
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA designed DNA binding motif that recognizes extended sites and spans two adjacent major grooves†
Jéssica
Rodríguez
,
Jesús
Mosquera
,
Rebeca
García-Fandiño
 ,
M. Eugenio
Vázquez
* and
José L.
Mascareñas
*
,
M. Eugenio
Vázquez
* and
José L.
Mascareñas
*
Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: joseluis.mascarenas@usc.es; Fax: +34 981 595 012; Tel: +34 981576541-14405
First published on 5th February 2016
Abstract
We report the rational design of a DNA-binding peptide construct composed of the DNA-contacting regions of two transcription factors (GCN4 and GAGA) linked through an AT-hook DNA anchor. The resulting chimera, which represents a new, non-natural DNA binding motif, binds with high affinity and selectivity to a long composite sequence of 13 base pairs (TCAT-AATT-GAGAG).
Transcription Factors (TFs) are specialized proteins that bind to specific DNA regulatory sequences,1 and thereby promote or inhibit the transcription of particular genes.2 The recognition process typically requires the cooperative action of several modules, which are connected either in a covalent or non-covalent way. This natural strategy allows the recognition of relatively large DNA sites, which is fundamental to ensure the selective targeting of specific genes.3 Thus, the bZIP or bHLH families bind DNA in the form of leucine zipper-mediated homo- or heterodimers,4 and the Cys2His2 zinc finger TFs present multiple recognition modules that simultaneously interact with consecutive sites along the DNA major groove.5 Other TFs, such as the cro repressor, or the glucocorticoid nuclear receptor protein, interact to DNA as non-covalent dimers, inserting recognition helices in the same face of adjacent major grooves.6
Over the last few decades there have been many efforts to develop miniaturized synthetic DNA binders that reproduce the DNA recognition properties of these natural proteins;7,8 some of them have even shown potential for the artificial control of gene expression.9 Most designed DNA-binding peptides rely on the modification of monomeric DNA binding domains,10 or in the artificial dimerization of bZIP basic regions.11 Our group has demonstrated that appropriate conjugation of monomeric fragments of transcription factors with small DNA-binding agents, such as distamycin or pentamidine derivatives,12 or with short AT-hook peptide motives,13 also leads to high-affinity and selective DNA binders. However, these binary artificial constructs allow the specific recognition of relatively short stretches of DNA (up to of 9 base pairs), far from the typical extended DNA sites covered by the natural counterparts.3b This represents a serious limitation for future applications in the selective targeting of specific genes. While the desired targeting of long DNA sites has been successfully achieved by recombinant oligomeric zinc fingers,14 we are not aware of synthetic peptide constructs that address extended sites by using TF-based DNA binding modules. Dervan's polyamides are capable of targeting up to sixteen contiguous base pairs, however they interact to the DNA through the minor groove.15
Inspired by proteins like the cro repressor, we explored the possibility of achieving a selective recognition of relatively long DNA sites by a composite “miniprotein” designed to insert TF recognition fragments into two consecutive major grooves (Fig. 1). Herein we demonstrate that covalent tethering of monomeric TF fragments through a polyglycine linker does not produce effective binders. However, if the linkage is carried out by an AT-hook module, the resulting construct binds with high affinity and specificity to an extended consensus sequence spanning 13 bp (TCAT·AATT·GAGAG). This conjugate represents the first synthetic peptide chimera capable of binding specific DNA sites in a tripartite (major–minor–major groove) manner, and provides a novel DNA binding architecture that lacks natural counterparts.
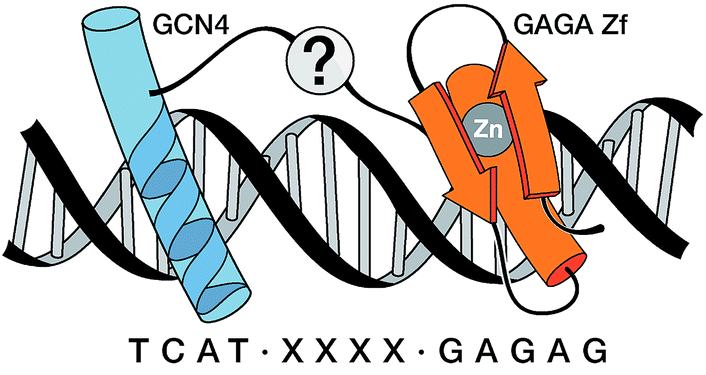 | ||
| Fig. 1 Cartoon representing the goal of this research, namely the recognition of extended DNA sites by inserting the basic region (BR) of GCN4 and the GAGA Zf in adjacent major grooves, and along one face of the double helix. The question mark intends to indicate the unknown nature of the connection that could allow the desired recognition. | ||
As constituent TF fragments, we selected the basic region (BR) of the yeast GCN4 bZIP protein, and the zinc finger of the GAGA factor of Drosophila melanogaster, which are both unable to bind to their target DNA sites as isolated monomers. In the case of GCN4, we chose the sequence between residues Asp226 and Gln248, which has been identified as the smallest peptide that retains specific DNA recognition properties when presented as a dimer,11a,16 or as a stapled derivative.17 With respect to GAGA, we chose a truncated 31-residue peptide of its zinc finger region (residues Ser28 to Phe58) that is non functional as an isolated peptide, but can bind to the DNA when conjugated to small DNA-binding agents.18
Using as reference the X-ray structures of the GCN4–DNA19 and GAGA–DNA complexes,20 we built a model for the simultaneous interaction of the GCN4 basic region and the GAGA zinc finger in contiguous DNA major grooves covering the same face of the DNA double helix (see the ESI†). Inspection of this coarse model suggested that the Arg245 in GCN4, and the Arg44 in GAGA could be suitable positions for tethering both DNA binding domains, and that the distance between both domains, of about 17 Å, could be effectively satisfied by a peptidic linker composed of nine glycines. After completing the solid-phase synthesis of the GCN4 basic region, the connector bearing a Gly9 linker was introduced through an orthogonally-deprotected Lys side chain that replaces the natural Arg245. We then added a bromoacetyl moiety that provides a reactive site for coupling the GAGA peptide fragment (Scheme 1).
