
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed cascade annulation of unsaturated α-bromocarbonyls with enynals: a facile access to ketones from aldehydes†
Chao
Che
,
Qianwen
Huang
,
Hanliang
Zheng
and
Gangguo
Zhu
*
Department of Chemistry, Zhejiang Normal University, 688 Yingbin Road, Jinhua 321004, China. E-mail: gangguo@zjnu.cn
First published on 4th March 2016
Abstract
A Cu-catalyzed cascade annulation of enynals with alkenyl or alkynyl α-bromocarbonyls for the synthesis of various cyclohexenone-fused polycyclic compounds is described. Up to six new C–C bonds and four new carbocycles can be established in a single reaction, highlighting the high efficiency and step-economics of this protocol. This reaction offers a novel and straightforward entry to the synthesis of ketones featuring the addition of carbon radicals to aldehydes.
Ketones are ubiquitous chemical entities in bioactive molecules, drugs and materials (Scheme 1).1 Typically, they are prepared by the addition of organometallic compounds to aldehydes, followed by oxidation, which requires the utilization of stoichiometric organometallic reagents and oxidants. Alternatively, the aldehydic C–H bond functionalization2–7 has become a powerful strategy for assembling ketones because of its outstanding advantages in atom and step efficiency. Among these, the radical reactions have attracted more and more attention.4–7 For example, a N-hydroxyphthalimide catalyzed radical hydroacylation of simple alkenes with aldehydes has been achieved by the Ishii group.4 More recently, Lei and co-workers reported an elegant synthesis of α,β-unsaturated ketones via the Cu-catalyzed oxidative coupling of terminal alkenes with aldehydes.5 It should be noted that most of these reactions depend on the generation and transformation of acyl radical A (type I) (Scheme 2a).7 However, the type II version, with aldehydes as acceptors for the addition of carbon radicals,8 has never been realized for the access of ketones (type II) (Scheme 2b). This may be ascribed to the higher dissociation energy of C–H bonds as compared to that of C–C bonds, and consequently, the alkoxyl radical B strongly prefers to proceed via the C–C β-scission, instead of the C–H β-scission.9 As such, the intermediate B is in favor of transforming back to aldehydes.
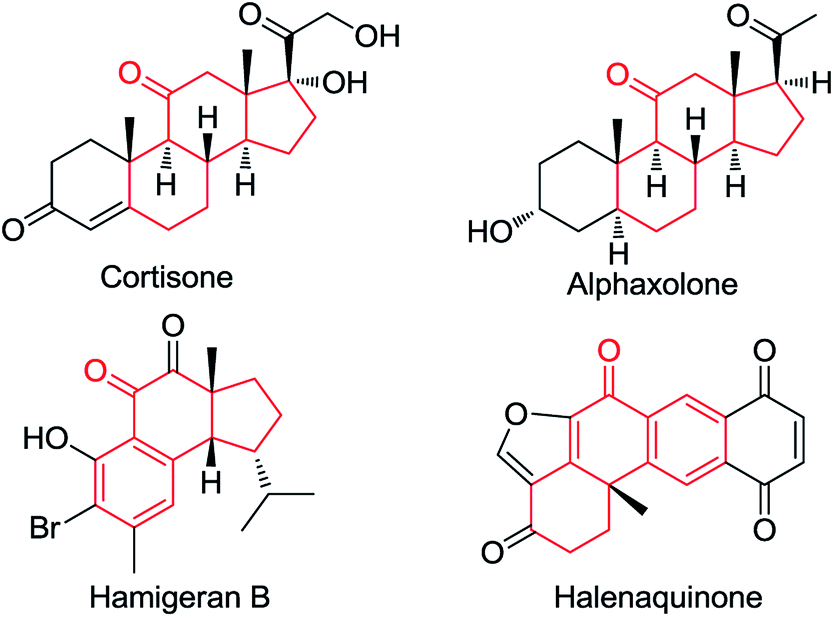 | ||
| Scheme 1 Examples of bioactive ketones. | ||
 | ||
| Scheme 2 Radical approaches to ketones from aldehydes and our proposal. | ||
Given the high efficiency of this transformation (type II), we decided to explore the feasibility. While pursuing our recent work on the Cu-catalyzed atom-transfer radical addition (ATRA) of alkynes,10–12 we envisaged that the direct conversion of aldehydes into ketones might be accomplished via a Cu-catalyzed redox-neutral pathway, which consists of the following steps: (1) a single-electron transfer (SET) between the Cu(I) catalyst and organohalides (R2X) produces a radical R2˙, together with the formation of Cu(II), (2) the alkoxyl radical B, resulting from the addition of R2˙ to R1CHO, undergoes a formal 1,2-H atom shift13 to afford the carbon-centered radical C, (3) another SET between C and Cu(II) species delivers a cationic intermediate D accompanied by the regeneration of the Cu(I) catalyst, and (4) deprotonation of D gives ketones as the final products. Herein, we describe a Cu-catalyzed cascade annulation of alkenyl or alkynyl α-bromocarbonyls with enynals, providing a variety of polycyclic ketones in moderate to excellent yields under mild reaction conditions. In this reaction, up to six new C–C bonds and four new rings can be assembled from the readily attained starting materials, highlighting the high efficiency and step-economics of this method.
