang
and
Lian
Duan
*
Key Lab of Organic Optoelectronics and Molecular Engineering of Ministry of Education, Department of Chemistry, Tsinghua University, Beijing 100084, P. R. China. E-mail: duanl@mail.tsinghua.edu.cn; Fax: +86-10-62795137; Tel: +86-10-62788802
First published on 12th February 2016
Abstract
The high driving voltage of blue organic light-emitting diodes (OLEDs) based on emitters with thermally activated delayed fluorescence (TADF) remains a constraint for their portable application. A major reason for this is that the high triplet (T1) of the host required to match the blue TADF emitters would always lead to inferiority in terms of carrier injection. Therefore, a suitable host should possess not only a high T1 but also a relatively low singlet (S1) for improved carrier injection, indicating that small singlet–triplet splittings (ΔESTs) are highly desired. Here, four carbazolyl benzonitrile derivatives are facilely prepared in a one-step approach with restrained conjugate lengths to maintain high triplet energies while their highly twisted structures spatially separate the frontier orbital distribution to achieve relatively low ΔESTs. Meanwhile, the charge transporting mobilities of these hosts are effectively tuned by the different linker types of the host moieties. Consequently, high-triplet-energy hosts with favorable carrier injection/transporting abilities are realized, endowing blue TADF devices with a maximum external quantum efficiency of 21.5%, a maximum power efficiency of 42.0 lm W−1 and an ultra-low onset voltage of 2.8 V. It is noteworthy that a driving voltage of 4.9 V is achieved at a practical luminance of 1000 cd m−2, which is the lowest among the doped blue TADF OLEDs reported until now. This work suggests that manipulation of the molecular topologies not only leads to the flexible and feasible design of novel bipolar host materials, but also affords a promising method for fine-tuning physical properties and thus obtaining state-of-the-art device performances.
Introduction
Conceptual advancements have led to exciting approaches to harness both the spin-symmetric and anti-symmetric molecular excitations formed under electrical excitations to achieve maximum luminous efficiencies in organic light-emitting diodes (OLEDs).1–6 One successful example of this is the utilization of phosphorescent materials, which incorporate heavy metals into the organic aromatic frameworks to facilitate the transition from triplets to the ground states to give out phosphorescence, though the noble metals used may increase the cost of the OLEDs.1,7–10 A potential alternative is to use materials with thermally activated delayed fluorescence (TADF), as proposed by Adachi et al.11 Non-radiative triplet excitons (T1) can be transformed to emissive singlet excitons (S1) in TADF molecules due to the small singlet–triplet energy splittings (ΔESTs), leading to internal quantum efficiencies (IQE) of unity, without using heavy metal atoms. Since then, highly efficient TADF emitters covering the whole of the visible spectrum have been reported,12–16 highlighting the potential of such materials as next-generation OLED emitters.To maximize the performances of TADF dopants in OLEDs, hosts with high T1 energies are required.17,18 Commonly used hosts for TADF emitters are conventional unipolar electron- or hole-transport materials, such as N,N′-dicarbazolyl-4,4′-biphenyl (CBP),12,13 1,3-bis(carbazolyl)benzene (mCP) or bis(2-(di(phenyl)phosphino)-phenyl)ether oxide (DPEPO),11,14,16 which have been used to achieve high external quantum efficiencies (EQE) above 10%. Nevertheless, due to the large energy gap between the S1 and T1 energies of those compounds, a high T1 is always accompanied with an even higher S1, leading to a mismatch in the frontier energy levels with the adjacent function layers, and consequently, high device operation voltages.19,20 This influence is even more significant for true-blue TADF emitters with emission peaks shorter than 470 nm, in virtue of their higher triplet energies of over 2.6 eV. Therefore, the high driving voltage of blue TADF OLEDs has been one of the constraints for their portable application. For instance, devices based on a highly efficient blue TADF emitter, 10,10′-(sulfonylbis(4,1-phenylene))bis(9,9-dimethyl-9,10-dihydroacridine) (DMAC-DPS), showed a onset voltage of 3.7 V using DPEPO as the host.21 Lee et al. also reported blue TADF OLEDs based on thiazine/carbazole derivatives with onset voltages as high as ≈4 V.14 To reduce the voltage of the blue TADF devices, Xu and Wei et al. developed multiphosphine-oxide hosts and an onset voltage as low as 2.8 V was achieved.22 However, at a practical luminance of 1000 cd m−2, the operation voltage is still as high as 5.9 V. Therefore, further development of hosts for blue TADF devices is still highly desired to promote their performance, especially to reduce the driving voltages.