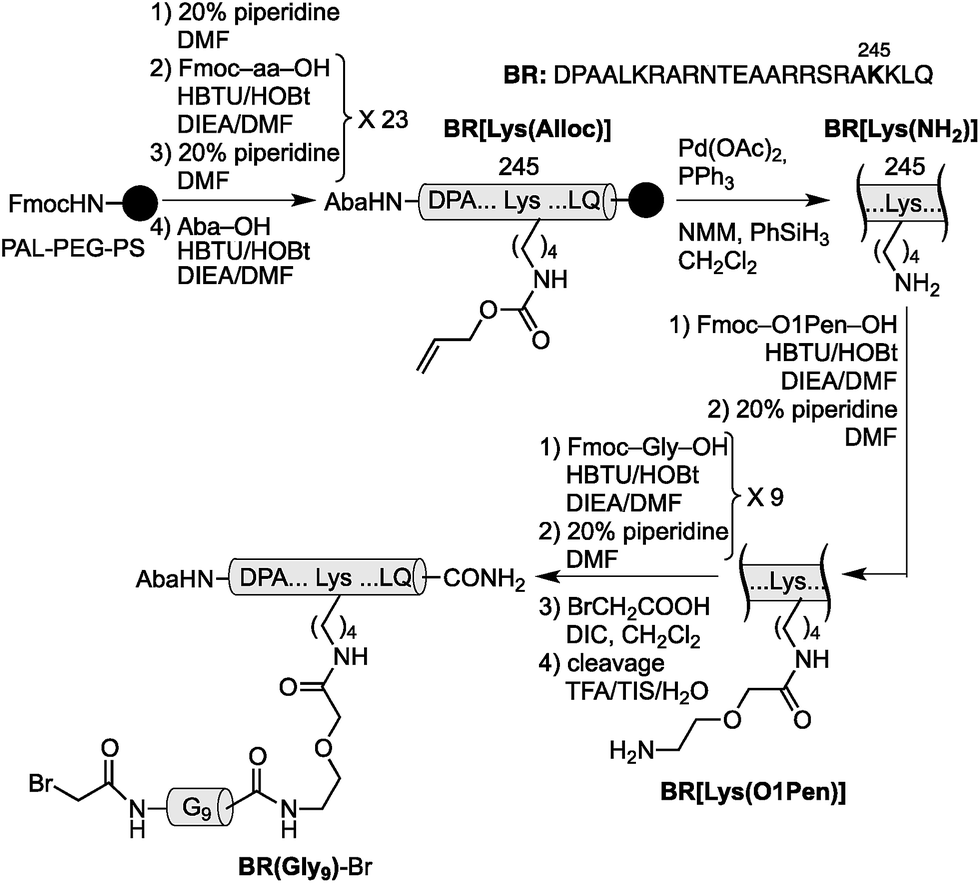 | ||
| Scheme 1 Strategy used for the synthesis of the GCN4/Gly9 chimera BR(Gly9)-Br. The 4-acetamidobenzoic acid (Aba) chromophore is introduced at the N-terminus of the GCN4 basic region as a spectroscopic reporter. O1Pen: 5-amino-3-oxapentanoic acid. | ||
The resulting peptide BR(Gly9)-Br was isolated in an approximate 20% yield after standard cleavage and HPLC purification. The GAGA fragment was engineered to incorporate a Cys residue in the side chain of Lys44 (which replaces the native Arg). The expected peptide GAGA-SH was obtained in good yield after the standard deprotection/resin cleavage step (Scheme 2). The key coupling reaction between the two peptide fragments was carried out by dissolving the peptide GAGA-SH in phosphate buffer at pH 7.5 in presence of 1.5 equiv. of ZnSO4, and 2 equiv. of BR(Gly9)-Br (see the ESI†). After 1 h at room temperature, we obtained the desired conjugate BR(Gly9)GAGA in an approximate 45% yield. The chemoselectivity of this reaction (only the Cys in the side chain of Lys44 is modified, and no alkylation of Cys36 and Cys39 is observed) stems from the coordination of the Zn cation by these other two Cys that support the zinc finger fold.21
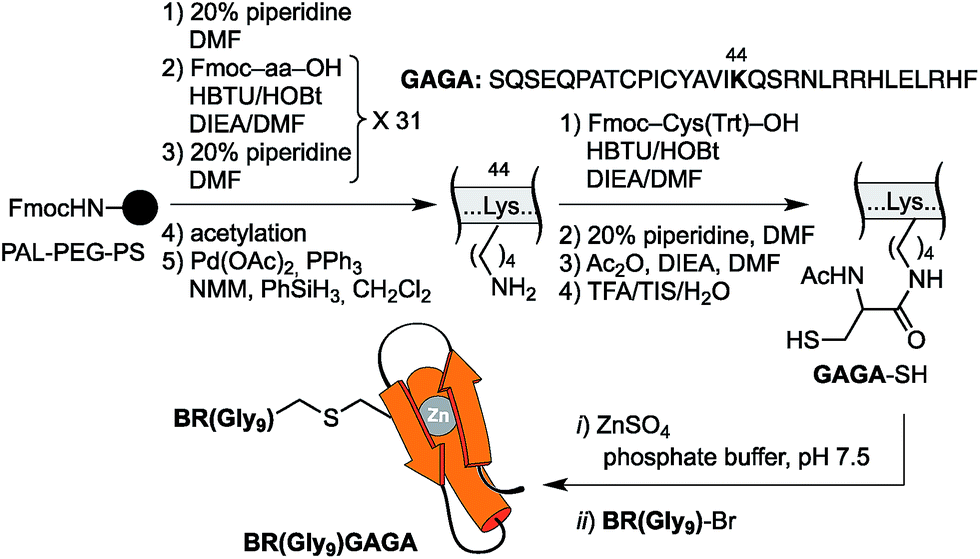 | ||
| Scheme 2 Strategy used for the synthesis of the GCN4/Gly9/GAGA chimera, BR(Gly9)GAGA, by chemoselective modification of the GAGA Cys2His2 peptide in solution. | ||
With the conjugate at hand, we analyzed its DNA binding properties using standard non-denaturing EMSA assays in polyacrylamide gel, and double-stranded (ds) oligonucleotides featuring composite sites comprising the GCN4 basic region and the GAGA Zf target sequences separated by four (dsDNA A and B), or five base pairs (dsDNA C). These particular base pair spacers were selected so that the interaction of both binding regions of the conjugate could take place through the same face of the DNA double helix. As shown in Fig. 2, incubation of these dsDNAs with BR(Gly9)GAGA failed in all cases to rise stable DNA peptide complexes, and we observed only faint slow-migrating bands that suggest the formation of low affinity complexes.