To test this hypothesis, the reaction between 2-ethynylbenzaldehyde (1a) and diethyl α-allyl-α-bromomalonate (2a) was conducted in MeCN. Using 10 mol% of CuBr as the catalyst, 20 mol% of pentamethyldiethylenetriamine (L1) as the ligand, and 1 equivalent of K2CO3 as the base, tricyclic ketone 3aa was isolated in 42% yield, after being heated at 80 °C for 10 h (Table 1, entry 1). Encouraged by this result, we further screened the reaction parameters. To our satisfaction, using diethylazodicarboxylate (DEAD) as the reducing reagent for in situ generation of the Cu(I) catalyst, the reaction afforded 3aa in 86% yield (entry 4). Employment of other ligands such as L2–L4 and L5 resulted in decreased yields (entries 6–9). Replacing DEAD with either azodiisobutyrodinitrile (AIBN) or 2,2′-azobis(2,4-dimethylvaleronitrile) (V65) led to inferior results (entries 10 and 11). As for the solvent, MeCN demonstrated better performance than other solvents such as THF, toluene and DMF (entries 14–16).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | [Cu] | Ligand | Additive | Base | Solvent | Yield (%) |
| a Reaction conditions: 1a (0.25 mmol), 2a (0.30 mmol), [Cu] (10 mol%), ligand (20 mol%), additive (20 mol%), base (0.25 mmol), solvent (3 mL), under N2 80 °C, 10 h. Yields of the isolated products are given. | ||||||
| 1 | CuBr | L1 | — | K2CO3 | MeCN | 42 |
| 2 | CuBr2 | L1 | DEAD | K2CO3 | MeCN | 36 |
| 3 | Cu(acac)2 | L1 | DEAD | K2CO3 | MeCN | 83 |
| 4 | Cu(OAc)2 | L1 | DEAD | K2CO3 | MeCN | 86 |
| 5 | Cu(OAc)2 | L1 | — | K2CO3 | MeCN | 61 |
| 6 | Cu(OAc)2 | L2 | DEAD | K2CO3 | MeCN | 52 |
| 7 | Cu(OAc)2 | L3 | DEAD | K2CO3 | MeCN | 34 |
| 8 | Cu(OAc)2 | L4 | DEAD | K2CO3 | MeCN | 75 |
| 9 | Cu(OAc)2 | L5 | DEAD | K2CO3 | MeCN | 80 |
| 10 | Cu(OAc)2 | L1 | AIBN | K2CO3 | MeCN | 62 |
| 11 | Cu(OAc)2 | L1 | V65 | K2CO3 | MeCN | 65 |
| 12 | Cu(OAc)2 | L1 | DEAD | Cs2CO3 | MeCN | 47 |
| 13 | Cu(OAc)2 | L1 | DEAD | DBU | MeCN | 21 |
| 14 | Cu(OAc)2 | L1 | DEAD | K2CO3 | THF | Trace |
| 15 | Cu(OAc)2 | L1 | DEAD | K2CO3 | PhMe | Trace |
| 16 | Cu(OAc)2 | L1 | DEAD | K2CO3 | DMF | 14 |

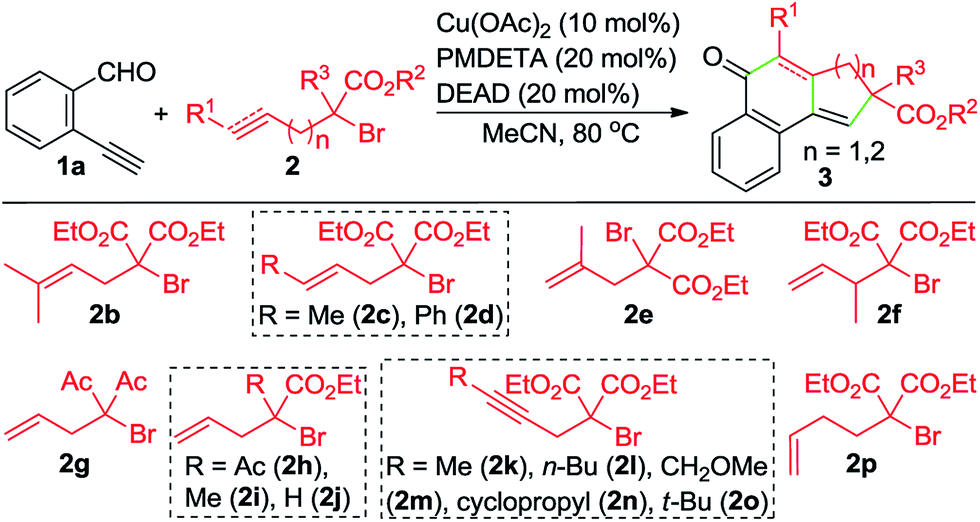
With the optimized reaction conditions in hand, we investigated the scope of this Cu-catalyzed domino annulation by varying enynals 1 and α-bromocarbonyls 2. As shown in Table 2, the standard conditions were well compatible with a variety of enynals, including 2-ethynylbenzaldehydes and pent-2-en-4-ynal derivatives. Substrates with different substituents on the aryl ring of 1 were successfully converted into polycyclic ketones in good to excellent yields, regardless of the electronic effects of the substituents (3ba–3ia). Halogen atoms such as F and Cl were well tolerated under the reaction conditions (3da–3fa), giving ample opportunities for further elaboration by the transition-metal-catalyzed coupling reactions. Intriguingly, the reaction of 1m–1o with 2a occurred uneventfully to provide tetracyclic ketones 3ma–3oa in high yields. Aldehyde 1p with the 2-thienyl group was transformed into the corresponding ketone 3pa in 68% yield. The process was extended to substrate 1q, bearing an amide group, providing 3qa in a good yield. Moreover, 2-ethynylcyclohex-1-enecarbaldehyde (1r) was also a competent substrate, and 3ra was synthesized without erosion of the reaction yield.