A suitable host material to reduce the device voltage should possess not only a high T1 to fit the excited level of the TADF emitters, but also a relatively low S1 for improved carrier injection, rendering a small ΔEST. However, ΔESTs of most hosts reported are in the range of 0.5–1.0 eV.23 In principle, small ΔESTs are usually obtained in molecules with charge transfer (CT) states resulting from spatially separated donor and acceptor units.11–16 The difficulty in designing hosts with small ΔESTs for blue TADF emitters is maintaining the high triplet energy, which is in conflict with the CT states on account of the significant decrease in the excited state energy of the CT states. Therefore, high triplet energy hosts with ΔESTs lower than 0.5 eV are rarely reported.19,24 Furthermore, beside from the frontier energy levels, it is noteworthy that the charge transporting mobilities of the hosts are also crucial in determining the device voltage, which should be finely tuned. In this work, four facilely prepared carbazolyl benzonitrile derivatives (CzBNs) were designed and synthesized. On one hand, restrained molecule conjugate lengths are fulfilled by utilizing the small-sized cyano unit as the electron-withdrawing moiety to maintain high triplet energies. On the other hand, highly twisted structures are adopted to spatially separate the frontier energy level distribution, resulting in relatively low ΔESTs, leading to favorable frontier energy levels matching the adjacent layers. It is also observed that the charge transporting mobilities of the hosts can be efficiently tuned by the different linker types of the moieties, attributed to the required efficient overlap of the frontier orbital levels of the adjacent molecules for improved charge transportation. Blue TADF devices based on the optimized hosts show a maximum EQE of 21.5% with a record-low operation voltage of 4.9 V at a practical luminance of 1000 cd m−2 among the doped blue TADF OLEDs reported up until now. The results ensure the validity of our host design concept and may shed light on developing more efficient hosts for blue TADF OLEDs.
Results and discussion
Synthesis and characterization
The compounds (as shown in Scheme 1) 2,4-di(9H-carbazol-9-yl)benzonitrile (2,4-2CzBN), 2,6-di(9H-carbazol-9-yl) benzonitrile (2,6-2CzBN), 3,5-di(9H-carbazol-9-yl)benzonitrile (3,5-2CzBN) and 2,4,6-tri(9H-carbazol-9-yl)benzonitrile (3CzBN), were facilely prepared through aromatic nucleophilic substitution reactions by using carbazolyl units as nitrogen nucleophiles and the cyano unit as the electrophile. All the synthetic routes were one-step reactions with yields higher than 90%. These compounds are cost effective since no palladium or other rare-earth-metal catalysts are required. All of the compounds were fully characterized using 1H NMR spectroscopy, mass spectrometry and elemental analysis (ESI†). The molecular structures of 2,4-2CzBN, 2,6-2CzBN and 3,5-2CzBN were further determined by single-crystal X-ray crystallographic analysis (Fig. S1†). | ||
| Scheme 1 The synthesis process of the CzBNs. | ||
In principle, to achieve hosts with a high T1 as well as small ΔESTs, limited molecule conjugate lengths and spatially separated donor and acceptor moieties should be fulfilled simultaneously. Molecules were designed with mCP as the prototype on account of its limited conjugate length with a high triplet energy of 3.0 eV.25 The small-sized cyano moiety, as an electron-withdrawing unit, is adopted to enhance the electron affinity at the same time as restraining the conjugate length of the hosts. The planar structures of the carbazolyl units induce large steric hindrance, resulting in highly twisted structures with dihedral angles between the carbazole unit and the benzene ring of 41 °C and 68 °C for 2,4-2CzBN, 54 °C and 89 °C for 2,6-2CzBN and 50 °C and 65 °C for 3,5-2CzBN, respectively, which can be observed from the single-crystal structures. Such structures facilitate the spatially separated frontier energy levels demonstrated later. In the meantime, to obtain sufficient electron transportation, the cyano moieties of different molecules should facilely achieve mutual contact with each other, which may be hindered by the surrounding carbazolyl units. Therefore, the molecule linkage type is adjusted with carbazolyl units attached at the 2,4-, 2,6- and 3,5-positions of benzonitrile, respectively, to tune the charge transport mobilities of the CzBNs.
Theoretical calculations
To understand the structure–property relationship of the compounds at the molecular level, the geometrical and electronic properties of the compounds were studied using density functional theory (DFT) and time-dependent DFT (TDDFT) calculations with the B3LYP hybrid functional. As can be seen from Fig. 1, highly twisted structures with dihedral angles between the carbazolyl units and the benzene ring in the range of 49–70° were observed from the optimized molecular structures, corresponding to the experimental values obtained from the single-crystal structures. The highly twisted structures lead to spatially separated frontier orbital distributions with the highest occupied molecular orbitals (HOMOs) being mainly delocalized over the carbazolyl moieties while the lowest unoccupied molecular orbitals (LUMOs) are mainly centered on the benzonitrile moieties. In principle, the energy difference between the T1 and S1 states is determined by the exchange energy (J) of the two unpaired electrons in the excited states, which is twice the value of J, as shown in eqn (1):23| ΔEST = ES − ET = 2J | (1) |
Meanwhile, the value of J can be calculated by eqn (2) as follows:23
 | (2) |
 | ||
| Fig. 1 Theoretically calculated spatial distributions and energies of the HOMO and LUMO levels of the hosts as well as their singlet and triplet energies and values of ΔEST. | ||
To evaluate the ΔEST values, the triplet and the singlet energies were theoretically calculated based on the molecule ground state geometry with triplet energies of 2.88 eV, 2.95 eV, 2.97 eV and 2.83 eV and singlet energies of 3.15 eV, 3.12 eV, 3.29 eV and 3.04 eV for 2,4-2CzBN, 2,6-2CzBN, 3,5-2CzBN and 3CzBN, respectively. The ΔESTs of these hosts can therefore be calculated to be 0.27 eV, 0.17 eV, 0.32 eV and 0.21 eV for 2,4-2CzBN, 2,6-2CzBN, 3,5-2CzBN and 3CzBN, respectively. Besides, singlet and triplet energies based on optimized excited state geometries were also calculated and the ΔESTs are 0.45 eV, 0.33 eV, 0.46 eV and 0.39 eV for 2,4-2CzBN, 2,6-2CzBN, 3,5-2CzBN and 3CzBN, respectively. For most organic molecules, S1 is considerably higher than T1 by 0.5–1.0 eV. Therefore, the ΔESTs of these hosts are relatively small, which arises from the spatially separated HOMO and LUMO levels of those hosts.