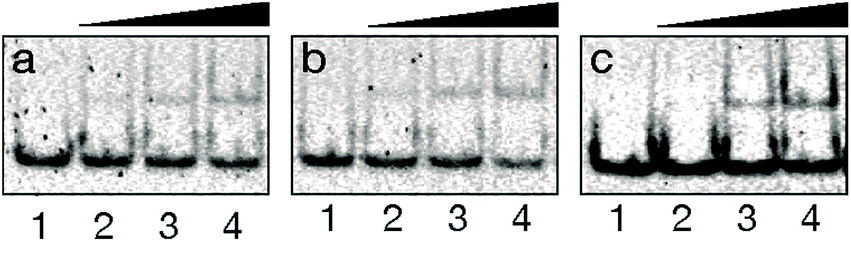 | ||
| Fig. 2 DNA binding studies of BR(Gly9)GAGA by EMSA. In all cases, lanes 1–4: [BR(Gly9)GAGA] = 0, 400, 600, 800 nM with (a) 75 nM of dsDNA A. (b) with 75 nM of dsDNA B. (c) with 75 nM of dsDNA C. Oligonucleotide sequences (only one strand shown): A 5′-CGCG TCATAATTGAGAG CGC-3′; B 5′-CGCG TCATCAGCGAGAG CGC-3′; C 5′-CGCG TCATAAATTGAGAG CGC-3′. Experiment was resolved by PAGE on a 10% nondenaturing polyacrylamide gel and 0.5× TBE buffer over 40 min at rt, and analyzed by staining with SyBrGold (Molecular Probes: 5 μL in 50 mL of 1× TBE) for 10 min, followed by fluorescence visualization. | ||
Although these poor results would advice against further pursuing this approach to achieve the desired bipartite major groove binding, we envisioned that using an AT-hook peptide instead of the (Gly9) connector might allow the formation of more stable DNA complexes. AT-hook motives are cationic short peptides present in HMG-I(Y) eukaryotic nuclear proteins that feature a central Arg–Gly–Arg core capable of deeply inserting in the minor groove of AT-rich sites.22 We reasoned that these peptides, in addition to providing stabilizing contacts with the DNA, might work as minor groove anchors to ensure the correct delivery of the TF fragments to their consensus sites.13 Therefore, using as reference the structure of the AT-hook motif RKPRGRPKK, bound to the PRDII sequence of the IFN-β promoter (see the ESI†), we designed a new conjugate, BR(Hk)GAGA, comprising three different DNA binding fragments of natural TFs (GCN4, AT-hook, GAGA). Whereas individually these fragments are not functional, they might cooperate to form a trivalent complex with a target composite DNA.