|
|
|---|
| a Reaction conditions: 1 (0.25 mmol), 2a (0.30 mmol), Cu(OAc)2 (10 mol%), L1 (20 mol%), DEAD (20 mol%), K2CO3 (0.25 mmol), MeCN (3 mL), under N2, 80 °C, 10 h. Yields of the isolated products are given. NPhth = phthalimidyl. |

|

By varying α-bromo γ,δ-unsaturated carbonyl compounds 2 with 1a as the coupling partner, further examples of tricyclic ketones (3ab–3ai) were synthesized (Table 3). The product 3ab, containing a gem-dimethyl subunit, was isolated in an excellent yield. Substitution of the terminal C–C double bond of 2 with a methyl group resulted in the production of 3ac in 80% yield and good diastereoselectivity (dr = 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12). In contrast, the Ph-substituted analogue 2d was not suitable for this Cu-catalyzed domino process (3ad). In the case of β-branched substrate 2f, the reaction produced 3af in a moderate yield. The reaction covered other activated organobromides, as exemplified by the construction of 3ag and 3ah. Compound 2i, a weakly activated substrate, was effective for the transformation, while no detectable product was observed when secondary bromide 2j was used as the coupling partner (3ai and 3aj). This reaction was well amenable to propargyl α-bromocarbonyls. For example, the coupling of 1a with 2k also took place, affording 3ak in 74% yield. Substitution of the terminal alkynyl carbon by primary alkyl groups led to the facile generation of tricyclic ketones (3ak–3am), whereas the cyclopropane-substituted counterpart 2n delivered the corresponding product in a lower yield (3an), potentially due to the increased steric hindrance. Meanwhile, an α-bromo δ,ε-unsaturated carbonyl such as 2p performed well in this Cu-catalyzed cascade annulation reaction, giving a direct and convenient access to the 6-6-6-tricyclic ketone 3ap. The structure of polycyclic ketones 3fa and 3ap was determined by the X-ray diffraction analysis.14
:12). In contrast, the Ph-substituted analogue 2d was not suitable for this Cu-catalyzed domino process (3ad). In the case of β-branched substrate 2f, the reaction produced 3af in a moderate yield. The reaction covered other activated organobromides, as exemplified by the construction of 3ag and 3ah. Compound 2i, a weakly activated substrate, was effective for the transformation, while no detectable product was observed when secondary bromide 2j was used as the coupling partner (3ai and 3aj). This reaction was well amenable to propargyl α-bromocarbonyls. For example, the coupling of 1a with 2k also took place, affording 3ak in 74% yield. Substitution of the terminal alkynyl carbon by primary alkyl groups led to the facile generation of tricyclic ketones (3ak–3am), whereas the cyclopropane-substituted counterpart 2n delivered the corresponding product in a lower yield (3an), potentially due to the increased steric hindrance. Meanwhile, an α-bromo δ,ε-unsaturated carbonyl such as 2p performed well in this Cu-catalyzed cascade annulation reaction, giving a direct and convenient access to the 6-6-6-tricyclic ketone 3ap. The structure of polycyclic ketones 3fa and 3ap was determined by the X-ray diffraction analysis.14