For all CzBNs, the carbazolyl units act as the hole-transporting moieties while the benzonitrile acts as the electron-transporting unit, respectively, in consideration of the frontier orbital distributions. The HOMO energy levels of the compounds are calculated to be 1.80 eV, 1.82 eV, 1.82 eV and 1.94 eV while the LUMO levels are 5.62 eV, 5.63 eV, 5.73 eV and 5.65 eV for 2,4-2CzBN, 2,6-2CzBN, 3,5-2CzBN and 3CzBN, respectively. The differences in the HOMOs and LUMOs of the CzBNs are quite small, which is ascribed to the identical carrier transport units used in all CzBNs.
Thermal properties
The good thermal stabilities of the compounds are indicated by their high decomposition temperatures (Fig. S2†), Td (corresponding to 5% weight loss), which are 350 °C, 360 °C, 346 °C and 401 °C for 2,4-2CzBN, 2,6-2CzBN, 3,5-2CzBN and 3CzBN, respectively, determined through thermogravimetric analysis (TGA). Their glass-transition temperatures (Tg), determined through differential scanning calorimetry, are 101 °C, 104 °C, 96 °C and 153 °C for 2,4-2CzBN, 2,6-2CzBN, 3,5-2CzBN and 3CzBN, respectively. The high Tgs can be attributed to the highly twisted structures of those materials. Compared with the other three hosts, the Td and Tg values of 3CzBN are much higher, resulting from the relatively larger molecular weight of 3CzBN. These values indicate that these compounds can form uniform amorphous films upon evaporation, which is highly important in improving the efficiencies and lifetimes of OLEDs.Carrier transporting properties
A time-of-flight (TOF) technique was utilized to measure the carrier mobilities of the hosts. The device used for TOF measurement was prepared through vacuum deposition of the tri-layer structure of ITO glass/host (2 μm)/Al (150 nm). Fig. 2 shows the hole (μh) and electron (μe) mobilities plotted as functions of the electric field (E), verifying that all CzBNs are bipolar hosts with μes higher than μhs. Although the electron-withdrawing units are identical for all hosts, their μes are diverse, with μe values of 3.21 × 10−4, 2.4 × 10−5, 1.87 × 10−3 and 5.53 × 10−5 cm2 V−1 s−1 for 2,4-2CzBN, 2,6-2CzBN, 3,5-2CzBN and 3CzBN at 670 (V cm−1)1/2, respectively. The μes follow the sequence of 3,5-2CzBN > 2,4-2CzBN > 2,6-2CzBN ≈ 3CzBN. The reason for this can be revealed by the different spatial frontier orbital distributions resulting from the linker type of the moieties. The dominant electron transport mechanism in the host materials is electron hopping between adjacent molecules, which requires sufficient orbital overlap between the LUMOs of the molecules. For these hosts, the electron transporting moieties are the benzonitrile moieties, which have the LUMO levels mainly localized on them. This indicates that the cyano units of adjacent molecules should facilely achieve mutual contact with each other to achieve efficient electron transport. Since the cyano moieties are relatively small, the moieties surrounding it will induce a significant steric hindrance effect, which may prevent contact between the cyano units, and thus lead to low electron transporting mobility. For 3,5-2CzBN, there are no carbazolyl units around the cyano units, while there is one for 2,4-2CzBN and two for 2,6-2CzBN and 3CzBN. Therefore, the influence of the steric effect of the carbazolyl units on the electron transport in 