The construct BR(Hk)GAGA was made following the same synthetic scheme as described for BR(Gly9)GAGA, involving the independent synthesis of an electrophilic GCN4/AT-hook module (BR(Hk)-Br), and its chemoselective coupling with a Cys side chain of the GAGA fragment not involved in the zinc finger complexation (see Scheme 3 below and the ESI†). The desired conjugate was obtained after reverse-phase HPLC purification in a reasonable overall yield of approximately 10%, and identified by ESI-MS.
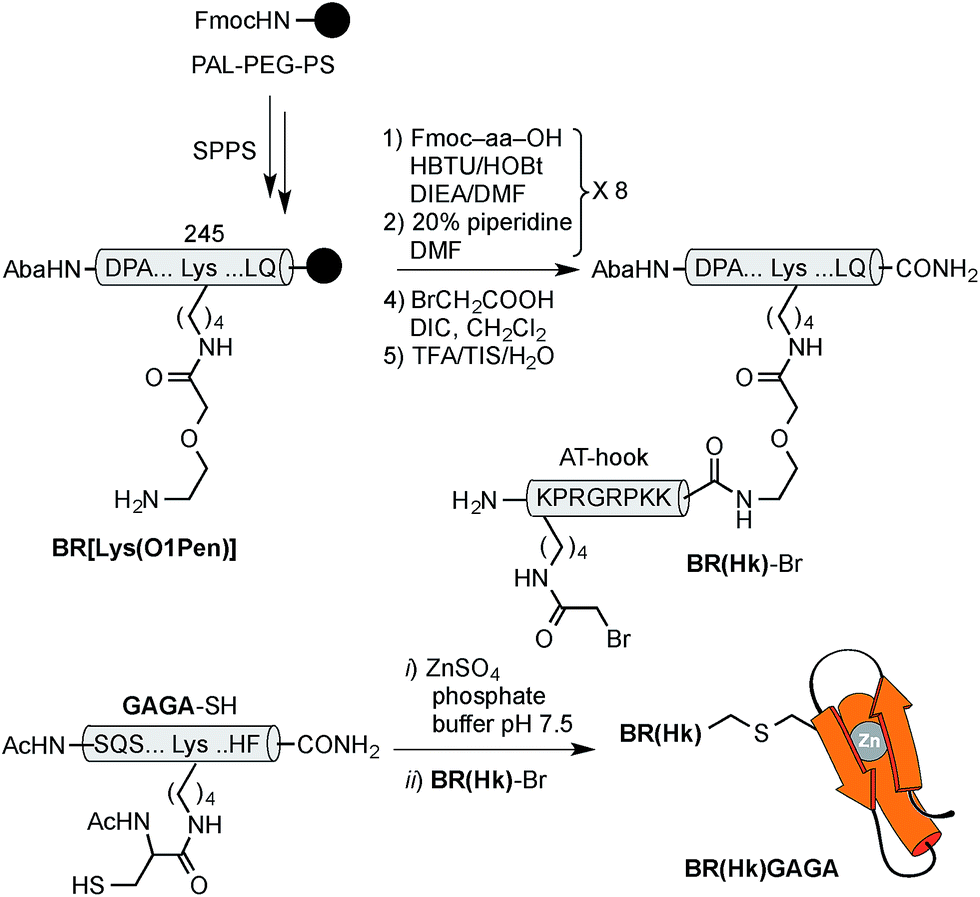 | ||
| Scheme 3 Top: synthesis of the GCN4/AT-hook chimera BR(Hk)-Br. See Scheme 1 for solid phase peptide synthesis of BR[Lys(O1Pen)]. Also note that the bromoacetyl unit was introduced in the side chain of the N-terminal lysine of the AT-hook, using Boc-Lys(Fmoc)-OH as orthogonal amino acid. Bottom: chemoselective modification of the GAGA Cys2His2 peptide in solution. | ||
In contrast with the results obtained with our original oligoglycine design, incubation of BR(Hk)GAGA with a ds-oligonucleotide featuring a composite sequence comprising the binding sites for the GCN4, the AT-hook, and the GAGA fragment (dsDNA A), led to clear EMSA slow-migrating bands (Fig. 3 top, panel a). This is fully consistent with the formation of a highly-stable peptide–DNA complex. However, incubation of BR(Hk)GAGA with a control DNA that does not contain the consensus GAGA binding site (dsDNA D), shows faint bands that indicate the formation of low-affinity complexes, presumably arising from weak binary interactions involving the GCN4 and the AT-hook modules (Fig. 3 top, panel b).13 Likewise, incubation with a second control oligonucleotide lacking the GCN4 binding site (dsDNA E), leads also to faint retardation bands, and only at high concentrations of the conjugate (Fig. 3 top, panel c). Therefore, these results confirm that the trifunctional construct presents and excellent selectivity for its composite tripartite site over potential bipartite competitors. A control oligonucleotide (dsDNA B), lacking the central A/T-rich, also gave rise to weaker complexes than with the consensus DNA A (Fig. 3 top, panel d), although the binding appears to be better than with DNAs D and E, probably because the highly charged AT-hook presents stabilizing electrostatic interactions with the DNA backbone.