|
|
|---|
| a Reaction conditions: 1a (0.25 mmol), 2 (0.30 mmol), Cu(OAc)2 (10 mol%), L1 (20 mol%), DEAD (20 mol%), K2CO3 (0.25 mmol), MeCN (3 mL), under N2, 80 °C, 10 h. Yields of the isolated products are given. |

|
Remarkably, the one-pot construction of pentacyclic diketones 3sa and 3sp was achieved by reacting enynal 1s with 2a and 2p, respectively (Scheme 3). Although the yield appears to be moderate, considering the formation of six new C–C bonds and four new rings in a single reaction, it still represents a highly attractive method for the synthesis of polycyclic ketones from readily accessible starting materials.
 | ||
| Scheme 3 Cu-catalyzed double cascade annulation. | ||
To gain insights into the reaction mechanism, a series of experiments were performed. First, the reaction between 1a and 2a was inhibited by adding 2 equivalents of 2,2,6,6-tetramethylpiperidinooxy (TEMPO), and instead, 4a was formed in 51% yield (eqn (1)). In the presence of butylated hydroxytoluene (BHT), no detectable 3aa was observed, and 4b was obtained in 35% yield (eqn (2)). Likewise, the addition of 1,1-diphenylethylene hindered the reaction between 1a and 2b and provided the Cu-catalyzed atom-transfer radical cyclization12a product 4c in 68% yield (eqn (3)). These results indicated that the Cu-catalyzed cascade annulation reaction might proceed via a radical mechanism. Furthermore, when compound 5 was employed as the starting material, alcohol 6a was obtained in 62% yield (eqn (4)), implying that the aldehydic hydrogen atom of 1 is essential for the ketone synthesis. Alcohol 6b, generated by the reduction of 3aa with NaBH4, was subjected to the optimized reaction conditions, and as a result, no formation of 3aa was observed (eqn (5)). It indicated that the formation of alcohol intermediate 6b followed by oxidation with Cu(II) reagents is less likely in this case.
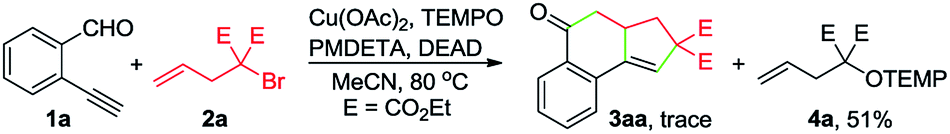 | (1) |
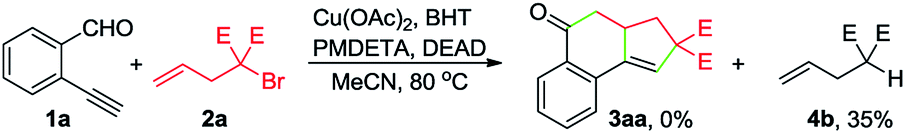 | (2) |
 | (3) |
 | (4) |
 | (5) |
Whereas the full mechanistic features of this Cu-catalyzed domino annulation are still under investigation, a working mechanism is proposed in Scheme 4, using 1a and 2a as representative starting materials. Initially, a radical I is formed by a SET process from 2a and Cu(I) catalyst, which is generated in situ by the reduction of Cu(OAc)2 with DEAD. The isolation of adduct 4a confirmed the formation of radical I. The radical I adds to the C–C triple bond of 1a to deliver an alkenyl radical II, which is converted to the alkyl radical species IIIvia a 5-exo-trig cyclization. Then, an intramolecular addition of carbon radical to the aldehyde group generates the alkoxy radical IV, followed by a formal 1,2-H shift13 to give the benzyl radical V. Subsequently, a second SET between V and Cu(II) produces the cationic intermediate VI with the regeneration of Cu(I) catalyst. Finally, VI is deprotonated to afford the tricyclic ketone 3aa with the aid of K2CO3.