3,5-2CzBN is quite limited, manifesting in the largest μe. On the contrary, the electron transport for 2,6-2CzBN will be hindered by the surrounding carbazolyl units, resulting in the smallest μe. Since the cyano moieties of 3CzBN are also being surrounded by the carbazole units, its μe is similar to 2,6-2CzBN, verifying our hypothesis. The linker type also demonstrates an influence on the hole transporting abilities of the hosts. Compared to 2,4-2CzBN and 3,5-2CzBN with μh values of 8.45 × 10−5, 8.8 × 10−5 at 707 (V cm−1)1/2, respectively, the μh value of 2,6-2CzBN is much lower, and is 2.32 × 10−6 at 806 (V cm−1)1/2.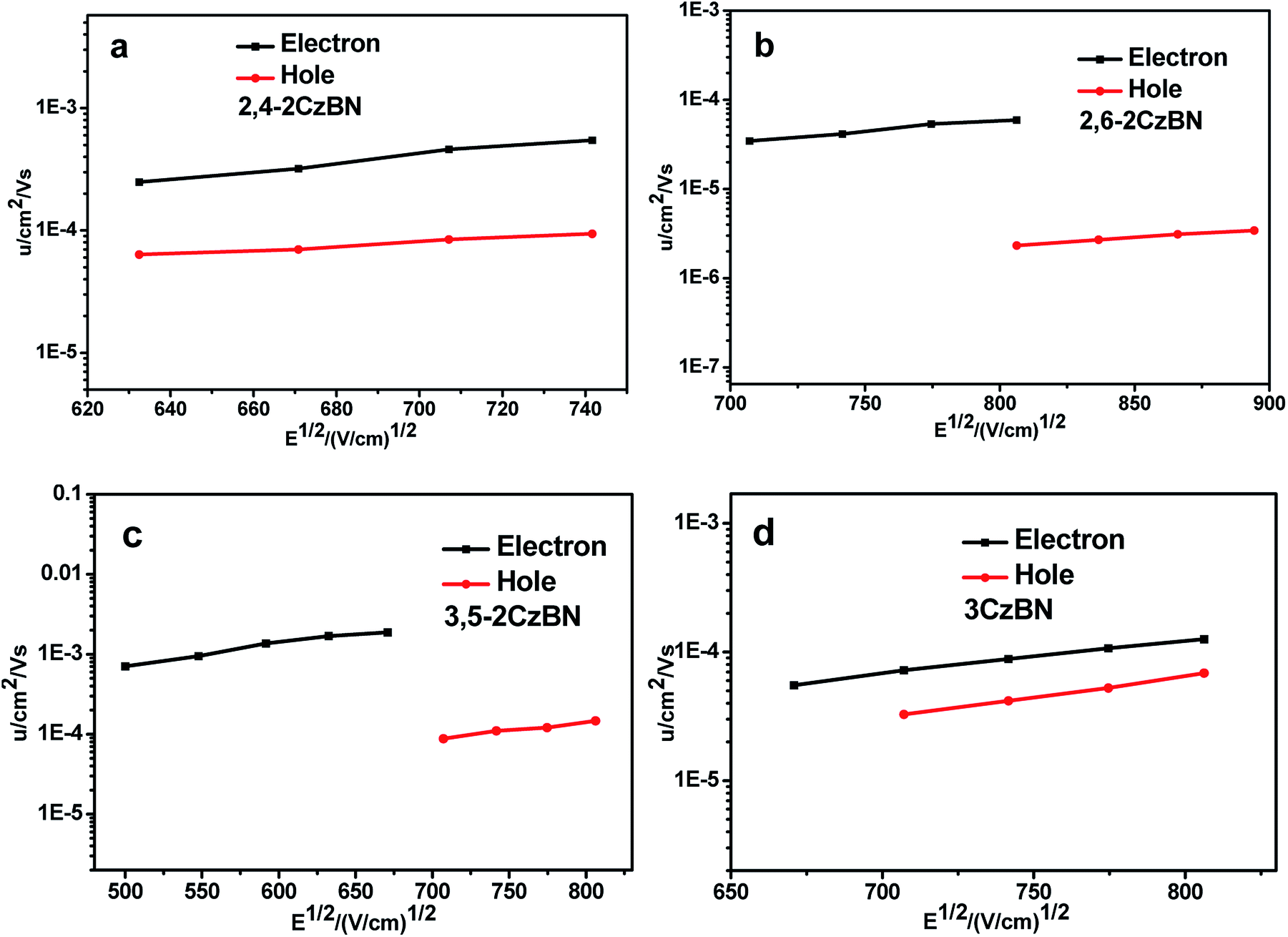 | ||
| Fig. 2 The electron and hole mobilities of (a) 2,4-2CzBN, (b) 2,6-2CzBN, (c) 3,5-2CzBN and (d) 3CzBN. | ||
Photophysical and electrochemical properties
To reveal the origin of the absorption peaks, the absorption spectra of the hosts were measured (Fig. 3a) with that of mCP for comparison. The absorption spectra of all the compounds exhibit similar patterns with three absorption bands centered at 250–350 nm, which are assigned to the n–π* and π–π* transitions of the carbazolyl units. Compared with mCP, a longer-wavelength absorption at about 360 nm was observed for CzBNs, due to CT states being formed by charge transfer from the carbazolyl units to the benzonitrile moiety. The absorption intensities of the CT states of 2,6-2CzBN and 3CzBN are much higher than that of 2,4-2CzBN and 3,5-2CzBN, indicating more efficient charge transfer. The energy gaps (Egs) for the hosts can be obtained from the absorption spectra, which are 3.31 eV, 3.19 eV, 3.34 eV and 3.10 eV for 2,4-2CzBN, 2,6-2CzBN, 3,5-2CzBN and 3CzBN, respectively, all smaller than that of mCP (3.55 eV). The smallest Eg also leads to the longest emission wavelength of 3CzBN, which is centered at 431 nm in the solution of toluene. The emission peaks of 2,4-2CzBN, 2,6-2CzBN and 3,5-2CzBN are also measured to be 405 nm, 404 nm and 396 nm, respectively. It has been reported that for a CT state without any vibronic structure, its S1 should be calculated from the onset of its broad emission band.26 Therefore, the singlet energies are calculated to be 3.34 eV, 3.27 eV, 3.39 eV and 3.20 eV, while the triplet energies, determined by the highest-energy vibronic sub-band of the phosphorescence spectra at 77 K, are 2.95 eV, 3.03 eV, 3.03 eV and 2.99 eV for 2,4-2CzBN, 2,6-2CzBN, 3,5-2CzBN and 3CzBN, respectively (Fig. S3†). The triplet energies of the CzBNs approach the one of mCP, indicating that the introduced CT states show quite a limited influence on the triplet energies of the compounds. Meanwhile, the ΔESTs of the CzBNs can be calculated to be 0.39 eV, 0.24 eV, 0.40 eV and 0.21 eV for 2,4-2CzBN, 2,6-2CzBN, 3,5-2CzBN and 3CzBN, respectively, which are relatively small given that the ΔESTs of most hosts reported are in the range of 0.5–1.0 eV. Considering that high triplet energies and small ΔESTs are achieved simultaneously in CzBNs, our molecular design concept discussed above is thereby demonstrated to be successful. | ||
| Fig. 3 (a) The absorption spectra of the compounds. (b) The absorption and emission spectra of 4TCzBN and the emission spectra of the CzBNs. (c) The emission spectra of the doped films. (d) The PL transient decay curves of the films observed at 470 nm. | ||