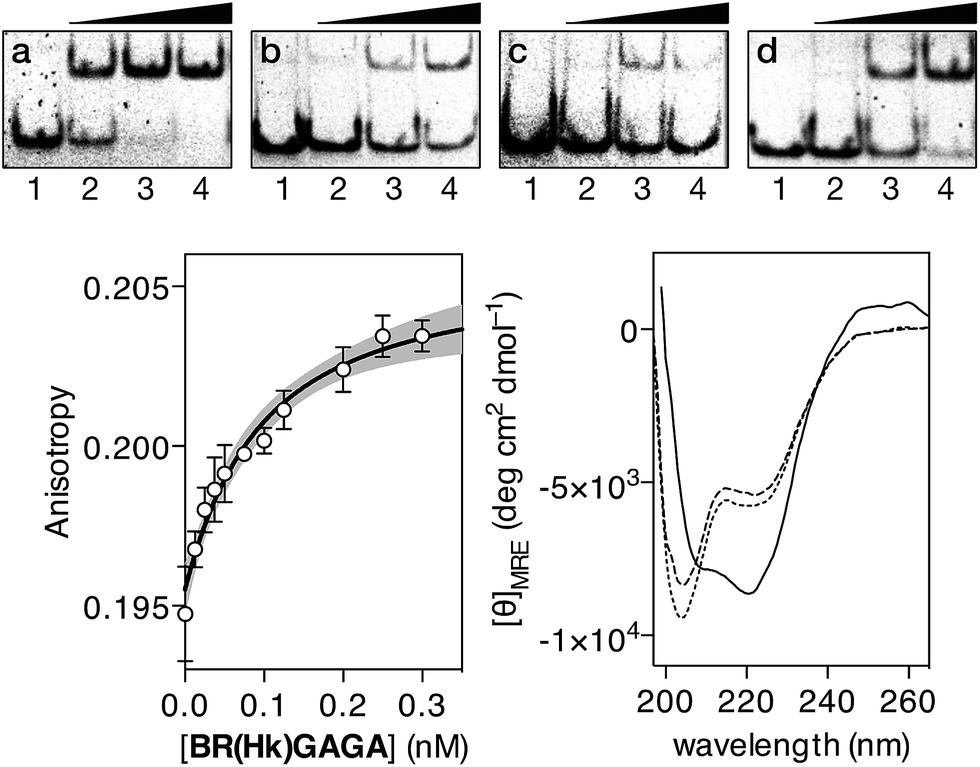 | ||
Fig. 3 EMSA DNA binding studies of BR(Hk)GAGA. In all cases, lanes 1–4: [BR(Hk)GAGA] = 0, 400, 600, 800 nM with (a) 75 nM of dsDNA A; (b) 75 nM of dsDNA D; (c) 75 nM of dsDNA E; (d) 75 nM of dsDNA B. Oligonucleotide sequences (only one strand shown): A: 5′-CGCG TCATAATTGAGAG CGC-3′; D: 5′-CGCG TCATAATTCGCGA CGC-3′; E: 5′-CGCG TGCTAATTGAGAG CGC-3′; B: 5′-CGCG TCATCAGCGAGAG CGC-3′. Experiments were carried out by PAGE on a 10% non-denaturing gel and 0.5× TBE buffer over 40 min at rt, and analyzed by staining with SyBrGold (Molecular Probes: 5 μL in 50 mL of TBE) for 10 min, followed by fluorescence visualization. Bottom left: fluorescence anisotropy titration of a 25 nM solution of TMR-A in the presence of competing non-specific calf thymus DNA (50 μM) and with increasing concentrations of BR(Hk)GAGA. The best fit to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model and the 95% confidence band of the fit (in grey) are also shown. Bottom right: circular dichroism of a 5 μM solution of BR(Hk)GAGA (dotted line), of the same solution after the addition of 1 equiv. of ZnSO4 (dashed line), and after the subsequent addition of 1 equiv. of the target dsDNA A (10 mM phosphate buffer pH 7.5 and 100 mM of NaCl; the contribution of the DNA to the CD spectrum has been subtracted for clarity). :1 binding model and the 95% confidence band of the fit (in grey) are also shown. Bottom right: circular dichroism of a 5 μM solution of BR(Hk)GAGA (dotted line), of the same solution after the addition of 1 equiv. of ZnSO4 (dashed line), and after the subsequent addition of 1 equiv. of the target dsDNA A (10 mM phosphate buffer pH 7.5 and 100 mM of NaCl; the contribution of the DNA to the CD spectrum has been subtracted for clarity). | ||
In order to quantify the DNA binding of our construct, we carried out fluorescence anisotropy titrations with selected fluorescently-labeled oligonucleotides. Thus, titrations using a tetramethyl rhodamine (TMR)-labeled ds-oligonucleotide containing the target composite site (TMR-A) confirmed formation of a high affinity complex (KD ≈ 28 nM at rt), even in the presence of excess of competing calf thymus DNA (Fig. 3 bottom left). Importantly, titration with the ds-oligonucleotide B (TMR-B), which promoted the appearance of electrophoresis retarded bands at high concentrations, revealed a much weaker interaction (more than 500 times lower affinity, see the ESI†). This confirms that in the presence of excess of non-specific DNA, the designed hybrid shows exquisite selectivity for its target 13 base-pair composite tripartite binding site (TCAT·AATT·GAGAG). In agreement with the results obtained by EMSA, circular dichroism experiments revealed that addition of 1 equiv. of the target oligonucleotide A to a 5 μM solution of BR(Hk)GAGA promotes a significant increase in the ellipticity of the negative bands at 208 and 222 nm, consistent with the α-helical folding of the GCN4 BR (Fig. 3, bottom right).23,24
While the relatively large size of the synthetic construct might hinder its cellular internalization, we reasoned that the oligocationic character of its basic region and AT-hook units could be beneficial for the cellular transport.25 Indeed, a preliminary test with mammalian Vero cells using a tetramethylrhodamine (TMR) derivative of BR(Hk)GAGA (see the ESI†), led to bright emission inside cells, in a pattern consistent with endosomal localization (Fig. 4).26 This efficient cell internalization opens the door for cellular applications of these peptide chimeras.