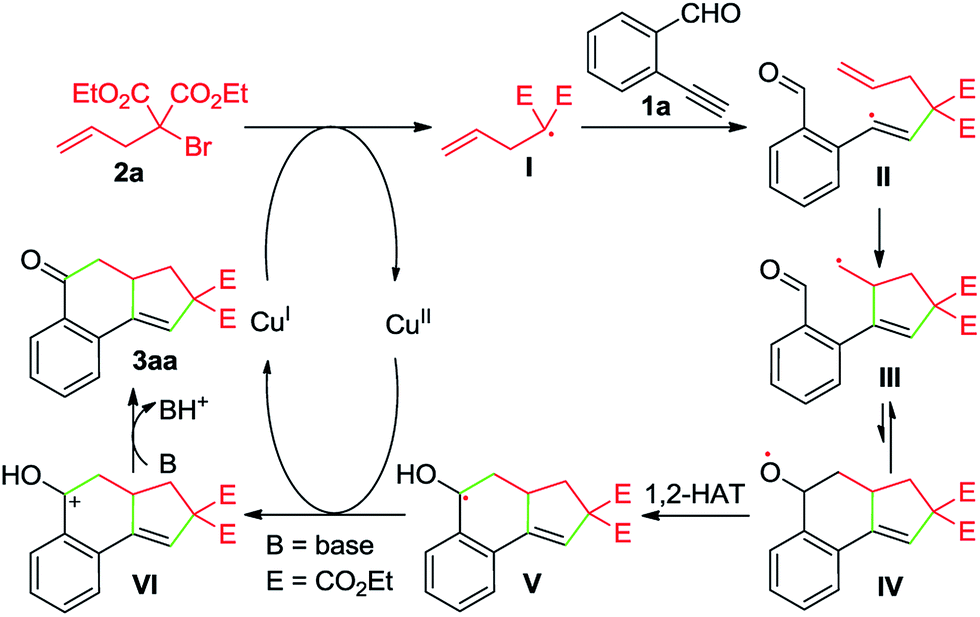 | ||
| Scheme 4 A possible mechanism. | ||
The synthetic utility of this reaction was also explored (Scheme 5). Treatment of 3aa with LiCl and H2O in DMSO at reflux15 resulted in the production of 80% yield of 3aj, a product that was not able to be synthesized via the Cu-catalyzed cascade annulation (Table 3, 3aj). Obviously, the decarbalkoxylation procedure offered a good complementary method to the domino annulation. Epoxidation of 3fa with meta-chloroperbenzoic acid (m-CPBA) gave rise to a single diastereoisomer, 7b, in 76% yield.14 Furthermore, the one-pot synthesis of α-bromo diketone 7c could be accomplished through the exposure of 3aa to a combination of N-bromosuccinimide (NBS) and NH4OAc in Et2O.16 By treating 3ak with NaBH4 in a 1:1 mixture of MeOH and THF, the 1,6-addition product 7d was obtained in 92% yield, which constitutes a new efficient access to polysubstituted 1-naphthols.17
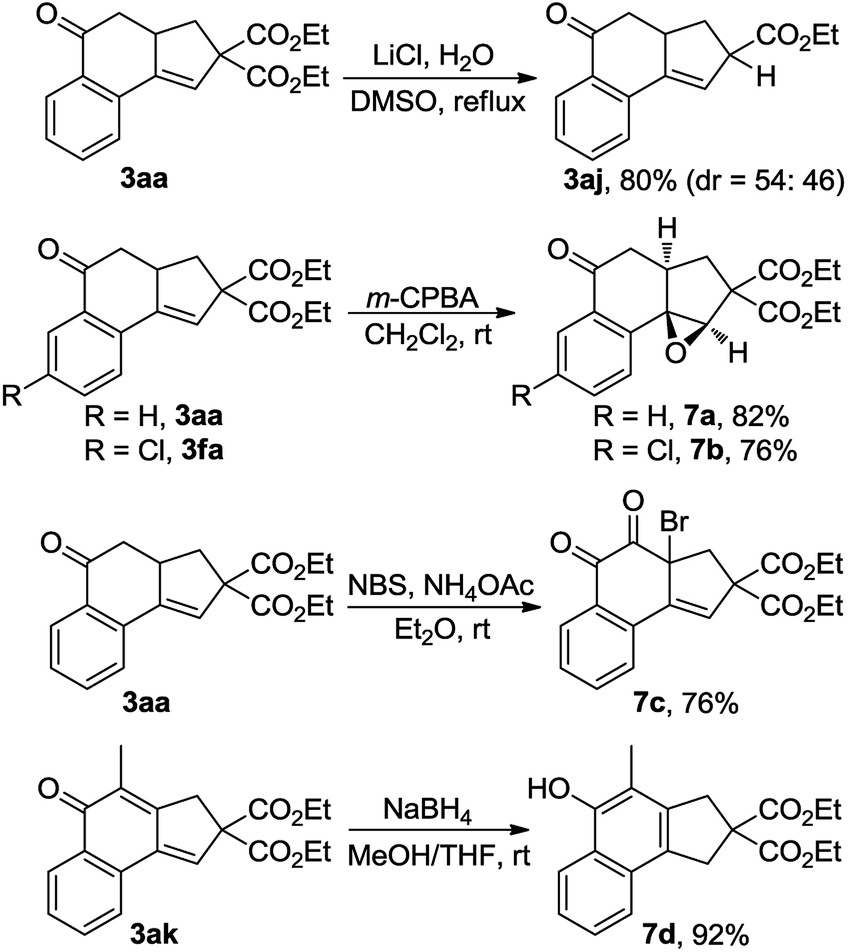 | ||
| Scheme 5 Synthetic utility of cascade annulation. | ||
Conclusions
We have developed a Cu-catalyzed cascade annulation of enynals with alkenyl or alkynyl α-bromocarbonyls, yielding various cyclohexenone-fused polycyclic compounds under mild reaction conditions. Up to six new C–C bonds and four new rings can be established in a single reaction, highlighting the high efficiency of this protocol. A wide range of functional groups such as F, Cl, OMe, CF3, CO2Et, Ac, amide, thienyl and alkyl substituents are well tolerated. This reaction represents a novel method for the one-step synthesis of ketones featuring the addition of carbon radicals to aldehydes. Further investigations on the reaction mechanism and application to bioactive ketones are currently underway in our laboratory.Acknowledgements