Smaller ΔESTs usually lead to proper matching of the frontier energy levels with the adjacent layers. To demonstrate this, the electrochemical properties of the compounds were probed by cyclic voltammetry. As can be seen from Fig. S4,† all compounds except 2,6-2CzBN show a reversible reduction reaction, demonstrating that the cyano unit is electrochemically stable. On the contrary, all of the oxidation reactions of the compounds are irreversible, which has been reported as being due to the active sites of carbazole (3,6-positions). The HOMO energy levels were determined from the onsets of the oxidation diagrams, while the LUMO energy levels were calculated from the peaks of the reduction diagrams, except for the reduction potential of 2,6-2CzBN, which was determined from the onset of the reduction diagram. The HOMO and LUMO energies are summarized in Table 1. Compared with mCP, the HOMOs of the CzBNs are much shallower while the LUMOs are relatively deeper, facilitating the hole and electron injection from the adjacent layers, which corresponds to our inspiration above.
| Compounds | Fluorescencea (nm) | Phosa (nm) | S1 (eV) | T1 (eV) | ΔEST (eV) | HOMO (eV) | LUMO (eV) | T g (°C) | τ d (μs) | ϕ PL | μ e (cm2 V−1 s−1) | μ h (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Measured in toluene solution. b At 670 (V cm−1)1/2. c At 707 (V cm−1)1/2. d At 806 (V cm−1)1/2. | ||||||||||||
| 2,4-2CzBN | 405 | 420 | 3.34 | 2.95 | 0.41 | 5.74 | 2.45 | 101 | 3.9 | 0.72 | 3.21 × 10−4 | 8.45 × 10−5c |
| 2,6-2CzBN | 404 | 409 | 3.27 | 3.03 | 0.27 | 5.73 | 2.47 | 104 | 3.9 | 0.77 | 2.4 × 10−5 | 2.32 × 10−6d |
| 3,5-2CzBN | 396 | 409 | 3.39 | 3.03 | 0.4 | 5.78 | 2.54 | 96 | 3.9 | 0.75 | 1.87 × 10−3 | 8.80 × 10−5c |
| 3CzBN | 415 | 432 | 3.20 | 2.87 | 0.23 | 5.75 | 2.61 | 153 | 3.1 | 0.50 | 5.53 × 10−5 | 3.27 × 10−5c |
A high triplet energy, bipolar transporting abilities and suitable frontier energy levels of CzBNs establish the basis for being potentially ideal hosts for blue TADF emitters. A blue TADF emitter, 2,3,5,6-tetrakis(3,6-di-tert-butyl-9H-carbazol-9-yl)benzonitrile (4TCzBN), which was also a carbazolyl benzonitrile derivative, was chosen.27 As can be seen in Fig. 3b, extensive overlap between the photoluminescence (PL) emission of the hosts and the absorption spectra of 4TCzBN was observed, guaranteeing efficient energy transfer from the host to the guest. The spectra of the doped films are slightly different according to Fig. 3c, which is ascribed to the different polarity of the hosts. The emission of TADF emitters is intrinsically originated from their charge transfer (CT) states. In a polar matrix, the PL emissions of the emitters would red-shift since the singlet states of the CT states can be stabilized by the polar environment. The delayed emission of 4TCzBN in the hosts was further confirmed by transient PL measurement of 10% 4TCzBN doped films of the hosts. As can be seen from Fig. 3d, the transient decay curves of all the doped films show a delayed component, indicating efficient TADF emission from 4TCzBN. The lifetime of the delayed part of the transient decay curve of the 3CzBN doped film is shorter than those of the other three hosts. Combined with the lowest PL quantum yield (PLQY) of the 3CzBN doped film, the conclusion can be made that the low triplet energy of 3CzBN may quench the emission of 4TCzBN.