 | ||
| Fig. 4 Fluorescence micrography of Vero cells. Brightfield images are superimposed to the red emission channel after incubation with 5 μM TMR-BR(Hk)GAGA for 30 min at 37 °C. | ||
To gain some structural insight in the complex between the conjugate BR(Hk)GAGA and the target DNA, we carried out a computational study using molecular mechanics (MM) calculations with the obminimize utility script of OpenBabel 2.3.1,27 and the UFF force field.28 Building on the structural data available for the DNA interaction of the parent GCN4 and GAGA proteins, we obtained a model for the interaction of the conjugate with the target sequence: TCAT·AATT·GAGAG. The resulting model is fully consistent with a tripartite major–minor–major groove interaction that involves a relatively large binding surface covering one face of the DNA (see Fig. 5). This type of non-natural DNA binding has not been previously described, and its discovery should open new and important opportunities in the field.
 | ||
| Fig. 5 Model obtained using MM calculations of the interaction between the tripartite construct and the target composite DNA sequence. The image on the right shows the interaction of the three modules along the DNA axis covering one side of the double helix. | ||
Conclusions
In summary, we have developed a novel DNA binding motif consisting of two DNA binding fragments of natural TFs connected via an AT-hook linker, which allows the selective recognition of designed, extended DNA sequences (up to 13 bp). The success of this design relies on the ability of the AT-hook moiety to act as a bidentate minor groove-anchoring device that delivers the DNA binding TF fragments to appropriate positions for insertion in their respective major grooves. The peptidic nature of the AT-hook allowed an easy installation of each of the DNA binding peptides at the C- and N-terminus of the anchor.The construct represents the first demonstration of an engineered synthetic DNA binder that reaches two consecutive major grooves across the minor groove. The tripartite (major–minor–major groove) recognition introduces a novel DNA binding motif that lacks a natural counterpart. This approach promises to be applicable to other DNA binding TF fragments addressing different sites, and introduces a novel way of targeting specific and long DNA sequences.
Acknowledgements
We are thankful for the support given by the Spanish grants SAF2013-41943-R, CTQ2013-43264-R and CTQ2013-49317-EXP, the Xunta de Galicia GRC2013-041, the ERDF, and the European Research Council (Advanced Grant No. 340055). Support of COST Action CM1105, COST CM1306 and the orfeo-cinqa network are kindly acknowledged. J. R. thanks the Xunta de Galicia for her PhD fellowship. All calculations were carried out at the CESGA. We thank Dr José Couceiro for his help with the cell internalization experiments.Notes and references
- C. W. Garvie and C. Wolberger, Mol. Cell, 2001, 8, 937 CrossRef CAS PubMed.
- (a) D. S. Latchman, Eukaryotic Transcription Factors, Elsevier, London, 2004 Search PubMed; (b) M. Ptashne, A Genetic Switch, Cell Press & Blackwell, 1992 Search PubMed.
- (a) H. C. Nelson, Curr. Opin. Genet. Dev., 1995, 5, 180 CrossRef CAS PubMed; (b) R. Moretti and A. Z. Ansari, Biochimie, 2008, 90, 1015 CrossRef CAS PubMed; (c) D. J. Segal and C. F. Barbas, Curr. Opin. Chem. Biol., 2000, 4, 34 CrossRef CAS PubMed; (d) L. Chen, Curr. Opin. Struct. Biol., 1999, 9, 48 CrossRef CAS PubMed.