We thank the Science Technology Department of Zhejiang Province (2015C31030), Natural Science Foundation of Zhejiang Province (LR12B02001), and National Natural Science Foundation of China (21172199) for the financial support.Notes and references
- (a) L. Gyermek and L. F. Soyka, Anesthesiology, 1975, 42, 331 CrossRef CAS PubMed; (b) K. C. Nicolaou, D. Gray and J. Tae, Angew. Chem., Int. Ed., 2001, 40, 3675 CrossRef CAS; (c) M. A. Kienzler, S. Suseno and D. Trauner, J. Am. Chem. Soc., 2008, 130, 8604 CrossRef CAS PubMed.
- (a) A. J. Wommack, D. C. Moebius, A. L. Travis and J. S. Kingsbury, Org. Lett., 2009, 11, 3202 CrossRef CAS PubMed; (b) B.-X. Tang, R.-J. Song, C.-Y. Wu, Y. Liu, M.-B. Zhou, W.-T. Wei, G.-B. Deng, D.-L. Yin and J.-H. Li, J. Am. Chem. Soc., 2010, 132, 8900 CrossRef CAS PubMed; (c) Y.-X. Liao and Q.-S. Hu, J. Org. Chem., 2010, 75, 6986 CrossRef CAS PubMed; (d) T. Fukuyama, H. Okamoto and I. Ryu, Chem. Lett., 2011, 40, 1453 CrossRef CAS; (e) Y. Wu, B. Li, F. Mao, X. Li and F. Y. Kwong, Org. Lett., 2011, 13, 3258 CrossRef CAS PubMed; (f) Z. Shi, N. Schröder and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 8092 CrossRef CAS PubMed; (g) W. Ai, Y. Wu, H. Tang, X. Yang, Y. Yang, Y. Li and B. Zhou, Chem. Commun., 2015, 51, 7871 RSC; (h) R. Kuppusamy, P. Gandeepan and C.-H. Cheng, Org. Lett., 2015, 17, 3846 CrossRef CAS PubMed; (i) S. Tang, L. Zeng, Y. Liu and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 15850 CrossRef CAS PubMed; for a review, see: (j) C. Pan, X. Jia and J. Cheng, Synthesis, 2012, 677 CAS.