The performances of TADF OLEDs
To evaluate the electroluminescence (EL) performance of the CzBNs, OLEDs were fabricated with structures of ITO/HATCN (5 nm)/NPB (30 nm)/TCTA (10 nm)/mCP (10 nm)/EML (30 nm)/DPEPO (10 nm)/Bphen (30 nm)/LiF (0.5 nm)/Al (150 nm), where HATCN, NPB, TCTA and Bphen are dipyrazino[2,3-f:2′,3′-h]quinoxaline-2,3,6,7,10,11-hexacarbonitrile, N,N′-bis(1-naphthalenyl)-N,N′-diphenyl-[1,1′-biphenyl]-4,4′-diamine, N,N,N-Tris(4-(9-carbazolyl)phenyl)amine and 4,7-diphenyl-1,10-phenanthroline, respectively, as shown in Fig. 4. High triplet energy materials of mCP and DPEPO were used as exciton-blocking layers to confine the triplet excitons on the dopant. The doping concentrations of the four emitters were optimized to be 30%. Besides, as a commonly used host for blue TADF emitters, mCP was also chosen for comparison. | ||
| Fig. 4 The energy diagrams of the devices and the molecular structures of the compounds used in the devices. | ||
Only the emission of 4TCzBN was observed from the EL spectra of the devices in Fig. 5a, indicating that energy transfer from the host to the guest was complete. The CIE coordinates of the device based on 2,4-2CzBN is (0.16, 0.26), the same as the device based on mCP as the host. But the emission of the device based on 3,5-2CzBN is slightly blue-shifted with a CIE of (0.16, 0.23). On the contrary, the emissions of the devices based on 2,6-2CzBN and 3CzBN as hosts are slightly red-shifted with CIEs of (0.17, 0.29) and (0.17, 0.28), respectively. The reasons for the differences in the emission peaks can be attributed to the different polarity of the host materials, corresponding to the PL behaviors of the doped films. Quantum efficiency–luminance and power efficiency–luminance curves of the devices are shown in Fig. 5b and c. The highest EQE of 21.5% was achieved for the device based on 2,4-2CzBN as the host. At the same time, the EQE of the device based on 3,5-2CzBN was also high, as 20.1%. To our knowledge, devices based on true-blue TADF emitters with emission peaks at 470 nm rarely achieve EQEs above 20%.14,21,22
 | ||
| Fig. 5 (a) The EL spectra of the devices. (b) The EQE–brightness characteristics of the devices. (c) The power efficiency–brightness characteristics of the devices. (d) The brightness–voltage characteristics of the devices. | ||
Those values are among the highest for devices based on true-blue TADF emitters. Devices based on the other two hosts also achieved EQEs as high as 13.0% and 14.8% for 2,6-2CzBN and 3CzBN, respectively. The lowest EQE was obtained from the device with 2,6-2CzBN as the host, though the PLQY of the 2,6-2CzBN doped film is the highest, which may be attributed to the most unbalanced charge mobilities of 2,6-2CzBN compared with the other hosts. Therefore, it is evident that both the PLQY of the EML and the transport properties of the host should be taken in to consideration when fabricating high performance OLEDs. All the devices based on the CzBN hosts show much higher efficiencies than the device based on mCP as the host, which possesses a maximum EQE of only 10.5%. The reason for the higher EQEs of devices based on CzBN than mCP may be attributed to the bipolar transporting abilities, as well as the proper frontier energy levels of CzBNs, which may lead to more balanced charges in the emitting layers. The huge differences between the EL performances of the devices reflect the crucial effects of the host matrixes in TADF diodes, revealing the importance of host engineering. The EQEs at a practical luminance of 1000 cd m−2 are still 11.4 and 11.1 for the devices based on 2,4-2CzBN and 3,5-2CzBN, respectively. The power efficiencies of the devices are also summarized in Table 2. The highest value of 42 lm W−1 was achieved for the 2,4-2CzBN based device, owing to the high EQE and low voltage of the device. The performances of the devices are summarized in Table 2.