- C. Vinson, A. Acharya and E. J. Taparowsky, Biochim. Biophys. Acta, 2006, 4, 1759 Search PubMed.
- A. Klug, Ann. Rev. Biochem., 2010, 79, 213 CrossRef CAS PubMed.
- (a) N. M. Luscombe, S. E. Austin, H. M. Berman and J. M. Thornton, Genome Biol., 2000, 1, 1 CrossRef PubMed; (b) B. F. Luisi, W. X. Xu, Z. Otwinowski, L. P. Freedman, K. R. Yamamoto and P. B. Sigler, Nature, 1991, 352, 497 CrossRef CAS PubMed.
- (a) M. E. Vázquez, A. M. Caamaño and J. L. Mascareñas, Chem. Soc. Rev., 2003, 32, 338 RSC; (b) E. Pazos, J. Mosquera, M. E. Vázquez and J. L. Mascareñas, ChemBioChem, 2011, 12, 1958 CrossRef CAS PubMed; (c) C. Y. Majmudar and A. K. Mapp, Curr. Opin. Chem. Biol., 2005, 9, 467 CrossRef CAS PubMed.
- (a) P. P. Pandolfi, Oncogene, 2001, 20, 3116 CrossRef CAS PubMed; (b) R. Pollock, M. Giel, K. Linher and T. Clackson, Nat. Biotechnol., 2002, 20, 729 CrossRef CAS PubMed; (c) C. Denison and T. Kodadek, Chem. Biol., 1998, 5, R129 CrossRef CAS PubMed; (d) A. K. Mapp, Org. Biomol. Chem., 2003, 1, 2217 RSC.
- (a) J. W. Højfeldt, A. R. Van Dyke and A. K. Mapp, Chem. Soc. Rev., 2011, 40, 4286 RSC; (b) H.-D. Arndt, K. E. Hauschild, D. P. Sullivan, K. Lake, P. B. Dervan and A. Z. Ansari, J. Am. Chem. Soc., 2003, 125, 13322 CrossRef CAS PubMed.
- (a) N. J. Zondlo and A. Schepartz, J. Am. Chem. Soc., 1999, 121, 6938 CrossRef CAS; (b) J. K. Montclare and A. Schepartz, J. Am. Chem. Soc., 2003, 125, 3416 CrossRef CAS PubMed; (c) M. Zhang, B. Wu, J. Baum and J. W. Taylor, J. Pept. Res., 2000, 55, 398 CrossRef CAS PubMed; (d) T. Morii, S.-I. Sato, M. Hagihara, Y. Mori, K. Imoto and K. Makino, Biochemistry, 2002, 41, 2177 CrossRef CAS PubMed.
- (a) R. V. Talanian, C. J. McKnight and P. S. Kim, Science, 1990, 249, 769 CAS; (b) C. R. Palmer, S. S. Sloan, J. C. Adrian, B. Cuenoud, D. N. Paolella and A. Schepartz, J. Am. Chem. Soc., 1995, 117, 8899 CrossRef CAS; (c) M. Ueno, A. Murakami, K. Makino and T. Morii, J. Am. Chem. Soc., 1993, 115, 12575 CrossRef CAS; (d) T. Morii, J. Yamane, Y. Aizawa, K. Makino and Y. Sugiura, J. Am. Chem. Soc., 1996, 118, 10011 CrossRef CAS; (e) T. Morii, Y. Saimei, M. Okagami, K. Makino and Y. Sugiura, J. Am. Chem. Soc., 1997, 119, 3649 CrossRef CAS; (f) A. Mazumder, A. Maiti, K. Roy and S. Roy, ACS Chem. Biol., 2012, 7, 1084 CrossRef CAS PubMed; (g) J. Mosquera, A. Jiménez-Balsa, V. I. Dodero, M. E. Vázquez and J. L. Mascareñas, Nat. Commun., 2013, 4, 1874 CrossRef PubMed; (h) Y. Ruiz Garcia, J. Zelenka, Y. V. Pabon, A. Iyer, M. Buděšínsky, T. Kraus, C. I. E. Smith and A. Madder, Org. Biomol. Chem., 2015, 13, 5273 RSC; (i) G. A. Bullen, J. H. R. Tucker and A. F. A. Peacock, Chem. Commun., 2015, 51, 8130 RSC; (j) L. L. G. Carrette, T. Morii and A. Madder, Eur. J. Org. Chem., 2014, 2883 CrossRef CAS.
- (a) J. B. Blanco, M. E. Vazquez, L. Castedo and J. L. Mascareñas, ChemBioChem, 2005, 6, 2173 CrossRef CAS PubMed; (b) M. E. Vazquez, A. M. Caamaño, J. Martinez-Costas, L. Castedo and J. L. Mascareñas, Angew. Chem., Int. Ed., 2001, 40, 4723 CrossRef CAS; (c) M. I. Sanchez, J. Mosquera, M. E. Vázquez and J. L. Mascareñas, Angew. Chem., Int. Ed., 2014, 53, 9917 CrossRef CAS PubMed; (d) J. B. Blanco, O. Vázquez, J. Martínez-Costas, L. Castedo and J. L. Mascareñas, Chem.–Eur. J., 2005, 11, 4171 CrossRef CAS PubMed; (e) J. B. Blanco, J. Marínez-Costas, L. Castedo and J. L. Mascareñas, Chem. Biol., 2003, 10, 713 CrossRef CAS PubMed.