- For selected reviews on ketone synthesis via hydroacylation, see: (a) M. C. Willis, Chem. Rev., 2010, 110, 725 CrossRef CAS PubMed; (b) J. C. Leung and M. J. Krische, Chem. Sci., 2012, 3, 2202 RSC; (c) S. K. Murphy and V. M. Dong, Chem. Commun., 2014, 50, 13645 RSC.
- S. Tsujimoto, T. Iwahama, S. Sakaguchi and Y. Ishii, Chem. Commun., 2001, 2352 RSC.
- J. Wang, C. Liu, J. Yuan and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 2256 CrossRef CAS PubMed.
- For other radical versions, see: (a) K. Yoshikai, T. Hayama, K. Nishimura, K.-I. Yamada and K. Tomioka, J. Org. Chem., 2005, 70, 681 CrossRef CAS PubMed; (b) V. Chudasama, R. J. Fitzmaurice, J. M. Ahern and S. Caddick, Chem. Commun., 2010, 46, 133 RSC; (c) V. Chudasama, R. J. Fitzmaurice and S. Caddick, Nat. Chem., 2010, 2, 592 CrossRef CAS PubMed; (d) W. Liu, Y. Li, K. Liu and Z. Li, J. Am. Chem. Soc., 2011, 133, 10756 CrossRef CAS PubMed; (e) M.-B. Zhou, R.-J. Song, X.-H. Ouyang, Y. Liu, W.-T. Wei, G.-B. Deng and J.-H. Li, Chem. Sci., 2013, 4, 2690 RSC; (f) X.-H. Ouyang, R.-J. Song, C.-Y. Wang, Y. Yang and J.-H. Li, Chem. Commun., 2015, 51, 14497 RSC; (g) J.-Y. Luo, H.-L. Hua, Z.-S. Chen, Z.-Z. Zhou, Y.-F. Yang, P.-X. Zhou, Y.-T. He, X.-Y. Liu and Y.-M. Liang, Chem. Commun., 2014, 50, 1564 RSC; (h) X. Liu, L. Yu, M. Luo, J. Zhu and W. Wei, Chem.–Eur. J., 2015, 21, 8745 CrossRef CAS PubMed; (i) M. Okada, K. Yamada, T. Fukuyama, D. Ravelli, M. Fagnoni and I. Ryu, J. Org. Chem., 2015, 80, 9365 CrossRef CAS PubMed; (j) X. Mi, C. Wang, M. Huang, Y. Wu and Y. Wu, J. Org. Chem., 2015, 80, 148 CrossRef CAS PubMed; (k) D. Leifert, C. G. Daniliuc and A. Studer, Org. Lett., 2013, 15, 6286 CrossRef CAS PubMed.
- For selected reviews, see: (a) C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef CAS PubMed; (b) C. Liu, D. Liu and A. Lei, Acc. Chem. Res., 2014, 47, 3459 CrossRef CAS PubMed; (c) D. Liu, C. Liu and A. Lei, Chem.–Asian J., 2015, 10, 2040 CrossRef CAS PubMed.
- For selected reports on the synthesis of alcohols featuring the addition of carbon radicals to aldehydes, see: (a) A. L. J. Beckwith and B. P. Hay, J. Am. Chem. Soc., 1989, 111, 2674 CrossRef CAS; (b) S.-Y. Chang, Y.-F. Shao, S.-F. Chu, G.-T. Fan and Y.-M. Tsai, Org. Lett., 1999, 1, 945 CrossRef CAS; (c) P. Devin, L. Fensterbank and M. Malacria, Tetrahedron Lett., 1999, 40, 5511 CrossRef CAS; (d) M. Tiecco, L. Testaferri, F. Marini, S. Sternativo, C. Santi, L. Bagnoli and A. Temperini, Tetrahedron, 2007, 63, 5482 CrossRef CAS; (e) T. Kawamoto, T. Fukuyama and I. Ryu, J. Am. Chem. Soc., 2012, 134, 875 CrossRef CAS PubMed.