| Device | Maximum | 500 cd m−2 | 1000 cd m−2 | CIE | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| V (V) | EQE (%) | PE (lm W−1) | V (V) | EQE (%) | PE (lm W−1) | V (V) | EQE (%) | PE (lm W−1) | ||
| a At 1 cd m−2. | ||||||||||
| 2,4-2CzBN | 2.8 | 21.5 | 42.0 | 4.8 | 11.4 | 14.0 | 5.4 | 10.6 | 10.7 | (0.16, 0.26) |
| 2,6-2CzBN | 3.3 | 13.0 | 23.3 | 5.5 | 9.5 | 11.0 | 6.2 | 8.9 | 9.0 | (0.17, 0.29) |
| 3,5-2CzBN | 2.8 | 20.1 | 34.5 | 4.5 | 11.1 | 13.7 | 4.9 | 10.1 | 10.4 | (0.16, 0.23) |
| 3CzBN | 2.8 | 14.8 | 29.5 | 4.9 | 10.0 | 11.9 | 5.6 | 9.1 | 9.6 | (0.17, 0.28) |
| mCP | 3.2 | 10.5 | 18.0 | 5.0 | 6.7 | 7.6 | 5.8 | 5.6 | 5.5 | (0.16, 0.26) |
A most remarkable observation from Fig. 5d is that ultra-low operation voltages were achieved for devices based on CzBNs. The onset voltages (at 1 cd m−2) for the devices utilizing 2,4-2CzBN, 3,5-2CzBN and 3CzBN as hosts were almost the same at about 2.8 V, which were much lower than that of the device using mCP as the host (3.2 eV). For the device with 3,5-2CzBN as the host, an ultra-low voltage of 4.9 V was observed at a practical luminance of 1000 cd m−2, which is the lowest for blue TADF doped devices reported until now to our knowledge. The reason for the low voltage can be attributed to, on the one hand, the frontier energy levels of the hosts matching properly with the adjacent layer, which is beneficial to the relatively small ΔEST; on the other hand, it can be attributed to the enhanced carrier transportation through adjustment of the linker type of the moieties of the hosts. Compared with commonly used hosts, the proper frontier energy levels of the CzBNs lead to more excitons being formed in the emitting layers under the same voltage, and thus higher luminance under a low driving voltage. Surprisingly, although the ΔEST of 2,6-2CzBN is even lower than 2,4-2CzBN or 3,5-2CzBN, the device based on 2,6-2CzBN shows the highest operational voltage of 3.3 V, even higher than the one based on mCP, which is unaligned with the discussion above. What should be kept in mind is that not only the injection barriers between the emitting layers and the adjacent layers, but also the carrier mobilities of the hosts show a great influence on the device voltage.
The carrier mobility of 2,6-2CzBN is the lowest in all hosts, even lower than that of mCP,25 manifesting the high voltage of the device based on 2,6-CzBN. These results reveal that both carrier mobilities and proper frontier energy levels are crucially important to reduce the operational device voltage. Notably, mCP and DPEPO, which work against obtaining low voltages due to their low carrier mobilities, are adopted in the devices.21 Therefore, it can be anticipated that even lower operation voltages can be obtained with further optimization. The performances of the reported blue TADF doped devices are summarized in Table 3. Compared with others, the efficiency of our devices are among the highest and the operational voltages of our devices are significantly reduced, manifesting the CzBNs as promising hosts for blue TADF devices.
| V on (V) | V (V) | EQE | CIE | |
|---|---|---|---|---|
| a At 1000 cd m−2. | ||||
| CzTPN17 | 4.8 | 11.9 | (0.17, 0.40) | |
| DCzIPN28 | 3.5 | 16.4 | (0.17, 0.19) | |
| CC2BP29 | 4.4 | 14.3 | (0.17, 0.27) | |
| DDCzTrz14 | ∼4 | 18.9 | (0.16, 0.22) | |
| DMAC-DPS21 | 3.7 | 19.5 | (0.16, 0.20) | |
| DMAC-DPS30 | 4.6 | 22.6 | (0.16, 0.23) | |
| DMAC-DPS22 | 2.8 | 5.9 | 23.0 | (0.16, 0.21) |
| This work | 2.8 | 5.4 | 21.5 | (0.16, 0.26) |
| This work | 2.8 | 4.9 | 20.1 | (0.16, 0.23) |
Conclusions
In conclusion, high triplet hosts based on CzBNs were designed for blue TADF emitters to facilitate carrier injection to reduce device driving voltages. Proper frontier energy levels that match with the adjacent layers are fulfilled by reducing the ΔEST of the hosts while the high triplet energies were maintained by restraining the molecule conjugate length. Furthermore, it is observed that the transporting mobilities of the hosts were greatly influenced by the linker type of the compounds, and thus the device performances. Blue TADF OLEDs based on CzBNs as hosts achieved a highest EQE of 21.5%, which is one of the highest values for blue TADF devices with an emission peak at 470 nm. The most notable thing is that the lowest voltage of 4.9 V was achieved at a practical luminance of 1000 cd m−2. This work reveals the crucial role of host engineering in developing high-performance TADF OLEDs, and the strategy proposed here may be general for designing more efficient high triplet hosts with small ΔESTs.Acknowledgements
We would like to thank the National Natural Science Foundation of China (Grant No. 51525304) and the National Key Basic Research and Development Program of China (Grant No. 2015CB655002) for financial support. This work was also supported by the CAS Interdisciplinary Innovation Team.References
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750–753 CrossRef CAS PubMed.
- C.-J. Chiang, A. Kimyonok, M. K. Etherington, G. C. Griffiths, V. Jankus, F. Turksoy and A. P. Monkman, Adv. Funct. Mater., 2013, 23, 739–746 CrossRef CAS.
- M. Segal, M. Singh, K. Rivoire, S. Difley, T. V. Voorhis and M. A. Baldo, Nat. Mater., 2007, 6, 374–378 CrossRef CAS PubMed.
- D. D. Zhang, L. Duan, C. Li, Y. L. Li, H. Y. Li, D. Q. Zhang and Y. Qiu, Adv. Mater., 2014, 26, 5050–5055 CrossRef CAS PubMed.