- J. Rodríguez, J. Mosquera, J. R. Couceiro, M. E. Vázquez and J. L. Mascareñas, Chem. Sci., 2015, 6, 4767 RSC.
- C. A. Gersbach, T. Gaj and C. F. Barbas III, Acc. Chem. Res., 2014, 47, 2309 CrossRef CAS PubMed.
- (a) P. B. Dervan and B. S. Edelson, Curr. Opin. Struct. Biol., 2003, 13, 284 CrossRef CAS PubMed; (b) J. W. Trauger, E. E. Baird and P. B. Dervan, Chem. Biol., 1996, 3, 369 CrossRef CAS PubMed; (c) J. J. Kelly, E. E. Baird and P. B. Dervan, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 6981 CrossRef CAS PubMed.
- R. V. Talanian, C. J. McKnight, R. Rutkowski and P. S. Kim, Biochemistry, 1992, 31, 6871 CrossRef CAS PubMed.
- A. Iyer, D. Van Lysebetten, Y. Ruiz García, B. Louage, B. G. De Geest and A. Madder, Org. Biomol. Chem., 2015, 13, 3856 CAS.
- (a) O. Vazquez, M. E. Vazquez, J. B. Blanco-Canosa, L. Castedo and J. L. Mascareñas, Angew. Chem., Int. Ed., 2007, 46, 6886 CrossRef CAS PubMed; (b) M. I. Sánchez, O. Vazquez, M. E. Vázquez and J. L. Mascareñas, Chem.–Eur. J., 2013, 19, 9923 CrossRef PubMed.
- (a) T. E. Ellenberger, C. J. Brandl, K. Struhl and S. C. Harrison, Cell, 1992, 71, 1223 CrossRef CAS PubMed (PDB ID: 1YSA); (b) P. Köning and T. J. Richmond, J. Mol. Biol., 1993, 233, 139 CrossRef PubMed.
- J. G. Omichinski, P. V. Pedone, G. Felsenfeld, A. M. Gronenborn and G. M. Clore, Nat. Struct. Biol., 1997, 4, 123 CrossRef (PDB ID: 1YUI).
- J. Rodríguez, J. Mosquera, O. Vázquez, M. E. Vázquez and J. L. Mascareñas, Chem. Commun., 2014, 50, 2258 RSC.
- (a) J. R. Huth, C. A. Bewley, M. S. Nissen, J. N. Evans, R. Reeves, A. M. Gronenborn and G. M. Clore, Nat. Struct. Mol. Biol., 1997, 4, 657 CrossRef CAS (PDB ID: 2EZD and 2EZF); (b) E. Fonfría-Subirós, F. Acosta-Reyes, N. Saperas, J. Pous, J. A. Subirana and J. L. Campos, PLoS One, 2012, 7, e37120 Search PubMed (PDB ID: 3UXW).
- A. D. Frankel, J. M. Berg and C. O. Pabo, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 4841 CrossRef CAS.
- A. Jiménez, E. Pazos, B. Martínez-Albardonedo, J. L. Mascareñas and M. E. Vazquez, Angew. Chem., Int. Ed., 2012, 51, 8825 CrossRef PubMed.
- (a) F. Milletti, Drug Discovery Today, 2012, 17, 85 CrossRef PubMed; (b) B. Gupta, T. S. Levchenko and V. P. Torchilin, Adv. Drug Delivery Rev., 2005, 57, 637 CrossRef CAS PubMed; (c) E. A. Goun, T. H. Pillow, L. R. Jones, J. B. Rothbard and P. A. Wender, ChemBioChem, 2006, 7, 149 CrossRef PubMed; (d) J. Mosquera, M. I. Sánchez, J. Valero, J. de Mendoza, M. E. Vázquez and J. L. Mascareñas, Chem. Commun., 2015, 51, 4811 RSC; (e) O. Vázquez, J. B. Blanco-Canosa, M. E. Vázquez, J. Martínez Costas, L. Castedo and J. L. Mascareñas, ChemBioChem, 2008, 9, 2822 CrossRef PubMed.
- A number of strategies are available for promoting endosomal escape of internalized compounds: (a) A. Erazo-Oliveras, N. Muthukrishnan, R. Baker, T. Y. Wang and J. P. Pellois, Pharmaceuticals, 2012, 5, 1177 CrossRef CAS PubMed; (b) A. Erazo-Oliveras, K. Najjar, L. Dayani, T.-Y. Wang, G. A. Johnson and J.-P. Pellois, Nat. Methods, 2014, 11, 861 CrossRef CAS PubMed.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Peptide synthesis, full experimental procedures and analytical data of the peptides and products obtained. See DOI: 10.1039/c6sc00045b |
| This journal is © The Royal Society of Chemistry 2016 |