- (a) S. Wilsey, P. Dowd and K. N. Houk, J. Org. Chem., 1999, 64, 8801 CrossRef CAS PubMed; (b) C. S. A. Antunes, M. Bietti, O. Lanzalunga and M. Salamone, J. Org. Chem., 2004, 69, 5281 CrossRef CAS PubMed; (c) M. Bietti, O. Lanzalunga and M. Salamone, J. Org. Chem., 2005, 70, 1417 CrossRef CAS PubMed; (d) M. Newcomb, P. Daublain and J. H. Horner, J. Org. Chem., 2002, 67, 8669 CrossRef CAS PubMed; (e) T. Nakamura, Y. Watanabe, S. Suyama and H. Tezuka, J. Chem. Soc., Perkin Trans. 2, 2002, 1364 RSC; (f) S. R. A. Marque and D. Siri, ChemPhysChem, 2012, 13, 703 CrossRef CAS PubMed; (g) J. Zhang, Y. Li, F. Zhang, C. Hu and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 1872 CrossRef CAS PubMed; for selected reviews, see: (h) J. Hartung, T. Gottwald and K. Špehar, Synthesis, 2002, 1469 CrossRef CAS; (i) M. Salamone and M. Bietti, Synlett, 2014, 1803 CAS and references therein.
- C. Che, H. Zheng and G. Zhu, Org. Lett., 2015, 17, 1617 CrossRef CAS PubMed.
- For Cu-catalyzed ATRA of alkyl halides to alkynes, see: (a) T. Xu and X. Hu, Angew. Chem., Int. Ed., 2015, 54, 1307 CrossRef CAS PubMed; (b) J. Lai, L. Tian, Y. Liang, Y. Zhang, X. Xie, B. Fang and S. Tang, Chem.–Eur. J., 2015, 21, 14328 CrossRef CAS PubMed; (c) M.-C. Belhomme, D. Dru, H.-Y. Xiong, D. Cahard, T. Besset, T. Poisson and X. Pannecoucke, Synthesis, 2014, 1859 Search PubMed.
- For recent reports on the Cu-catalyzed ATRA of alkyl halides to alkenes, see: (a) M. J. W. Taylor, W. T. Eckenhoff and T. Pintauer, Dalton Trans., 2010, 39, 11475 RSC; (b) M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, Chem.–Eur. J., 2012, 18, 7336 CrossRef CAS PubMed; (c) M. Knorn, T. Rawner, R. Czerwieniec and O. Reiser, ACS Catal., 2015, 5, 5186 CrossRef CAS; (d) T. Nishikata, Y. Noda, R. Fujimoto and T. Sakashita, J. Am. Chem. Soc., 2013, 135, 16372 CrossRef CAS PubMed; (e) T. Nishikata, K. Nakamura, K. Itonaga and S. Ishikawa, Org. Lett., 2014, 16, 5816 CrossRef CAS PubMed; (f) X. Zhang, H. Yi, Z. Liao, G. Zhang, C. Fan, C. Qin, J. Liu and A. Lei, Org. Biomol. Chem., 2014, 12, 6790 RSC; for selected reviews, see: (g) K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS PubMed; (h) T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087 RSC; (i) T. Pintauer, Eur. J. Inorg. Chem., 2010, 2449 CrossRef CAS; (j) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed.
- (a) B. C. Gilbert, R. G. G. Holmes, H. A. H. Laue and R. O. C. Norman, J. Chem. Soc., Perkin Trans. 2, 1976, 1047 RSC; (b) P. E. Elford and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1996, 2247 RSC.
- CCDC 1439849 (3fa), 1439850 (3ap), and 1439851 (7b).†.
- A. P. Krapcho, J. F. Weimaster, J. M. Eldridge, E. G. E. Jahngen Jr, A. J. Lovey and W. P. Stephens, J. Org. Chem., 1978, 43, 138 CrossRef CAS.
- K. Tanemura, T. Suzuki, Y. Nishida, K. Satsumabayashi and T. Horaguchi, Chem. Commun., 2004, 470 RSC.
- For a recent report on the synthesis of 1-naphthols, see: Y. Bai, J. Yin, Z. Liu and G. Zhu, J. Org. Chem., 2015, 80, 10226 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Data for all the products, experimental procedures. CCDC 1439849–1439851. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04980f |
| This journal is © The Royal Society of Chemistry 2016 |