- (a) H. C. Liu, Q. Bai, L. Yao, H. Y. Zhang, H. Xu, S. T. Zhang, W. J. Li, Y. Gao, J. Y. Li, P. Lu, H. Y. Wang, B. Yang and Y. G. Ma, Chem. Sci., 2015, 6, 3797–3804 RSC; (b) W. J. Li, Y. Y. Pan, R. Xiao, Q. Peng, S. T. Zhang, D. G. Ma, F. Li, F. Z. Shen, Y. H. Wang, B. Yang and Y. G. Ma, Adv. Funct. Mater., 2014, 24, 1609–1614 CrossRef CAS.
- K.-H. Kim, S. H. Lee, C.-K. Moon, S.-Y. Kim, Y.-S. Park, J.-H. Lee, J. W. Lee, J. Huh, Y. M. You and J. J. Kim, Nat. Commun., 2014, 5, 4769 CrossRef CAS PubMed.
- K. Li, G. Cheng, C. S. Ma, X. G. Guan, W. M. Kwok, Y. Chen, W. Liu and C. M. Che, Chem. Sci., 2013, 4, 2630–2644 RSC.
- G. Cheng, G. K. So, W. P. To, Y. Chen, C. C. Kwok, C. S. Ma, X. G. Guan, X. Y. Chang, W. M. Kowk and C. M. Che, Chem. Sci., 2015, 6, 4623–4635 RSC.
- D. D. Zhang, L. Duan, Y. L. Li, H. Y. Li, Z. Y. Bin, D. Q. Zhang, J. Qiao, G. F. Dong, L. D. Wang and Y. Qiu, Adv. Funct. Mater., 2014, 24, 3551–3561 CrossRef CAS.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef.
- (a) Q. S. Zhang, H. Kuwabara, W. J. Potscavage Jr, S. P. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS PubMed; (b) X. K. Chen, S. F. Zhang, J. X. Fan and A. M. Ren, J. Phys. Chem. C, 2015, 119, 9728–9733 CrossRef CAS; (c) T. J. Penfold, J. Phys. Chem. C, 2015, 119, 13535–13544 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- M. Kim, S. K. Jeon, S. Hwang and J. Y. Lee, Adv. Mater., 2015, 27, 2515–2520 CrossRef CAS PubMed.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- M. Liu, Y. Seino, D. C. Chen, S. Inomata, S. J. Su, H. Sasabe and J. Kido, Chem. Commun., 2015, 51, 16353–16356 RSC.
- T. Nishimoto, T. Yasuda, S. Y. Lee, R. Kondo and C. Adachi, Mater. Horiz., 2014, 1, 264–269 RSC.
- Y. J. Cho, K. S. Yook and J. Y. Lee, Adv. Mater., 2014, 26, 4050–4055 CrossRef CAS PubMed.
- D. H. Yu, F. C. Zhao, C. M. Han, H. Xu, J. Li, Z. Zhang, Z. P. Deng, D. G. Ma and P. F. Yan, Adv. Mater., 2012, 24, 509–514 CrossRef CAS PubMed.
- D. D. Zhang, L. Duan, D. Q. Zhang, J. Qiao, G. F. Dong, L. D. Wang and Y. Qiu, Org. Electron., 2013, 14, 260–266 CrossRef CAS.
- Q. S. Zhang, B. Li, S. P. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- J. Zhang, D. X. Ding, Y. Wei, F. Q. Han, H. Xu and W. Huang, Adv. Mater., 2016, 28, 479–485 CrossRef CAS PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. H. Li, R. F. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- H. Sasabe, N. Toyota, H. Nakanishi, T. Ishizaka, Y. J. Pu and J. Kido, Adv. Mater., 2012, 24, 3212–3217 CrossRef CAS PubMed.
- (a) D. D. Zhang, L. Duan, Y. L. Li, D. Q. Zhang and Y. Qiu, J. Mater. Chem. C, 2014, 2, 8191–8197 RSC; (b) J. H. Jou, W. B. Wang, S. Z. Chen, J. J. Shyue, M. F. Hsu, C. W. Lin, S. M. Shen, C. J. Wang, C. P. Liu, C. T. Chen, M. F. Wu and S. W. Liu, J. Mater. Chem., 2010, 20, 8411–8416 RSC.
- Q. S. Zhang, J. Li, K. Shizu, S. P. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- D. D. Zhang, M. H. Cai, Y. G. Zhang, D. Q. Zhang and L. Duan, Mater. Horiz., 2016 10.1039/C5MH00258C.
- Y. J. Cho, K. S. Yook and J. Y. Lee, Sci. Rep., 2015, 5, 7859 CrossRef CAS PubMed.
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402 CrossRef CAS PubMed.
- W. Song, I. H. Lee, S. H. Hwang and J. Y. Lee, Org. Electron., 2015, 23, 138 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis and characterization of the hosts, single-crystal structures. CCDC 1441236–1441238. TGA and DSC, emission spectra at 77 K, CV curves, as well as device performances. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04755b |
| This journal is © The Royal Society of Chemistry 2016 |